

Abstract
Insulin has been used to treat diabetes for almost 100 years, yet current rapid-acting insulin formulations do not have sufficiently fast pharmacokinetics to maintain tight glycemic control at mealtimes. Dissociation of the insulin hexamer, the primary association state of insulin in rapid-acting formulations, is the rate limiting step that leads to delayed onset and extended duration of action. A formulation of insulin monomers would more closely mimic endogenous post-prandial insulin secretion, but monomeric insulin is unstable in solution using present formulation strategies and rapidly aggregates into amyloid fibrils. Here, we implement high-throughput controlled radical polymerization techniques to generate a large library of acrylamide carrier/dopant copolymer (AC/DC) excipients designed to reduce insulin aggregation. Our top performing AC/DC excipient candidate enabled the development of an ultrafast-absorbing insulin lispro (UFAL) formulation, which remains stable under stressed aging conditions for 25 ± 1 hours compared to 5 ± 2 hours for commercial fast-acting insulin lispro formulations (Humalog). In a porcine model of insulin-deficient diabetes, UFAL exhibited peak action at 9 ± 4 minutes whereas commercial Humalog exhibited peak action at 25 ± 10 minutes. These ultrafast kinetics make UFAL a promising candidate for improving glucose control and reducing burden for patients with diabetes.
Overline: DIABETES
One Sentence Summary:
Monomeric insulin stabilized with polymeric excipient exhibits increased stability and faster pharmacokinetics than current rapid-acting insulins.
Introduction
Over 40 million patients live with type 1 diabetes worldwide and rely on insulin replacement therapy through daily subcutaneous insulin injections or insulin infusion pumps. These patients are unable to produce the insulin required to promote cellular glucose uptake in response to meals and must deliver calculated insulin boluses at mealtimes to prevent glycemic excursions. Unfortunately, the pharmacokinetics of current insulin formulations do not mimic endogenous insulin secretion, which can reach peak concentrations in 30 minutes in a non-diabetic individual (1–3). Even current rapid-acting insulin analogues designed for mealtime boluses exhibit delayed onset of action of 20–30 minutes, peak action at 60–90 minutes, and a total duration of action of 3–4 hours (4–6). These kinetics are an outcome of the mixed association states of the insulin molecules in formulation. Commercial insulin formulations typically contain a mixture of insulin hexamers, dimers, and monomers. Whereas monomers are rapidly absorbed into the bloodstream after injection, dimers and hexamers are absorbed more slowly on account of their size, and must dissociate into monomers to become active (Fig. 1) (7–9). Further, the extended duration of insulin action can make controlling post-prandial glycemic excursions difficult and increases the risk of hypoglycemia, as insulin may remain on board even after the mealtime glucose load passes (10, 11).
Fig. 1. Scheme of absorption kinetics of the various association states of insulin.
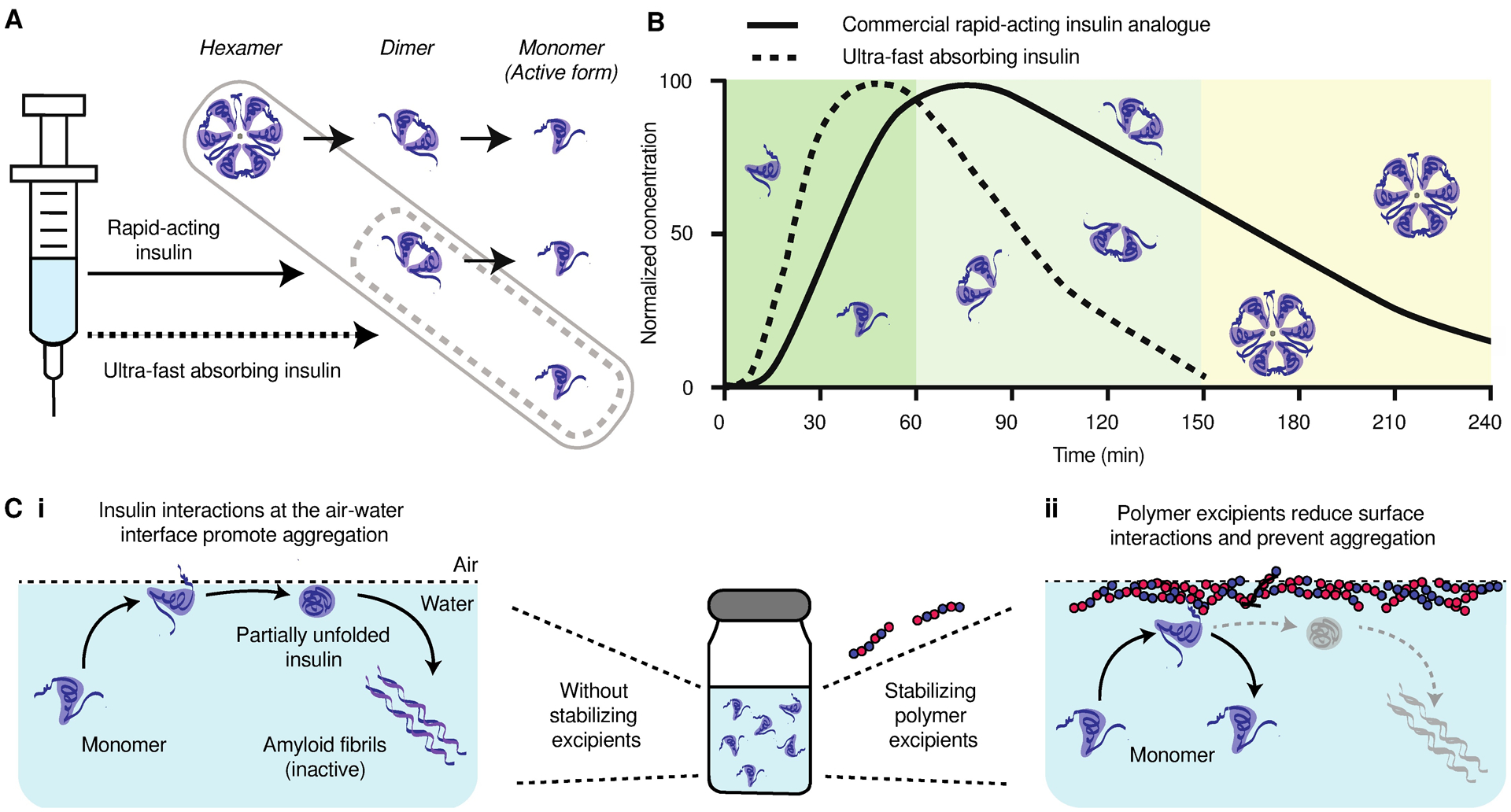
(A) Commercial rapid-acting insulin formulations contain a mixture of insulin hexamers, dimers, and monomers. Only the monomeric form of insulin is active, thus, the dissociation from the hexamer to the monomer is rate limiting for therapeutic action. An ultrafast insulin formulation would contain primarily insulin monomers and no insulin hexamers for rapid insulin absorption after subcutaneous administration. (B) A schematic illustrating the contribution of insulin hexamers, dimers, and monomers in commercial rapid-acting insulin formulations to the observed duration of insulin action when delivered subcutaneously in humans. Insulin monomers are absorbed in about 5–10 minutes, dimers are absorbed in 20–30 minutes, and hexamers can take 1–2 hours to be absorbed and result in prolonged insulin action accordingly. A primarily monomeric insulin formulation would reduce time to onset and result in shorter duration of insulin action for better management of blood glucose at mealtimes. (C) A hexamer-free ultrafast insulin formulation will face stability challenges due to the propensity for insulin monomers to aggregate into amyloid fibrils. (i) At the interface, exposure of hydrophobic domains during insulin-insulin interaction nucleate amyloid fiber formation. (ii) Stabilizing polymer excipients are drawn to the air-liquid interface, impeding the interfacial nucleation of insulin amyloidosis.
An insulin formulation that is absorbed rapidly from the subcutaneous space to more closely mimic endogenous post-prandial insulin secretion is needed to better control mealtime blood glucose. A monomeric insulin formulation would enable both faster onset and shortened duration of action, thus reducing the risk of post-prandial hypoglycemia by eliminating the subcutaneous depot of insulin hexamers (Fig. 1B). However, monomeric insulin is unstable in formulation and rapidly aggregates into amyloid fibrils, which are both inactive and immunogenic (12–14). Presently, zinc and phenolic preservatives are commonly used as excipients in insulin formulations because their propensity to promote insulin hexamer formation enables them to act as stabilizing agents (15, 16). It is critical to develop a new class of excipients that can improve insulin stability in the monomeric state to enable a viable ultrafast-acting insulin formulation.
Insulin aggregation typically is initiated at hydrophobic interfaces, such as the air-liquid interface, where monomers undergo partial unfolding upon adsorption and can nucleate amyloid fibril formation (Fig. 1C) (17–19). Hydrophobic moieties responsible for aggregation are typically shielded in the dimeric and hexameric association states, making the monomeric state most susceptible to aggregation (20). Current zinc-free methods for monomeric insulin stabilization have relied on shielding hydrophobic interactions by covalently or non-covalently attaching hydrophilic polymers such as poly(ethylene glycol) (PEG) or trehalose glycopolymers directly to insulin (21–24). These methods stabilize insulin in formulation but they lead to increased circulation time in vivo, which is undesirable for an ultrafast-acting insulin formulation.
An alternative approach to insulin stabilization exploits the propensity of amphiphilic polymers to occupy the interface, preventing insulin-interface interactions (Fig. 1C). Poloxamers are an example of polymer surfactants that have been used to improve the stability of commercial insulin formulations (Insuman U400, Sanofi-Aventis). Yet, these current excipients comprise a limited chemical space, exhibit a propensity to form micro-structures such as micelles in solution, and are susceptible to transitioning into gels at high concentrations, and as such, a stable ultrafast monomeric insulin formulation is still evasive. In this study, we aimed to synthesize a distinct class of excipients that can be used to enable the stable formulation of an ultrafast-acting monomeric insulin. These excipients are synthetic copolymers composed of a water-soluble carrier monomer, chosen to aid polymer solubility, and a functional dopant monomer, which affords the ability to screen a wide chemical space unexplored in current surfactant excipients. The dopant monomer is hypothesized to promote polymer-interface interactions, reducing insulin-insulin interactions at the interface and thus improving insulin stability. We used precision high-throughput synthesis with reverse additional fragmentation transfer (RAFT) polymerization to generate a library of over 100 acrylamide carrier/dopant copolymer (AC/DC) excipients. Here we demonstrate that top AC/DC excipient candidates enable the stable formulation of monomeric insulin lispro and that this ultrafast-absorbing lispro (UFAL) formulation exhibits pharmacokinetics that are 2-fold faster than commercial fast-acting insulin formulations in a porcine model of insulin-deficient diabetes.
Results
High-throughput synthesis of polyacrylamide library
A library of AC/DC excipients was synthesized combinatorially through statistical copolymerizations of water-soluble carrier monomers and functional dopant monomers (Fig. 2). The carrier monomers were the predominant species and were responsible for both maintaining solubility and providing an inert barrier to prevent insulin-insulin interactions. The functional dopants copolymerized at lower weight percentages were incorporated statistically throughout the resulting copolymer. These dopants are selected by design to promote either polymer-interface interactions or polymer-insulin interactions. The library targets a degree of polymerization (DP) of 50 for the copolymers, resulting in molecular weights similar to insulin and well below the glomerular filtration threshold for synthetic polymers (25).
Fig. 2. Scheme of polymer excipient library design.
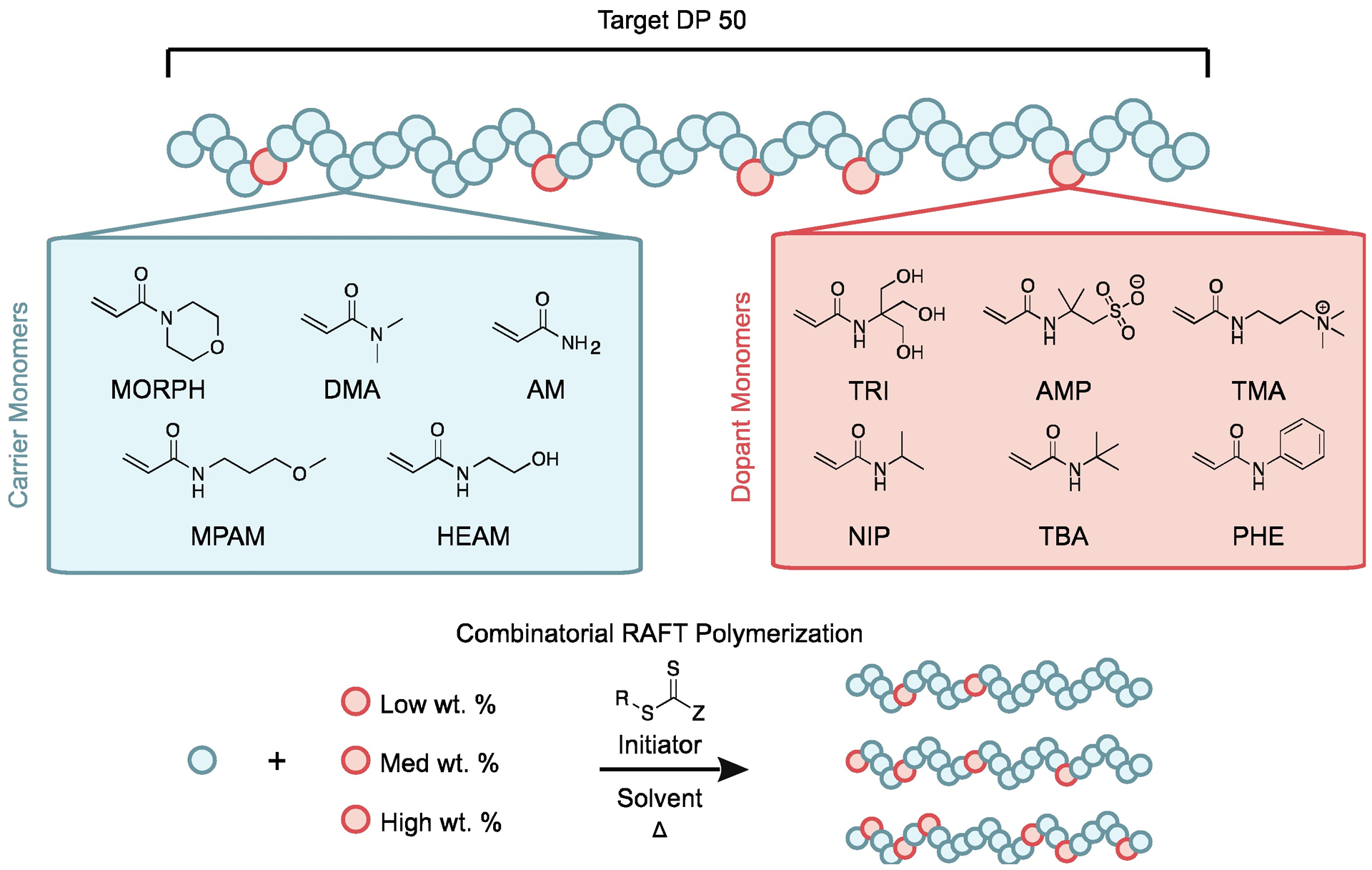
A library of statistical acrylamide copolymers with a target degree of polymerization (DP) of 50 was synthesized through controlled copolymerization using RAFT. Copolymer combinations consist of one carrier monomer: 4-acryloylmorpholine (MORPH), N-(3-methoxypropyl)acrylamide (MPAM), N,N-dimethylacrylamide (DMA), N-hydroxyethyl acrylamide (HEAM), or acrylamide (AM). Each copolymer also contains one dopant monomer: N-[tris(hydroxymethyl)-methyl]acrylamide (TRI), 2-acrylamido-2-methylpropane sulfonic acid (AMP), (3-acrylamidopropyl)trimethylammonium chloride (TMA), N-isopropylacrylamide (NIP), N-tert-butylacrylamide (TBA), or N-phenylacrylamide (PHE). Each carrier-dopant combination was repeated at low, medium, and high dopant loadings: NIP at 6.7, 13.3, and 20 wt.%; TRI at 5, 10, and 15 wt.%; and AMP, TMA, TBA, and PHE at 3.3, 6.7, and 10 wt.%.
The library was generated through parallel synthesis with a Chemspeed Swing XL Auto Synthesizer, a liquid handling robot in an inert environment. RAFT polymerization was implemented because it affords precise copolymerization stoichiometry, low dispersity, and controlled molecular weights for a wide scope of monomers. Polyacrylamide derivatives were used for both the carrier and dopant monomers due to the scope and availability of commercial water-soluble monomers (carriers) and functional monomers (dopants) and polymeric stability. Although monomeric acrylamide derivatives often exhibit acute toxicities, when properly purified from their monomeric precursors, polyacrylamide derivatives demonstrate a high degree of biocompatibility (26, 27). Moreover, the reactivity ratios between the various acrylamide monomers are close to 1, yielding copolymers with little to no dopant gradient composition. Carrier monomers included acrylamide (AM), N-hydroxyethyl acrylamide (HEAM), N,N-dimethylacrylamide (DMA), 4-acryloylmorpholine (MORPH), and N-(3-methoxypropyl)acrylamide (MPAM) because they are nonionic and water soluble (ordered in increasing hydrophobicity). Dopant monomers included N-[tris(hydroxymethyl)methyl]acrylamide (TRI), 2-acrylamido-2-methylpropane sulfonic acid (AMP), (3-acrylamidopropyl)trimethylammonium chloride (TMA), N-isopropylacrylamide (NIP), N-tert-butylacrylamide (TBA), and N-phenylacrylamide (PHE). These functional dopants could be further classified into hydrogen bonding (TRI), ionic (AMP, TMA), hydrophobic (NIP, TBA), and aromatic (PHE) monomers on the basis of their chemical composition.
A library of 90 AC/DC excipients was synthesized through the combinatorial copolymerization of carrier and dopant monomers at each of three different compositions for a given carrier-dopant pair. NIP was copolymerized at 6.7, 13.3, or 20 weight percentage (wt.%). TRI was copolymerized at 5, 10, or 15 wt.%. AMP, TMA, TBA, and PHE were copolymerized at 3.3, 6.7, or 10 wt.%. These values were selected to maximize dopant loading while yielding functional copolymers with lower critical solution temperature (LCST) values above 37 °C to ensure they would remain soluble at all relevant temperatures. Polymers were characterized by nuclear magnetic resonance (NMR) and size exclusion chromatography (SEC) (tables S1 and S2, figs. S1 and S2). Although RAFT polymerization affords many synthetic advantages, it yields polymers with a reactive trithiocarbonate chain transfer agent (CTA) attached at the Z-terminus. Accordingly, the CTA moiety on the synthesized AC/DC excipients was removed prior to utilization of the copolymers in subsequent assays to ensure their inertness.
High-throughput screen for insulin stabilizing excipient
After generating the library of AC/DC excipients, each polymer’s potential as a stabilizing excipient for insulin was evaluated using an absorbance-based stressed aging assay, in which destabilized insulin aggregates scatter light and increase the absorbance of the solution. Time to aggregation in these assays is defined as a 10% increase in absorbance of the formulation (21). Recombinant insulin was formulated in phosphate buffered saline (PBS) at standard formulation concentrations (100 U/mL; 3.4 mg/mL) and tested with (i) no polymer excipients, (ii) pluronic L-61 (the commercially available polymer that is most similar both chemically and physically to Poloxamer 171 used in InsumanU400), (iii) 1 mg/mL AC/DC excipients, or (iv) 10 mg/mL AC/DC excipients. Recombinant insulin controls without polymer excipient aggregated in 13 ± 8 hours in this assay. Formulation with pluronic L-61 (1 mg/mL) prolonged aggregation to 27 ± 2 hours, demonstrating efficacy of the commercial polymer as an excipient to prevent insulin aggregation. The use of water-soluble carrier homopolymer excipients (1 mg/mL) had no impact on insulin stability (Fig. 3A), demonstrating that free hydrophilic polymers are not sufficient to prevent insulin aggregation. This finding is supported by previous work showing that other hydrophilic polymers such as PEG do not improve insulin stability (21).
Fig. 3. Recombinant insulin stability screen with polymer excipient library.
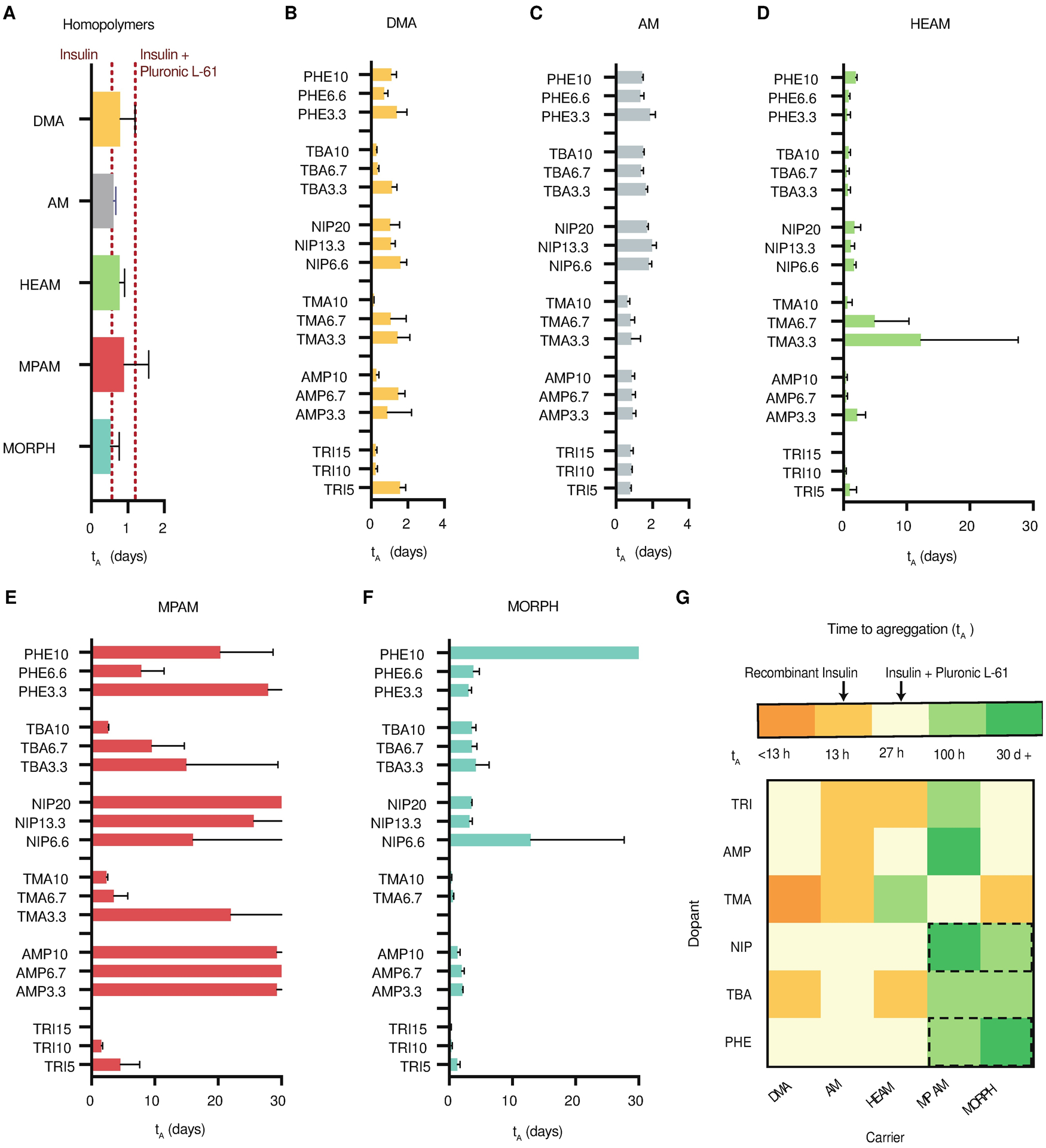
Time to aggregation (tA) of recombinant insulin (100 U/mL) formulated with (A) carrier homopolymers (0.1 wt.%) or a Pluronic L-61 control (0.1 wt.%), which has similar composition to Poloxamer 171 used in commercial insulin formulations. Carrier-dopant polymers (0.1 wt.%) with (B) DMA, (C) AM, (D) HEAM, (E) MPAM, and (F) MORPH carriers. Dopants and target weight percentages are listed on the y-axis. (G) Heatmap of the top performing excipient (the largest tA) for each carrier-dopant combination. Dotted black rectangles indicate the top dopant-carrier combinations, which were selected for further screening. These assays assess the aggregation of proteins in formulation over time during stressed aging (continuous agitation at 37 °C by monitoring changes in absorbance at 540 nm). Data shown are average time to aggregation (n=3; mean ± s.d.) where aggregation is defined as a 10% increase in absorbance.
Insulin stability when formulated with AC/DC excipients was highly chemistry-dependent. Each AC/DC excipient was formulated with insulin and stability was tested for up to one month (Fig. 3, B to G, table S3). Formulations comprising AC/DC excipients with MPAM and MORPH carrier chemistries demonstrated the overall highest improvement of insulin stabilization, especially when combined with NIP, TBA, and PHE dopants. Although many carrier-dopant combinations demonstrated long-term stability at 1 wt.% formulation concentrations, we sought to engineer copolymers capable of stabilizing insulin at minimal concentrations in formulation. AC/DC excipients comprising MPAM-PHE, MPAM-TBA, MPAM-TRI, and MORPH-TBA (0.1 wt.%) stabilized insulin for over 100 hours of stressed aging (Fig. 3G). These formulations are therefore 7-fold more stable than recombinant insulin alone and 3-fold more stable than formulations containing pluronic L-61. Moreover, AC/DC excipients comprising MPAM-NIP, MPAM-AMP, and MORPH-PHE (0.1 wt.%) stabilized insulin for 30 days of stressed aging (Fig. 3G), at which point the assay was terminated. These formulations are 50-fold more stable than insulin alone, and 24-fold more stable than formulations containing pluronic L-61. These select carriers and dopants are the most hydrophobic amongst the monomers screened, suggesting that amphiphilic water-soluble copolymers are most effective at preventing insulin aggregation.
Stabilization of monomeric insulin with refined screen
Based on the initial recombinant insulin stability screen, copolymers comprising MPAM or MORPH carriers with NIP or PHE dopants demonstrated the most promise as candidates for stabilizing monomeric insulin. Previous work by our group demonstrated that the equilibrium between insulin association states can be shifted by altering formulation excipients, where a formulation that is approximately 70% monomers can be achieved with formulation of zinc-free lispro with glycerol and phenoxyethanol (24). This formulation favors the insulin monomer and completely dissociates the insulin hexamer. Representative SEC traces and multiangle light scattering (MALS) of predominantly hexameric Humalog and predominantly monomeric zinc-free UFAL demonstrate the association states of insulin in formulation (fig. S3, Fig. 4A). However, insulin monomers are unstable in formulation and require additional stabilizing excipients to be viable for translation. Further, it will likely be prudent to use the lowest concentration of copolymer excipient possible to reduce chronic exposure to the excipient with frequent insulin use typical of diabetes management.
Fig. 4. Stabilized ultrafast-absorbing lispro (UFAL) formulation using top performing AC/DC polymer excipients.
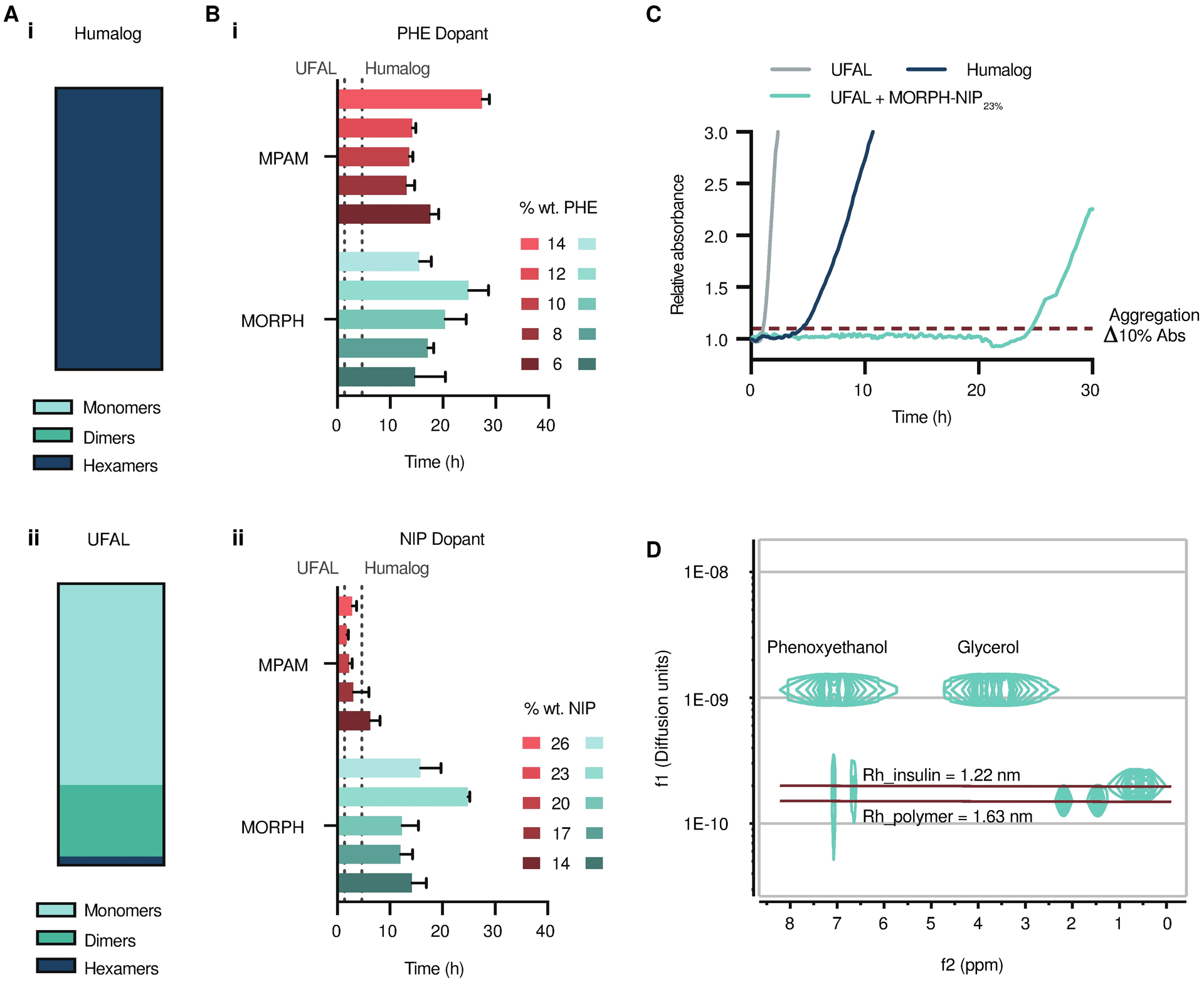
(A) Insulin association states in (i) Humalog and (ii) UFAL as determined by multiangle light scattering (MALS). (B) UFAL stability screen with polymer excipient library. Time to aggregation of UFAL (100 U/mL) formulated with polymer excipients from the second screen (0.01 wt.%). AC/DC polymer excipients were comprised of MPAM and MORPH carrier polymers with varied weight percent of dopants (i) PHE or (ii) NIP. (C) Representative absorbance traces showing UFAL stability when formulated with top performing candidate MORPH-NIP23% compared to controls of UFAL with no polymer excipient and Humalog. These assays assess the aggregation of proteins in formulation over time during stressed aging (continuous agitation at 37°C) by monitoring changes in transmittance at 540 nm. Data shown are average time to aggregation (n=3; mean ± s.d.) where aggregation is defined as a 10% increase in absorbance. (D) Diffusion-ordered NMR spectroscopy (DOSY) of UFAL with top performing polymer excipient MORPH-NIP23%. DOSY provides insight into the insulin association state and the insulin and polymer rates of diffusion in formulation.
To address this need, a second library of AC/DC excipients was synthesized to evaluate additional carrier-dopant ratios with our top performing candidate monomers: (i) MPAM and MORPH as carriers, and (ii) NIP and PHE as dopants. Standard synthesis practices were implemented to generate this secondary library, which consisted of copolymers at DP 50 with MORPH or MPAM as carriers and either (i) NIP loaded at 14, 17, 20, 23, or 26 wt.%, or (ii) PHE loaded at 6, 8, 10, 12, or 14 wt.%., respectively, via SEC and 1H NMR spectroscopy (fig. S4, table S4).
Using the AC/DC excipients synthesized in the second screen, UFAL formulations were prepared with 0.01 wt.% (0.1 mg/mL) copolymer excipient and insulin aggregation was assessed under stressed conditions using the same assay as the initial screen (table S5). Humalog, the commercial formulation of insulin lispro, aggregated under these conditions within 6 hours. UFAL without AC/DC excipients aggregated in 1.3 ± 0.3 hours, demonstrating the severe instability of the insulin monomer in solution. All UFAL formulations stabilized with MORPH-PHE or MPAM-PHE AC/DC excipients exhibited stabilities to stressed aging at least equivalent to commercial Humalog. Copolymers comprising MPAM with 14 wt.% PHE (MPAM-PHE14%) and MORPH with 12 wt.% PHE (MORPH-PHE12%) were among the top candidates, extending UFAL formulation stability to 27 ± 2 hours and 25 ± 5 hours, respectively (Fig. 4, B and C). MPAM-NIP copolymers demonstrated limited efficacy in stabilizing the monomeric insulin; however, MORPH-NIP copolymers extended monomeric insulin stability compared to Humalog. Indeed, copolymers comprising MORPH with 23 wt.% NIP (MORPH-NIP23%) extended UFAL formulation stability to over 25 ± 1 hours. The top candidate AC/DC excipients after the second screen were MPAM-PHE14%, MORPH-PHE12%, and MORPH-NIP23%. While these copolymers demonstrated high efficacy, MPAM-PHE14% and MORPH-PHE12% also demonstrated decreased solubility and LCST-like phase separation behavior at physiological temperature when present at higher concentrations. Thus, MORPH-NIP23% was chosen as the top candidate used to stabilize our UFAL formulation in subsequent in vivo studies. In vitro and in vivo bioactivity assays were used to corroborate the transmittance data and confirm UFAL integrity before and after aging. UFAL showed no loss in activity after 12 h of stressed aging in either the cellular assay for phosphorylation of Ser473 on AKT or when lowering blood glucose concentration in diabetic rats (fig. S5).
To verify that formulation with MORPH-NIP23% does not alter the lispro association state equilibrium away from the monomer form, NMR diffusion ordered spectroscopy (DOSY) was used (Fig. 4D). NMR DOSY indicates the diffusion rate of lispro under formulation conditions (100 U/mL lispro, 2.6 wt.% glycerol, 0.85 wt.% phenoxyethanol, and 0.1 wt.% MORPH-NIP23%) has a diffusion rate of 2.0 × 10−10 m2 s−1. This diffusion rate corresponds to a hydrodynamic radius of 1.2 nm, which is in agreement with the experimental hydrodynamic radius of the insulin monomer previously reported (28). NMR DOSY also provides insight into the stabilization mechanism of our polymer excipients. MORPH-NIP23% diffuses at a slower rate than insulin, suggesting that the mechanism of stabilization is not related to excipient-insulin complexation and co-diffusion. These data support our hypothesis that copolymer-interface interactions are the primary mechanism driving monomeric insulin stabilization in formulation.
Cytotoxicity and biocompatibility of polymer excipients
Because Morph-NIP23% is a new chemical entity, we sought to assess potential toxicity of the polymer and biocompatibility of the UFAL formulation. Cytotoxicity experiments performed in NIH/3T3 mouse fibroblasts demonstrate our new copolymer excipient is not toxic at doses an order of magnitude higher than those used in insulin formulations (fig. S6). Initial biocompatibility studies with UFAL compared to Humalog in diabetic rats also corroborate the cytotoxicity results, indicating that UFAL should be well tolerated (fig. S7). Two groups of diabetic rats (n=5) received a single daily dose of either Humalog (1.5 U/kg) or UFAL (1.5 U/kg) for 7 consecutive days. Blood was taken on day 0 and on day 7 and blood chemistry was evaluated to assess common markers of liver and kidney health. Liver toxicity was assessed through measurement of alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphotase (ALP), and bilirubin. Kidney toxicity was evaluated by examining creatinine and blood urea nitrogen (BUN) levels. Some markers elevated outside of the normal range for healthy rats were attributable to the diabetic phenotype. No differences were observed between Humalog and UFAL groups at either the Day 0 or Day 7 timepoint.
Pharmacokinetics and pharmacodynamics of UFAL formulation in diabetic pigs
To assess the ultrafast potential of the monomeric insulin formulations we conducted pharmacokinetic studies in a porcine model of insulin-deficient diabetes. Fasted diabetic pigs were treated with either commercial Humalog or UFAL (100 U/mL lispro, 2.6 wt.% glycerol, 0.85 wt.% phenoxyethanol, and 0.1 wt.% MORPH-NIP23%) at a dose of 2 to 4 U of insulin lispro, depending on the insulin sensitivity of each pig. Pigs had a starting blood glucose concentration between 330–430 mg/dL and insulin doses were chosen to reduce blood glucose to about 100 mg/dL. The insulin dose given to each pig was consistent between treatment groups and blood glucose depletion was similar in both Humalog and UFAL treatments (Fig. 5A, fig. S8). Plasma concentrations of lispro were measured over time by enzyme-linked immunosorbent assay (ELISA) to assess pharmacokinetics following subcutaneous injection of each of the treatment groups. No difference in overall exposure from the area under the curve (AUC210) between groups was observed (Fig. 5C). Percent exposure at various time points was analyzed by looking at the AUCt/AUC210. This analysis shows increased exposure for UFAL compared to Humalog at 10 min and 20 min timepoints (Fig. 5, D to I).
Fig. 5. Pharmacokinetics and pharmacodynamics of monomeric insulin in diabetic pigs.
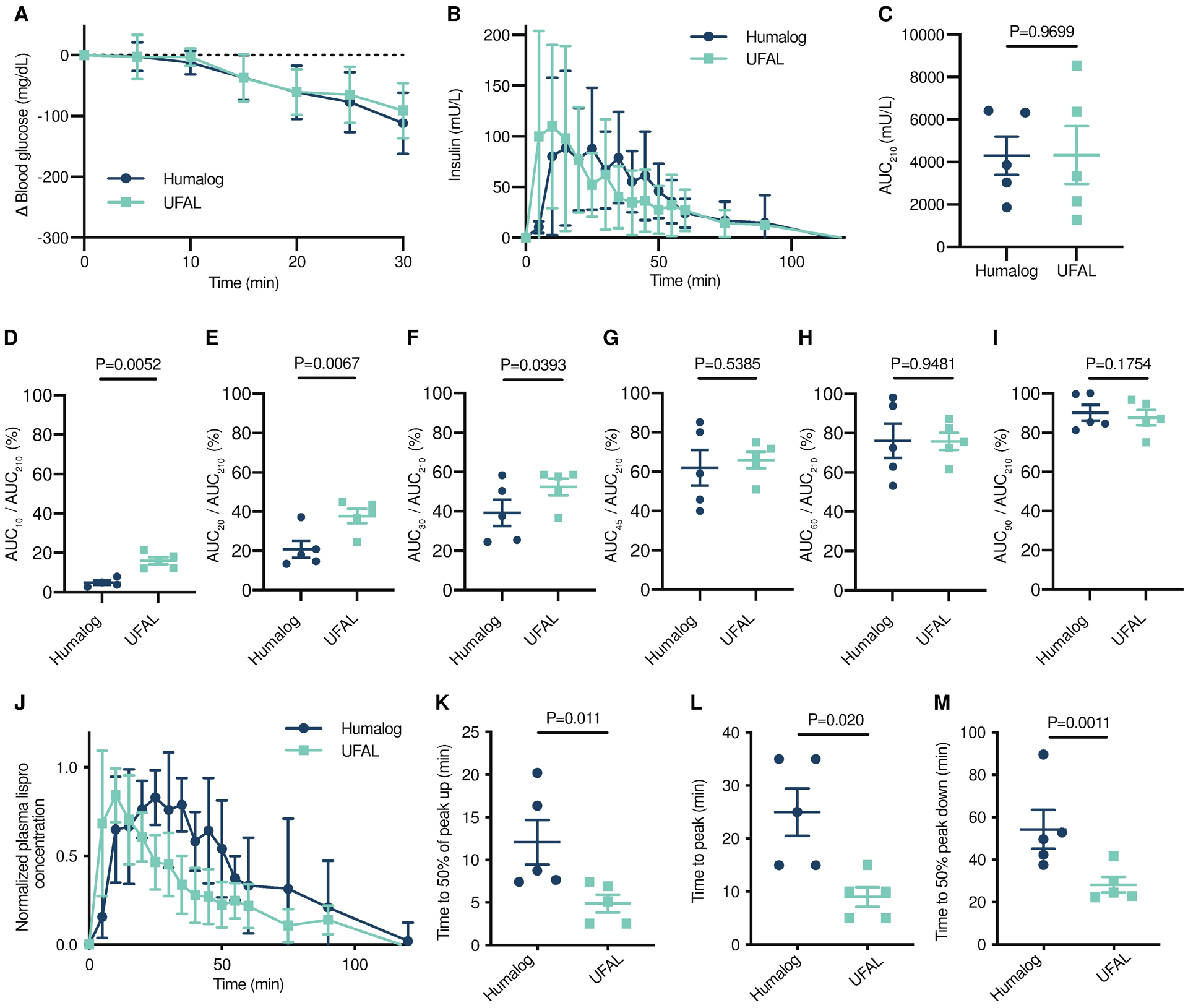
Diabetic female pigs received subcutaneous administration of therapies comprising either commercial Humalog or UFAL formulated with polymer. Pigs were dosed with insulin according to their individual insulin sensitivities to decrease their blood glucose by about 200 mg/dL. (A) Blood glucose measurements in pigs after insulin dosed subcutaneously. (B) Pharmacokinetics of insulin lispro in mU/L following s.c. injection. (C) Total exposure represented by area under the curve for 210 minutes. (D-I) Percent exposure at various time points (AUCt/AUC210). (J) Pharmacokinetics for each pig were individually normalized to peak concentrations and normalized values were averaged for lispro concentration for each treatment group. (K) Time to reach 50% of peak lispro concentration (onset). (L) Time to reach peak lispro concentration. (M) Time for lispro depletion to 50% of peak concentration. (A,B,J) Error bars indicate mean ± s.d. with n=5 for all groups. (D-I) Error bars indicate mean ± s.e.m. with n=5 for all groups. Bonferroni post-hoc tests were performed to account for comparisons of multiple individual exposure timepoints and significance and alpha was adjusted (alpha = 0.008). (C, K-M) Error bars indicate mean ± s.e.m. with n=5 for all groups (alpha=0.05). Statistical significance was determined by restricted maximum likelihood (REML) repeated measures mixed model.
Mean residence time (MRT) is a commonly reported for formulation pharmacokinetics. MRT is commonly described as area under the moment curve (AUMC) divided by area under the curve (AUC) for intravenous injections; however, when drugs are administered subcutaneously the mean absorption time (MAT) must also be considered. When there is an absorption phase, AUMC/AUC = MAT + MRT. Here we would be interested in the contribution of MAT, as this would indicate differences in absorption rate. The ratio of the area under the moment curve (AUMC) to the area under the curve for the pharmacokinetic plot (AUMC/AUC) was calculated and plotted, showing no difference between UFAL and Humalog treatment (fig. S9). This is not surprising, as we would expect the clearance rate from the blood to be similar for both Humalog and UFAL (both are insulin lispro) and the magnitude of MAT in comparison to MRT would be small, thereby masking differences between formulations.
Alternatively, exposure metrics are commonly reported for fast-acting insulin formulations to describe the formulation pharmacokinetics (5, 29–32). The “time-to-onset” rate of fast-acting insulins is often determined using two metrics: (i) time-to-50% of the normalized peak height on the way up following administration (denoted “50%-up”), and (ii) time-to-peak insulin plasma concentration. Normalized plasma concentration measurements were used to compare the time-to-peak lispro concentrations between commercial Humalog and UFAL treatment groups (Fig. 5, J to M). Pigs exhibited almost 2-fold faster Humalog pharmacokinetics compared to humans. UFAL demonstrated faster absorption than Humalog, whereby UFAL time to 50% of peak up (5 ± 2 min) was 2.4-fold faster than Humalog (12 ± 6 min), and UFAL time to peak (9 ± 4 min) was 2.8-fold faster than Humalog (25 ± 10 min). The exposure duration, defined as the time to 50% of the normalized peak height on the way down following peak exposure concentrations (time to 50% peak down), for UFAL (28 ± 8 min) was 1.9-fold shorter than for Humalog (54 ± 21 min).
Modeling UFAL pharmacokinetics in humans
To better understand how the fast onset and short duration demonstrated by UFAL in pigs would translate to humans, we adapted a pharmacokinetic model from Wong et al. (33) to approximate UFAL pharmacokinetics in humans. The model is constructed such that rapid-acting insulin analogues (Humalog) injected into the subcutaneous space (Iinj) dissociate and diffuse with a rate constant, k1, into the interstitium (Ieq); absorb with a rate constant, k2,into the plasma (Ip); and are subsequently cleared by several mechanisms that can nonetheless be approximated by a single elimination constant, k3 (Fig. 6A). We assume that k2 and k3 are species-dependent, k1 is formulation- and species-dependent, and the ratio of k1 between formulations is species-independent. Because the UFAL formulation is composed of insulin monomers and dimers, the time necessary to reach equilibrium in the interstitium is expected to be appreciably lower than for Humalog. Indeed, when fitting the experimental pig pharmacokinetics for subcutaneous administration of UFAL, we found that k1 trended towards infinity, meaning that UFAL effectively bypasses the first model compartment and the insulin monomers reach equilibrium in the subcutaneous space immediately (table S6). The fits for the pig pharmacokinetic data for both UFAL and Humalog are presented in Fig. 6B and a comparison between the model predictions and experimental data with relevant pharmacokinetic metrics is presented in fig. S10. The infinitely large k1 determined for UFAL in pigs was translated to a human pharmacokinetic model and used to estimate UFAL pharmacokinetics while maintaining k2 and k3 values reported in the literature (Fig. 6C) (33).
Fig. 6. Pharmacokinetic modelling of UFAL in humans.
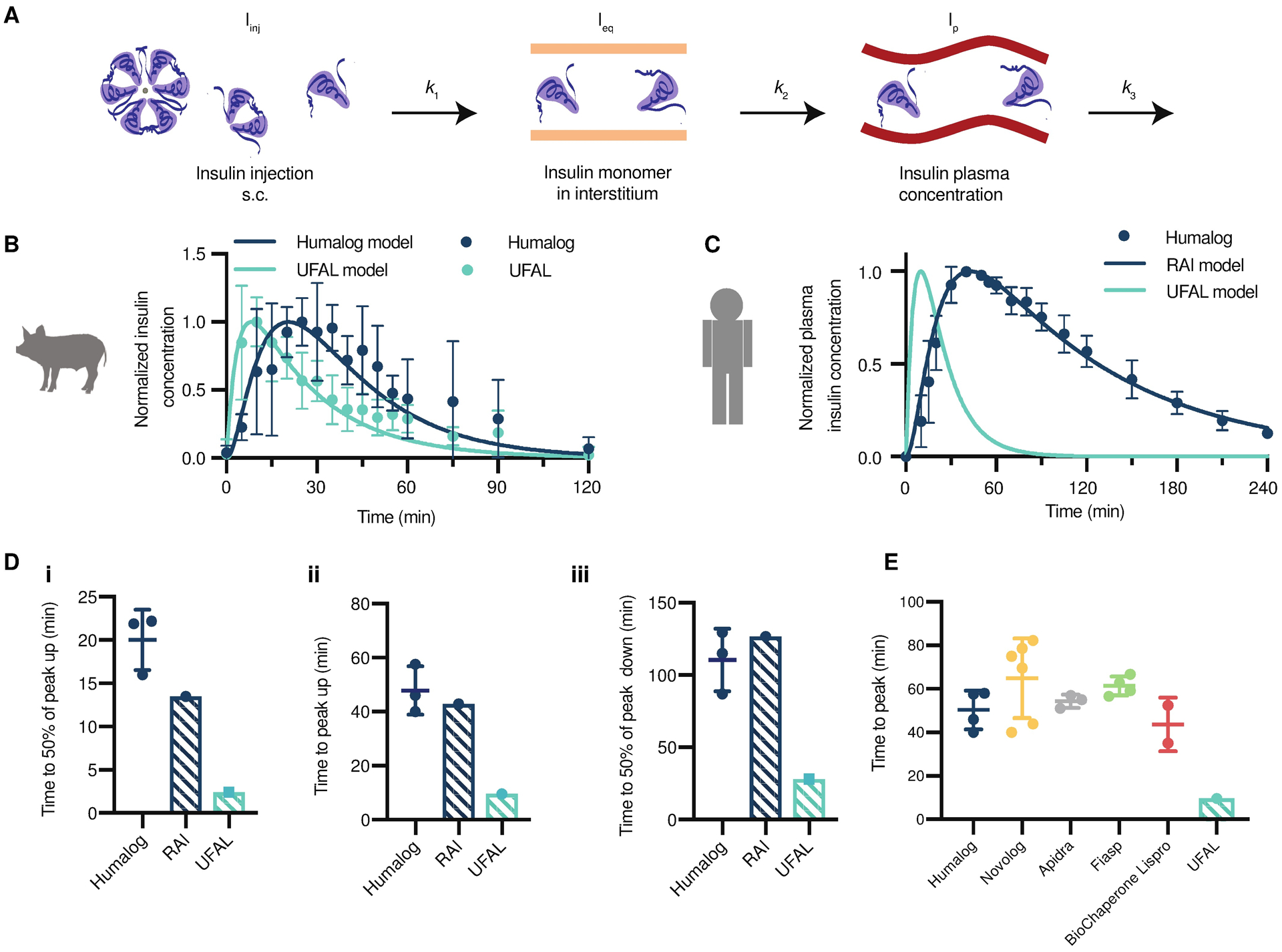
(A) A model of insulin plasma concentrations after injection in human patients was adapted from Wong et al (33). Rapid-acting insulin analogues are injected into the subcutaneous space (Iinj), then dissociate and diffuse (k1) into the interstitium (Ieq) where they are then absorbed (k2) into the plasma (Ip) and ultimately cleared (k3). (B) Normalized pharmacokinetic data for Humalog and UFAL in diabetic pigs (n=5 pigs per group) modeled using a least squares fit to determine k1, k2, and k3 in pigs (fig. S10, table S6). (C) Human clinical Humalog pharmacokinetic data (n=3; number of external studies analyzed) compared to modeled rapid-acting insulin (RAI) analogue kinetics (using known human parameters, table S6) (33), and the predicted kinetics of UFAL in humans. UFAL human pharmacokinetics were predicted by first fitting the pig pharmacokinetic data for Humalog and UFAL. The human UFAL pharmacokinetics was then plotted by using the estimated k1 with the known k2 and k3 parameters. (D) Model-predicted kinetics of RAI and UFAL compared to Humalog kinetics in published clinical studies (n=3; number of external studies analyzed) (29, 30, 34), (i) time to 50% of peak up, (ii) time to peak and duration of action, and (iii) time to 50% of peak down. (E) Comparison of the model-predicted time to peak for UFAL in humans compared to human clinical data for commercial rapid-acting insulin formulations (Humalog n=4; Novolog n=6; Apidra n=3; Fiasp n=4; BioChaperone Lispro n=2; where n is the number of external studies analyzed) (5, 29–32, 34–38). Error bars show s.d.
The model predicts human UFAL time to onset (50%-up) of 2.5 minutes, peak exposure at 10 minutes, and duration of exposure (50%-down) of 28 minutes (Fig. 6D). In comparison, using parameters reported in the literature, the model predicts rapid-acting insulin analogues (RAI), such as Humalog, to exhibit a time to onset of 14 minutes, peak exposure at 43 minutes, and a duration of exposure of 157 minutes (Fig. 6C). Although the RAI model underestimates the time to onset of exposure (t50% up), the predicted curve robustly captures published clinical pharmacokinetic data for peak and duration of Humalog exposure (29, 30, 34). The pharmacokinetic modeling, therefore, predicts UFAL to exhibit kinetics that are more than 4-fold faster than current rapid-acting insulin formulations. Further comparison to clinical data for rapid-acting insulin formulations demonstrates that UFAL is predicted to be faster than even second generation rapid-acting insulin formulations such as Fiasp (Novo-Nordisk) and BioChaperone Lispro (Adocia) (Fig. 6E) (5, 29, 31, 32, 35–38).
Discussion
In this study we report the development of a stable ultrafast-absorbing insulin lispro formulation using an acrylamide-based copolymer as a stabilizing excipient for monomeric insulin. PEG polymers have been traditionally used in drug delivery because of their water solubility and biocompatibility, but recent concerns around immunogenicity are beginning to limit their use (39). In contrast, acrylamide copolymers are emerging as alternative biomaterials for drug delivery systems (26, 39), as many such copolymers have been shown to be non-toxic and more stable under physiological conditions than PEG-based excipients (27, 39, 40). Using a high-throughput screen of a large library of combinatorial acrylamide-based copolymer excipients, we identified several candidate copolymers that act as simple stabilizing agents for insulin formulations. These copolymers enhance insulin formulation stability without any protein-modifying effects, thus eliminating concerns of reduced insulin activity or extended circulation times typically associated with covalent insulin modification (PEGylation). We hypothesized that statistical copolymers comprising a water-soluble carrier monomer and a functional dopant monomer would act as stabilizing excipients to reduce insulin interactions with the air-liquid interface, thus preventing insulin aggregation and enabling the stable formulation of insulin in its monomeric form and ultrafast kinetics in vivo. Indeed, a monomeric insulin formulation comprising our top-performing excipient, MORPH-NIP23%, is stable against stressed aging for over 24 hours, which is almost four-fold longer than commercial Humalog.
In diabetic pigs, this UFAL formulation exhibited ultrafast pharmacokinetics, with about two-fold faster time to onset and two-fold shorter duration of exposure than Humalog, a commercial rapid-acting insulin formulation using the same insulin molecule lispro. These results suggest that this UFAL formulation more closely mimics endogenous insulin secretion in healthy individuals and highlight that this formulation is promising for enhancing diabetes management. Even the incremental improvement in pharmacokinetics over current “fast-acting” insulin formulations observed for Fiasp, a faster-acting version of Novolog (commercial Aspart formulation), have shown numerous clinical benefits (41, 42). While Fiasp shows a modest 10 minute reduction in time to peak action and 15 minute reduction in duration of action over rapid-acting insulin formulations (43), Fiasp use nevertheless reduced post-prandial glucose excursions and reduced HbA1c concentrations in patients with diabetes (41). In contrast, in diabetic pigs, where insulin pharmacokinetics are twice as fast as in humans, UFAL reduced time to peak exposure by 16 minutes and reduced duration of exposure by 26 minutes compared to Humalog. The results observed in diabetic pigs, combined with the model-predicted human UFAL pharmacokinetics, suggest that UFAL may have absorption kinetics that are unprecedented in an injectable insulin formulation. If realized in human clinical studies, these kinetics would be approaching the ultrafast kinetics of Afrezza, the commercially available inhalable insulin (44). However, unlike Afrezza, UFAL is an injectable formulation, which enables more accurate dosing regimens and compatibility with pump and closed-loop systems, providing UFAL the potential to improve post-prandial glycemic control in patients with diabetes.
Taken together these studies identify a promising new copolymer excipient for protein stabilization and show its utility in stabilizing an ultrafast-absorbing insulin formulation. This study is limited in that there is no way to know the true pharmacokinetics of UFAL in humans beyond predictive modeling until it is translated to the clinic. Pigs are a good model for subcutaneous pharmacokinetics; however, insulin still exhibits shorter duration of action in pigs than is typically observed in humans (2 hours in pigs vs. 4 hours in humans) (45–47). Ultimately, it is possible that these faster absorption kinetics in pigs may mask differences in blood glucose depletion between Humalog and UFAL, and may account for the lack of observed pharmacodynamic differences (blood glucose) between formulations in this study. Further, while initial cytotoxicity and biocompatibility experiments performed in this study suggest our new copolymer excipient has a promising toxicity profile, clinical translation will require rigorous study of MORPH-NIP23% biocompatibility and check for formulation immunogenicity. Our stable ultrafast insulin formulation has the potential to improve diabetes management and reduce patient burden around mealtime glucose management.
Materials and Methods
Study design
The objective of this study was to assess an insulin lispro formulation, UFAL, and compare its pharmacokinetics to a commercial insulin lispro formulation (Humalog). To stabilize a UFAL formulation, a library of polyacrylamide derivative excipients was synthesized and characterized. These excipients were evaluated for their ability to prevent recombinant and monomeric insulin aggregation under accelerated aging conditions. The top performing excipient was selected for UFAL formulation. Blood glucose and plasma lispro concentrations were measured using a handheld blood glucose monitor or ELISA on collected blood samples after subcutaneous administration of either Humalog or UFAL in pigs. Randomization: 5 pigs were used for this study and each pig received each formulation once. The order in which the formulations were given in was randomized. Blinding: For analysis of pharmacokinetic parameters (t50% up, time to peak, t50% down) pharmacokinetic curves were coded, and were analyzed by a blinded researcher. Replication: 5 pigs were used in this study and each pig acted as its own control receiving each formulation (Humalog and UFAL) once.
Materials
Solvents N,N-dimethylformamide (DMF; HPLC Grade, Alfa Aeser, >99.7%), ethanol (EtOH; Certified ACS, Acros,>99.5%), acetone (Sigma, HPLC Grade, >99.9%), hexanes (Fisher, Certified ACS, >99.9%), ether (Sigma, Certified ACS, Anhydrous,>99%) and CDCl3 (Acros, >99.8%) were used as received. Monomers N,N-dimethylacrylamide (DMA; Sigma, 99%), N-(3-methoxypropyl)acrylamide (MPAM; Sigma, 95%), 4-acryloylmorpholine (MORPH; Sigma, >97%), acrylamide (AM; Sigma, >99%), and N-hydroxyethyl acrylamide (HEAM; Sigma, >97%) were filtered with basic alumina prior to use. Monomers N-phenylacrylamide (PHE; Sigma, 99%), N-tert-butylacrylamide (TBA; Sigma, 97%), N-isopropylacrylamide (NIP; Sigma, >99%), and N-[tris(hydroxymethyl)methyl]acrylamide (TRI; Sigma, 93%) were used as received. (3-acrylamidopropyl)trimethylammonium chloride (TMA; Sigma, 75%) was washed with ethyl acetate. 2-acrylamido-2-methylpropane sulfonic acid (AMP; Sigma, 99%) was converted to the sodium salt through equimolar mixing with sodium acetate in methanol and precipitated into acetone. RAFT chain transfer agents 2-cyano-2-propyl dodecyl trithiocarbonate (2-CPDT; Strem Chemicals, >97%) and 4-((((2-carboxyethyl)thio)carbonothioyl)thio)-4-cyanopentanoic acid (BM1433; Boron Molecular, >95%) were used as received. Initiator 2,2’-azobis(2-methyl-propionitrile) (AIBN; Sigma, >98%) was recrystallized from methanol (MeOH; Fisher, HPLC Grade, >99.9%) and dried under vacuum before use. Initiator 4,4-azobis(4-cyanovaleric acid)(ACVA; Sigma,>98%) was used as received. Z group removing agents lauroyl peroxide (LPO; Sigma, 97%) and hydrogen peroxide (H2O2; Sigma, 30%) were used as received.
Synthesis of first copolymer library via automated parallel synthesis
Copolymerizations of carriers and dopants were carried out using RAFT polymerization ([Total Monomer]/[CTA] = 50, [CTA]/[AIBN] = 0.2). MPAM, MORPH, and DMA carrier monomers copolymerized with AMP, TMA, NIP, TBA, or PHE dopant monomers were polymerized in DMF using 2-CPDT as the CTA and AIBN as the initiator. MPAM, MORPH, and DMA carrier monomers copolymerized with TRI dopant monomer were polymerized in a DMF/water mixture using BM1433 as the CTA and ACVA as the initiator. Total vinyl monomer molarity was held at 2.72M (MPAM copolymerizations), 2.86M (MORPH copolymerizations), and 3.84M (DMA copolymerizations) such that the homopolymerization of the carrier monomer in DMF would be carried at a constant 40 wt.%. HEAM carrier monomer copolymerized with AMP, TMA, NIP, TBA, or PHE dopant monomers were polymerized in DMF/EtOH mixture using 2-CPDT as the CTA and AIBN as the initiator. HEAM carrier monomer copolymerized with AMP, TMA, NIP, TBA, or PHE dopant monomers were polymerized in DMF/EtOH/water mixture using BM1433 as the CTA and ACVA as the initiator. Total vinyl monomer molarity was held at 2.58M (HEAM copolymerizations) such that the homopolymerization of HEAM in DMF would be carried at a constant 30 wt.%. AM carrier monomer copolymerized with AMP, TMA, NIP, TBA, or PHE dopant monomers were polymerized in DMF/water mixture using BM1433 as the CTA and ACVA as the initiator. AM carrier monomer copolymerized with TRI dopant monomer was polymerized in water using BM1433 as the CTA and ACVA as the initiator. Total vinyl monomer molarity was held at 4.05M (AM copolymerizations) such that the homopolymerization of AM in DMF would be carried at a constant 30 wt.%.
Reaction mixtures were prepared by combining stock solutions: (i) carriers, (ii) dopants, and (iii) CTA and initiator. The stock solutions of carrier monomers were HEAM (555 mg/mL inEtOH), AM (462 mg/mL in water), MPAM (818 mg/mL in DMF), DMA (no solvent dilution), and MORPH (no solvent dilution). The stock solutions of dopant monomers were TRI (181mg/mL in water), PHE (120 mg/mL in DMF), NIP (245 mg/mL in DMF), TBA (122 mg/mL in DMF), AMP (120 mg/mL in DMF), and TMA (124 mg/mL in DMF). Stock solutions of CTA and initiator were prepared such that [CTA]/[initiator] = 5 for AM (BM1433 at 310 mg/mL in water), HEAM and MPAM (BM1433 at 198 mg/mL in water, and 2-CPDT at 221 mg/mL in DMF), MORPH (BM1433 at 220 mg/mL in water and 2-CPDT at 247 mg/mL in DMF), and DMA (BM1433 at 220 mg/mL in water and 2-CPDT at 332 mg/mL in DMF). Reaction mixtures of HEAM, DMA, MPAM, and MORPH were diluted with DMF while reaction mixtures of AM were diluted with water to reach the desired vinyl monomer concentration.
Parallel syntheses of AC/DC excipients were conducted on a Chemspeed Swing XL automated synthesizer robot equipped with a 4-Needle Head tool and an iSynth reactor. The Reactions were performed in 8 mL disposable ISynth reactor vials. All aspirations and dispensing reagent solutions were performed using a the 4-Needle Head tool equipped with a 2 × 10 mL and 2 × 1 mL syringes fitted with septa piercing needles, with both the 1 mL and 10 mL syringes used in this particular experiment. All solvent lines were primed with 60 mL (6 strokes of syringe volume) of degassed DMF. Typical aspiration and dispense rates of the reagents were 10 mL/min for both the 1 mL syringes. An airgap of 50 μL and an extra volume of 50 μL was used for the 1 mL syringes, and an airgap of 50 μL and an extra volume of 100 μL was used for the 10 mL syringes during aspirations using the 4-Needle Head tool. The needles and lines were rinsed after each reagent dispense task with 3 mL inside and outside volume of the priming solvent for the 1 mL syringes and with 20 mL inside and outside volume of the priming solvent for the 10 mL syringes. The DMF reservoir was degassed by continuous nitrogen sparging. All stock solutions were prepared in septa capped reagent vials and degassed by sparging with argon for 15 minutes before transfer into the Chemspeed. The atmosphere within the Chemspeed was reduced to <1 % oxygen by purging with nitrogen while exhaust ports were closed. Reactor vials were exposed to nitrogen flow until the start of the reaction. The calculated aliquots of stock solutions and solvent were transferred to the reactors via the automated liquid handling system. Upon dispensing, reactor vials were manually sealed in the inert atmosphere, removed from the Chemspeed, manually shaken to combine reagents, and heated to 65 °C in an oven for 24 hours, after which, reaction vials were cooled to room temperature and exposed to air.
A procedure to remove the CTA Z groups from the AC/DC excipients containing MORPH, DMA, HEAM, and MPAM copolymers was adapted from the literature (48). The reaction vial was diluted to 6 mL with DMF. LPO (2 equivalents (eq.)) and AIBN (20 eq.) were added to the reaction mixture, which was sealed with a cap utilizing a PTFE seal. The reaction mixture was sparged with nitrogen gas for 10 minutes while heating at 90 °C and subsequently heated for 12 hours at 90 °C. A procedure to remove the CTA Z groups from the AC/DC excipients containing AM copolymers was adapted from the literature (49). The reaction vial was diluted to 5 mL with miliQ water. H2O2 (20 eq.) was added to the reaction vial, which was sealed and heated to 60 °C for 12 hours. The resulting copolymers were isolated by precipitation as outlined below.
AC/DC excipients synthesized with AM and HEAM carriers were precipitated twice from acetone. AC/DC excipients synthesized with DMA and MORPH were precipitated twice from diethyl ether. AC/DC excipients synthesized with MPAM were precipitated twice from diethyl ether and hexane (3:1 ratio) mixtures. The number (Mn) and weight (Mw) average molecular weights and dispersity for the AC/DC excipients containing MORPH, MPAM, DMA, and HEAM were determined using SEC in DMF with poly(ethyleneglycol) standards. Mn, Mw, and dispersity for the AC/DC excipients containing AM were determined using aqueous SEC-MALS.
Synthesis of second polymeric library
A typical procedure to synthesize a MORPH-NIP AC/DC excipient is as follows and is nearly identical for all other carrier/dopant combinations, where only the carrier/dopant selection and concentration are changed. MORPH (645 mg, 4.57 mmol, 41.5 eq.), NIP (105 mg, 0.93 mmol, 8.5 eq.), 2-CPDT (38 mg, 0.11 mmol, 1 eq.) and AIBN (3.6 mg, 0.02 mmol, 0.2 eq.) were combined and diluted with DMF to a total volume of 2.25 mL (33.3 w/v vinyl monomer concentration) in an 8 mL scintillation vial equipped with a PTFE septa. The reaction mixture was sparged with nitrogen gas for 10 minutes and then heated for 12 hours at 65 °C. To remove the Z-terminus of the resulting polymer, AIBN (360 mg, 2.2 mmol, 20 eq.) and LPO (88 mg, 0.22 mmol, 2 eq.) were added to the reaction mixture, which was then sparged with nitrogen gas for 10 minutes and heated for 12 hours at 90 °C (43). Z-group removal was confirmed by the ratio of the refractive index to UV (λ = 310 nm) intensity in SEC analysis. Resulting polymers were precipitated three times from ether, and dried under vacuum overnight. Resulting composition and molecular weights were determined via 1H NMR spectroscopy and SEC with poly(ethyleneglycol) standards.
Copolymer molecular weight characterization
Mn, Mw, and dispersity for copolymers with HEAM, DMA, MPAM, and MORPH carrier monomers were determined via SEC implementing poly(ethyleneglycol) standards (American Polymer Standards Corporation) after passing through two size exclusion chromatography columns [Re-solve Mixed Bed Low DVB, ID 7.8 mm, Mw range 200–600,000 g mol−1 (Jordi Labs)] in a mobile phase of N,N-dimethylformamide (DMF) with 0.1M LiBr at 35 °C and a flow rate of 1.0 ml min−1 (Dionex Ultimate 3000 pump, degasser, and autosampler (Thermo Fisher Scientific).
Mn, Mw, and dispersity for copolymers with AM were determined via SEC-MALS after passing through a size exclusion chromatography column [Superose 6 Increase 10/300 GL, 5,000–5,000,000 g mol−1 (GE Healthcare)] in a mobile phase of phsophate-buffered saline containing 300 ppm sodium azide. Detection consisted of an Optilab T-rEX (Wyatt Technology Corporation) refractive index detector operating at 658 nm and a TREOS II light scattering detector (Wyatt Technology Corporation) operating at 659 nm. The dn/dc value for AM copolymers were assumed to be 0.185 in this media.
In vitro insulin stability
Methods for aggregation assays for recombinant human insulin were adapted from Webber et al. (21). Briefly, formulation samples (3.4 mg/mL) were plated at 150 μL per well (n = 3/group) in a clear 96-well plate and sealed with optically clear and thermally stable seal (VWR). The plate was immediately placed into a plate reader and incubated with continuous shaking at 37°C. Absorbance readings were taken every 10 minutes at 540 nm for 100 h (BioTek Syner-gyH1 microplate reader). The aggregation of insulin leads to light scattering, which results in an increase in the measured absorbance. The time-to-aggregation (tA) was defined as the time at which a greater than 10% increase in absorbance from the absorbance at time zero was observed (21). After 100 h, the plate was removed from the plate reader and transferred to an incubator shaker plate where it was subjected to continued stressed aging. Absorbance readings were taken periodically for up to 30 days.
For the initial high-throughput stability screen, recombinant human insulin (Gibco) was formulated in phosphate buffered saline (0.9 wt.% NaCl) and AC/DC excipients were added at concentrations of 1 mg/mL or 10 mg/mL to the recombinant insulin formulation for a final insulin concentration of 3.4 mg/mL. Each plate contained a recombinant insulin control with no polymer added.
For the secondary stability screen with UFAL formulations, control groups included: (i) commercial Humalog (Eli Lilly), (ii) zinc-free lispro comprising phosphate buffer, glycerol (2.6 wt.%), and phenoxyethanol (0.85 wt.%). Zinc (II) was removed from commercial insulin formulations through competitive binding by addition of ethylenediaminetetraacetic acid (EDTA), which exhibits a dissociation binding constant approaching attomolar concentrations (KD ~ 10−18 M) (50). EDTA was added to formulations (1 eq. with respect to zinc) to sequester zinc from the formulation. Following zinc sequestration, PD MidiTrap G-10 gravity columns (GEHealthcare) were used to remove the zinc/EDTA complexes and other formulation excipients. Lispro was concentrated using Amino Ultra 3K centrifugal units (Millipore), and then reformulated at 100 U/mL with phosphate buffer (10 mM), glycerol (2.6 wt.%), phenoxyethanol (0.85 wt.%), and AC/DC excipient (0.01 wt.%).
NMR DOSY
1H 2D DOSY spectra were recorded at an insulin lispro concentration of 3.4 mg/mL with 40 wt.% D2O for UFAL formulation comprising phosphate buffer, glycerol (2.6 wt.%), phenoxyethanol (0.85 wt.%) and MORPH-NIP23% copolymer (0.1 wt.%). A Varian Inova 600 MHz NMR instrument was used to acquire the data. Magnetic field strengths ranging from 2 to 57 G cm−1. The DOSY time and gradient pulse were set at 132 ms (Δ) and 3 ms (δ) respectively. All NMR data were processed using MestReNova 11.0.4 software.
Streptozotocin-induced diabetes in pigs
Five female Yorkshire pigs (Pork Power) were used for our animal studies, which were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals and the Animal Welfare Act Regulations. All protocols were approved by the Stanford Institutional Animal Care and Use Committee. Type-1-like diabetes was induced in pigs (25–30 kg) using streptozotocin (STZ) (MedChemExpress). STZ was infused intravenously at a dose of 125 mg/kg and animals were monitored for 24 hours. Food and administration of 5% dextrose solution was given as needed to prevent hypoglycemia. Diabetes was defined as fasting blood glucose greater than 300 mg/dL.
In vivo pharmacokinetics and pharmacodynamics in diabetic pigs
Five diabetic pigs were fasted for 4–6 hours. Pigs were injected subcutaneously with a 2–4 U dose of the following formulations: (i) Humalog (100 U/mL, Eli Lilly) or (ii) ultrafast-absorbing lispro (UFAL) (100 U/mL Zn-free lispro, 2.6 wt.% glycerol, 0.85 wt.% phenoxyethanol, 0.01 wt.% MORPH-NIP23%). Doses were determined based on individual pig insulin sensitivity values with a target of a decrease in blood glucose of approximately 200 mg/dL. Individual pigs received the same dose for each treatment group. Pigs received each formulation once on separate days and the order of the treatment groups were randomized. Before injection, baseline blood was sampled from an intravenous catheter line and measured using a handheld glucose monitor (Bayer Contour Next). After injection, blood was sampled from the intravenous catheter line every 5 minutes for the first 60 minutes, then every 30 minutes up to 4 hours. Blood was collected in K2EDTA plasma tubes (Greiner-BioOne) for analysis with ELISA. Plasma lispro concentrations were quantified using an Insulin Lispro ELISA kit (Mercodia).
Pharmacokinetic modelling
The pharmacokinetic model used in this analysis was derived from literature reports (33). Insulin concentrations for injection (Iinj), equilibrium in the interstitium (Ieq), and the plasma (Ip) were numerically solved using a system of differential equations, outlined below, as a function of time using the SciPy (version 1.2.1) odeint function in Python (version 3.6.8).
| (1) |
| (2) |
| (3) |
Concentrations were initialized such that at t = 0 all insulin was present in Iinj. Kinetic rate constants were fit for the normalized pig pharmacokinetic curves by minimizing the sum of squared errors (SSE) between the generated, normalized insulin plasma concentrations derived from the model at the experimental time points from 0 to 90 minutes and the normalized pig plasma insulin concentrations for UFAL and Humalog. We assume that k2 and k3 are species dependent, while k1 is both species and formulation dependent. While minimizing the SSE, we observed that there was no upward bound for k1,UFAL,Pig; such that higher values of k1,UFAL,Pig resulted in increasingly marginally smaller SSEs for a given k2 and k3. Accordingly, k1,UFAL,Pig was then set at 100,000 min−1. The SSE was minimized by first employing a grid search using SciPy’s optimize brute function and subsequently refining the rate constants by employing SciPy’s optimize minimize function using the L-BFGS-B method. To solve for k1,UFAL,Human, we assume the following relationship;
Values for k1,Humalog,Human, k2,Human, and k3,Human were used as reported in the literature (33).
Statistical analysis
All results are expressed as a mean ± standard deviation unless specified otherwise. Fig. 5D–I and K–M are shown as mean ± standard error of the mean. All statistical analyses were performed as general linear models (GLMs) in JMP Pro Version 14. Comparisons between formulations (Fig. 5, D to I, K to M) were conducted using the restricted maximum likelihood (REML) repeated measures mixed model. Suitable transformations applied as needed to meet the assumptions of the methods (homogeneity of variance, normality of error, and linearity). Time to 50% of peak up, time to peak, and time to 50% peak down were log transformed for analyses to correct for non-homogeneity of variance. Pig was included as a variable in the model as a random effect blocking (control) factor to account for variation in individual pig response. Statistical significance was considered as p<0.05. For Fig. 5, D to I, post-hoc Bonferroni correction was applied to account for multiple comparisons and significance was adjusted to alpha=0.008.
Supplementary Material
Fig. S1. 1H NMR Spectroscopy and SEC traces to Validate SEC wt.% measurement.
Fig. S2. SEC traces of polymers from the initial copolymer library synthesis.
Fig. S3. Aqueous SEC elution profile for commercial Humalog and UFAL formulations.
Fig. S4. SEC traces of copolymers from the second screen targeting DP50.
Fig S5. In vitro and in vivo formulation activity.
Fig. S6. Cytotoxicity of leading AC/DC excipient MORPH-NIP23% with NIH/3T3 cells.
Fig. S7. Biocompatibility in diabetic rats.
Fig. S8. Blood glucose of monomeric insulin in diabetic pigs.
Fig. S9. AUMC/AUC for UFAL and Humalog in diabetic pigs.
Fig. S10. Pharmacokinetic outputs from model fitting compared to experimental pharmacokinetic data for Humalog and UFAL in diabetic pigs.
Table S1. Demonstration of work to validate SEC wt.% measurement.
Table S2. SEC and MALS characterization and analysis of polymers synthesized in initial AC/DC library.
Table S3. Days until aggregation for recombinant insulin formulated with AC/DC excipients at two excipient concentrations (1 mg/mL and 10 mg/ml).
Table S4. SEC and 1H NMR analysis of polymers synthesized during the second screen targeting DP50.
Table S5. Days until aggregation for UFAL formulated with AC/DC excipients at 0.1 mg/mL.
Table S6. Rate constants used for modeling PK curves in the manuscript.
Acknowledgments:
The authors thank the Stanford Veterinary Service Centre staff for their assistance with animal care and procedures as well as the Stanford Animal Diagnostic Lab for their technical assistance.
Funding: This work was funded in part by an NIDDK R01 (NIH grant #R01DK119254) and a Pilot & Feasibility seed grant from the Stanford Diabetes Research Center (NIH grant #P30DK116074) and the Stanford Child Health Research Institute, as well as the American Diabetes Association Grant (1-18-JDF-011) and a Research Starter Grant from the PhRMA Foundation. J.L.M. was supported by the Department of Defense NDSEG Fellowship and by a Stanford Graduate Fellowship. C.L.M. was supported by the NSERC Postgraduate Scholarship and the Stanford BioX Bowes Graduate Student Fellowship. A.A.A.S. was funded by grant NNF18OC0030896 from the Novo Nordisk Foundation and the Stanford Bio-X Program, and also funded by the Danish Council of Independent Research (Grant No. DFF5054-00215).
Footnotes
Supplementary Materials
Materials and Methods
Reference (51)
Competing interests: E.A.A., J.L.M., and C.L.M. are listed as inventors on a provisional patent application (63/011,928) filed by Stanford University describing the technology reported in this manuscript.
Data and materials availability: All data associated with this study are present in the paper or the Supplementary Materials.
References and Notes
- 1.Saad A, Dalla Man C, Nandy DK, Levine JA, Bharucha AE, Rizza RA, Basu R, Carter RE, Cobelli C, Kudva YC, Basu A, Diurnal pattern to insulin secretion and insulin action in healthy individuals, Diabetes 61, 2691–2700 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tillil H, Shapiro ET, Miller MA, Karrison T, Frank BH, Galloway JA, Rubenstein AH, Polonsky KS, Dose-dependent effects of oral and intravenous glucose on insulin secretion and clearance in normal humans, Am. J. Physiol 254, E349–57 (1988). [DOI] [PubMed] [Google Scholar]
- 3.Polonsky KS, Given BD, Van Cauter E, Twenty-four-hour profiles and pulsatile patterns of insulin secretion in normal and obese subjects, J. Clin. Invest 81, 442–448 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heptulla RA, Rodriguez LM, Bomgaars L, Haymond MW, The role of amylin and glucagon in the dampening of glycemic excursions in children with type 1 diabetes, Diabetes 54, 1100–1107 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Heise T, Zijlstra E, Nosek L, Rikte T, Haahr H, Pharmacological properties of faster-acting insulin aspart vs insulin aspart in patients with type 1 diabetes receiving continuous subcutaneous insulin infusion: A randomized, double-blind, crossover trial, Diabetes Obes. Metab 19, 208–215 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Senior P, Hramiak I, {Fast-Acting} Insulin Aspart and the Need for New Mealtime Insulin Analogues in Adults With Type 1 and Type 2 Diabetes: A Canadian Perspective, Can J Diabetes 43, 515–523 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Holleman F, Hoekstra JB, Insulin lispro N Engl. J. Med 337, 176–183 (1997). [DOI] [PubMed] [Google Scholar]
- 8.Gast K, Schüler A, Wolff M, Thalhammer A, Berchtold H, Nagel N, Lenherr G, Hauck G, Seckler R, {Rapid-Acting} and Human Insulins: Hexamer Dissociation Kinetics upon Dilution of the Pharmaceutical Formulation, Pharm. Res 34, 2270–2286 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mathieu C, Gillard P, Benhalima K, Insulin analogues in type 1 diabetes mellitus: getting better all the time, Nat. Rev. Endocrinol 13, 385–399 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Gingras V, Taleb N, Roy-Fleming A, Legault L, Rabasa-Lhoret R, The challenges of achieving postprandial glucose control using closed-loop systems in patients with type 1 diabetes, Diabetes Obes. Metab 20, 245–256 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cengiz E, Undeniable need for ultrafast-acting insulin: the pediatric perspective, J. Diabetes Sci. Technol 6, 797–801 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hua Q-X, Weiss MA, Mechanism of insulin fibrillation: the structure of insulin under amyloidogenic conditions resembles a protein-folding intermediate, J. Biol. Chem 279, 21449–21460 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Woods RJ, Alarcón J, McVey E, Pettis RJ, Intrinsic fibrillation of fast-acting insulin analogs, J. Diabetes Sci. Technol 6, 265–276 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu Y, Yan Y, Seeman D, Sun L, Dubin PL, Multimerization and aggregation of native-state insulin: effect of zinc, Langmuir 28, 579–586 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Derewenda U, Derewenda Z, Dodson EJ, Dodson GG, Reynolds CD, Smith GD, Sparks C, Swenson D, Phenol stabilizes more helix in a new symmetrical zinc insulin hexamer, Nature 338, 594–596 (1989). [DOI] [PubMed] [Google Scholar]
- 16.Weiss MA, in Vitamins & Hormones, (Academic Press, 2009), vol. 80, pp. 33–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sluzky V, Tamada JA, Klibanov AM, Langer R, Kinetics of insulin aggregation in aqueous solutions upon agitation in the presence of hydrophobic surfaces, Proc. Natl. Acad. Sci. U. S. A 88, 9377–9381 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sluzky V, Klibanov AM, Langer R, Mechanism of insulin aggregation and stabilization in agitated aqueous solutions, Biotechnol. Bioeng 40, 895–903 (1992). [DOI] [PubMed] [Google Scholar]
- 19.Nault L, Guo P, Jain B, Bréchet Y, Bruckert F, Weidenhaupt M, Human insulin adsorption kinetics, conformational changes and amyloidal aggregate formation on hydrophobic surfaces, Acta Biomater. 9, 5070–5079 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Muzaffar M, Ahmad A, The mechanism of enhanced insulin amyloid fibril formation by {NaCl} is better explained by a conformational change model, PLoS One 6, e27906 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Webber MJ, Appel EA, Vinciguerra B, Cortinas AB, Thapa LS, Jhunjhunwala S, Isaacs L, Langer R, Anderson DG, Supramolecular {PEGylation} of biopharmaceuticals, Proc. Natl. Acad. Sci. U. S. A 113, 14189–14194 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang C, Lu D, Liu Z, How PEGylation enhances the stability and potency of insulin: A molecular dynamics simulation, Biochemistry 50, 2585–2593 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Liu Y, Lee J, Mansfield KM, Ko JH, Sallam S, Wesdemiotis C, Maynard HD, Trehalose Glycopolymer Enhances Both Solution Stability and Pharmacokinetics of a Therapeutic Protein, Bioconjug. Chem 28, 836–845 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maikawa CL, Smith AAA, Zou L, Meis CM, Mann JL, Webber MJ, Appel EA, Stable Monomeric Insulin Formulations Enabled by Supramolecular PEGylation of Insulin Analogues, Adv. Ther. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bertrand N, Leroux J-C, The journey of a drug-carrier in the body: An anatomo-physiological perspective, J. Control. Release 161, 152–163 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Ting JM, Tale S, Purchel AA, Jones SD, Widanapathirana L, Tolstyka ZP, Guo L, Guillaudeu SJ, Bates FS, Reineke TM, High-Throughput Excipient Discovery Enables Oral Delivery of Poorly Soluble Pharmaceuticals, ACS Cent. Sci 2, 748–755 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amended Final Report on the Safety Assessment of Polyacrylamide and Acrylamide Residues in Cosmetics1, Int. J. Toxicol 24, 21–50 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Patil SM, Keire DA, Chen K, Comparison of {NMR} and Dynamic Light Scattering for Measuring Diffusion Coefficients of Formulated Insulin: Implications for Particle Size Distribution Measurements in Drug Products, AAPS J. 19, 1760–1766 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andersen G, Meiffren G, Lamers D, DeVries JH, Ranson A, Seroussi C, Alluis B, Gaudier M, Soula O, Heise T, Ultra-rapid {BioChaperone} Lispro improves postprandial blood glucose excursions vs insulin lispro in a 14-day crossover treatment study in people with type 1 diabetes, Diabetes Obes. Metab 20, 2627–2632 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Plank J, Wutte A, Brunner G, Siebenhofer A, Semlitsch B, Sommer R, Hirschberger S, Pieber TR, A Direct Comparison of Insulin Aspart and Insulin Lispro in Patients With Type 1 Diabetes, Diabetes Care 25, 2053–2057 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Heise T, Hövelmann U, Brøndsted L, Adrian CL, Nosek L, Haahr H, Faster-acting insulin aspart: earlier onset of appearance and greater early pharmacokinetic and pharmacodynamic effects than insulin aspart, Diabetes Obes. Metab 17, 682–688 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fath M, Danne T, Biester T, Erichsen L, Kordonouri O, Haahr H, Faster-acting insulin aspart provides faster onset and greater early exposure vs insulin aspart in children and adolescents with type 1 diabetes mellitus, Pediatr. Diabetes 18, 903–910 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Wong J, Chase JG, Hann CE, Shaw GM, Lotz TF, Lin J, Le Compte AJ, A Subcutaneous Insulin Pharmacokinetic Model for Computer Simulation in a Diabetes Decision Support Role: Model Structure and Parameter Identification, J. Diabetes Sci. Technol 2, 658–671 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pettis RJ, Hirsch L, Kapitza C, Nosek L, Hövelmann U, Kurth H-J, Sutter DE, Harvey NG, Heinemann L, Microneedle-based intradermal versus subcutaneous administration of regular human insulin or insulin lispro: pharmacokinetics and postprandial glycemic excursions in patients with type 1 diabetes, Diabetes Technol. Ther 13, 443–450 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Rave K, Klein O, Frick AD, Becker RHA, Advantage of premeal-injected insulin glulisine compared with regular human insulin in subjects with type 1 diabetes, Diabetes Care 29, 1812–1817 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Heise T, Meiffren G, Alluis B, Seroussi C, Ranson A, Arrubla J, Correia J, Gaudier M, Soula O, Soula R, DeVries JH, Klein O, Bode B, {BioChaperone} Lispro versus faster aspart and insulin aspart in patients with type 1 diabetes using continuous subcutaneous insulin infusion: A randomized euglycemic clamp study, Diabetes Obes. Metab (2018). [DOI] [PubMed] [Google Scholar]
- 37.Lindholm A, McEwen J, Riis AP, Improved postprandial glycemic control with insulin aspart. A randomized double-blind cross-over trial in type 1 diabetes, Diabetes Care 22, 801–805 (1999). [DOI] [PubMed] [Google Scholar]
- 38.Becker RHA, Frick AD, Clinical pharmacokinetics and pharmacodynamics of insulin glulisine, Clin. Pharmacokinet 47, 7–20 (2008). [DOI] [PubMed] [Google Scholar]
- 39.Kavitha A, Parambath A, in Engineering of Biomaterials for Drug Delivery Systems, Parambath A, Ed. (Woodhead Publishing, 2018), pp. 229–253. [Google Scholar]
- 40.Gorman M, Chim YH, Hart A, Riehle MO, Urquhart AJ, {Poly(N-acryloylmorpholine)}: a simple hydrogel system for temporal and spatial control over cell adhesion, J. Biomed. Mater. Res. A 102, 1809–1815 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Russell-Jones D, Bode BW, De Block C, Franek E, Heller SR, Mathieu C, Philis-Tsimikas A, Rose L, Woo VC, Østerskov AB, Graungaard T, Bergenstal RM , {Fast-Acting} Insulin Aspart Improves Glycemic Control in {Basal-Bolus} Treatment for Type 1 Diabetes: Results of a 26-Week Multicenter, {Active-Controlled}, {Treat-to-Target}, Randomized, {Parallel-Group} Trial (onset 1), Diabetes Care 40, 943–950 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Leelarathna L, Ashley D, Fidler C, Parekh W, The value of fast-acting insulin aspart compared with insulin aspart for patients with diabetes mellitus treated with bolus insulin from a {UK} health care system perspective, Ther. Adv. Endocrinol. Metab 9, 187–197 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haahr H, Pieber TR, Mathieu C, Gondolf T, Shiramoto M, Erichsen L, Heise T, Clinical Pharmacology of {Fast-Acting} Insulin Aspart Versus Insulin Aspart Measured as Free or Total Insulin Aspart and the Relation to {Anti-Insulin} Aspart Antibody Levels in Subjects with Type 1 Diabetes Mellitus, Clin. Pharmacokinet 58, 639–649 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heinemann L, Baughman R, Boss A, Hompesch M, Pharmacokinetic and Pharmacodynamic Properties of a Novel Inhaled Insulin, J. Diabetes Sci. Technol 11, 148–156 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Larsen MO, Rolin B, Use of the Göttingen minipig as a model of diabetes, with special focus on type 1 diabetes research, ILAR J. 45, 303–313 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Radziuk JM, Davies JC, Pye WS, Shields JE, DiMarchi RD, Chance RE, Bioavailability and bioeffectiveness of subcutaneous human insulin and two of its {analogs--LysB28ProB29-human} insulin and {AspB10LysB28ProB29-human} insulin--assessed in a conscious pig model, Diabetes 46, 548–556 (1997). [DOI] [PubMed] [Google Scholar]
- 47.Kildegaard J, Buckley ST, Nielsen RH, Povlsen GK, Seested T, Ribel U, Olsen HB, Ludvigsen S, Jeppesen CB, Refsgaard HHF, Bendtsen KM, Kristensen NR, Hostrup S, Sturis J, Elucidating the Mechanism of Absorption of {Fast-Acting} Insulin Aspart: The Role of Niacinamide, Pharm. Res 36, 49 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen M, Moad G, Rizzardo E, Thiocarbonylthio end group removal from RAFT-synthesized polymers by a radical-induced process, J. Polym. Sci., Part A Polym. Chem 47, 6704–6714 (2009). [Google Scholar]
- 49.Jesson CP, Pearce CM, Simon H, Werner A, Cunningham VJ, Lovett JR, Smallridge MJ, Warren NJ, Armes SP, H2O2 Enables Convenient Removal of RAFT End-Groups from Block Copolymer Nano-Objects Prepared via Polymerization-Induced Self-Assembly in Water, Macromolecules 50, 182–191 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berthon G, Ed., Handbook of metal-ligand interactions in biological fluids (Marcel Dekker, New York, 1995). [Google Scholar]
- 51.Wu KK, Huan Y, Streptozotocin-induced diabetic models in mice and rats, Curr. Protoc. Pharmacol Chapter 5, Unit 5.47 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. 1H NMR Spectroscopy and SEC traces to Validate SEC wt.% measurement.
Fig. S2. SEC traces of polymers from the initial copolymer library synthesis.
Fig. S3. Aqueous SEC elution profile for commercial Humalog and UFAL formulations.
Fig. S4. SEC traces of copolymers from the second screen targeting DP50.
Fig S5. In vitro and in vivo formulation activity.
Fig. S6. Cytotoxicity of leading AC/DC excipient MORPH-NIP23% with NIH/3T3 cells.
Fig. S7. Biocompatibility in diabetic rats.
Fig. S8. Blood glucose of monomeric insulin in diabetic pigs.
Fig. S9. AUMC/AUC for UFAL and Humalog in diabetic pigs.
Fig. S10. Pharmacokinetic outputs from model fitting compared to experimental pharmacokinetic data for Humalog and UFAL in diabetic pigs.
Table S1. Demonstration of work to validate SEC wt.% measurement.
Table S2. SEC and MALS characterization and analysis of polymers synthesized in initial AC/DC library.
Table S3. Days until aggregation for recombinant insulin formulated with AC/DC excipients at two excipient concentrations (1 mg/mL and 10 mg/ml).
Table S4. SEC and 1H NMR analysis of polymers synthesized during the second screen targeting DP50.
Table S5. Days until aggregation for UFAL formulated with AC/DC excipients at 0.1 mg/mL.
Table S6. Rate constants used for modeling PK curves in the manuscript.