

Abstract
97% of drug-indication pairs that are tested in clinical trials in oncology never advance to receive FDA approval. While lack of efficacy and dose-limiting toxicities are the most common causes of trial failure, the reason(s) why so many new drugs encounter these problems is not well-understood. Using CRISPR/Cas9 mutagenesis, we investigated a set of cancer drugs and drug targets in various stages of clinical testing. We show that – contrary to previous reports obtained predominantly with RNAi and small-molecule inhibitors – the proteins ostensibly targeted by these drugs are non-essential for cancer cell proliferation. Moreover, the efficacy of each drug that we tested was unaffected by the loss of its putative target, indicating that these compounds kill cells via off-target effects. By applying a genetic target-deconvolution strategy, we discovered that the mischaracterized anti-cancer agent OTS964 is actually a potent inhibitor of the cyclin-dependent kinase CDK11 and that multiple cancer types are addicted to CDK11 expression. We suggest that stringent genetic validation of the mechanism of action of cancer drugs in the preclinical setting may decrease the number of therapies tested in human patients that fail to provide any clinical benefit.
One-sentence summary:
CRISPR reveals that many cancer drug targets are dispensable for cell proliferation and identifies CDK11 as the target of one mischaracterized agent.
Introduction
Substantial progress has been made in the treatment of certain malignancies by targeting cancer ‘addictions’, or genetic dependencies that encode proteins required for the survival and/or proliferation of cancer cells (1). Therapeutic agents that block the function of a cancer dependency – like the kinase inhibitor lapatinib in HER2+ breast cancer – can trigger apoptosis and durable tumor regression (2). Discovering and characterizing druggable cancer dependencies is a key goal of preclinical research.
While screening cancer drug targets, we discovered that Maternal Embryonic Leucine Zipper Kinase (MELK), a protein previously reported to be essential in multiple cancer types, could be eliminated using CRISPR-mediated gene editing without any detectable loss in cancer cell fitness (3, 4). Additionally, we demonstrated that OTS167, a small-molecule inhibitor of MELK undergoing phase II clinical trials, continued to kill MELK-knockout (KO) cancer cells with no decrease in potency. These findings suggested that a drug tested in human cancer patients had been designed to target a non-essential cellular protein and that its putative inhibitor killed cells by interacting with proteins other than its reported target. We hypothesized that problems in drug development and inhibitor validation, as exemplified by MELK and OTS167, could potentially contribute to the high failure rate of new cancer therapies. In particular, drugs that target superfluous proteins may display limited efficacy in human patients, and if these drugs are active only via off-target effects, then this could potentially contribute to patient toxicity. Moreover, clinical trials that use a biomarker to select patients for trial inclusion are approximately twice as likely to succeed as those without one (5). Misidentifying a drug’s mechanism of action (MOA) could hamper efforts to uncover a biomarker capable of predicting therapeutic responses, further decreasing the success rate of clinical trials. To test whether other cancer drugs had similarly been designed against non-essential targets or had been assigned an incorrect MOA, we set out to systematically analyze multiple cancer drugs and drug targets that are undergoing clinical trials or in late-stage preclinical development.
Results
CRISPR competition assays to investigate several putative cancer dependencies
Based on an analysis of the literature, we chose drug targets that met several criteria (described in detail in the Materials and Methods). Notably, we selected drug targets that had been reported to play a cell-autonomous role in cancer growth, such that their loss or inhibition was reportedly sufficient to block cancer cell proliferation. Additionally, we selected drug targets that lacked a known mutation capable of conferring resistance to their targeted inhibitors, which we hypothesized represents the gold standard for proving a drug’s MOA. We identified 10 cancer drugs targeting six proteins that met these criteria (Table 1). Five of these proteins are reported to represent cancer dependencies (HDAC6, MAPK14/p38α, PAK4, PBK, and PIM1)(6–15). One protein (CASP3/caspase-3) is reported to induce apoptosis when activated by a small molecule (16, 17), and is discussed separately. Among the putative dependencies, over 180 different publications indicate that they are required for cancer cell proliferation or fitness (listed in data file S1). For each of these genes, the majority of evidence supporting their designation as cancer dependencies comes from RNAi studies, in which siRNA or shRNA-mediated knockdown was reported to impair cancer cell fitness. Additionally, each protein is targeted by one or more small-molecule drugs, which have been described to exhibit potent cell killing in vitro and in vivo. On the basis of these pre-clinical results, the drugs listed in Table 1 have been used in at least 29 different clinical trials, with an estimated enrollment of more than 1,000 patients.
Table 1.
Anti-cancer drugs and drug targets
| Target | Drug | # of Cancer Clinical Trials |
|---|---|---|
| CASP3 | 1541B | Pre-clinical |
| PAC-1 | 3 | |
| HDAC6 | Citarinostat | 5 |
| Ricolinostat | 10 | |
| MAPK14 (p38α) | Ralimetinib | 5 |
| SCIO-469 | 3 | |
| PAK4 | PF-03758309 | 1 |
| PBK (TOPK) | OTS514 | Pre-clinical |
| OTS964 | Pre-clinical | |
| PIM1 | SGI-1776 | 2 |
We first set out to validate the role of the putative dependencies targeted by these drugs in cancer cell fitness. To accomplish this, we applied a CRISPR/Cas9-based cell competition assay, in which cancer cells are infected at a low multiplicity of infection with GFP-expressing guide RNA (gRNA) vectors targeting a gene of interest (Fig. 1A)(18). If a CRISPR-induced mutation reduces cell fitness, then the untransduced cells within a population should outcompete the guide RNA-expressing cells, and the fraction of GFP+ cells should decrease over time. To verify this approach, we designed guide RNAs against pan-essential genes and against several confirmed cancer drug targets. In breast cancer, colorectal cancer, lung cancer, and melanoma cell lines, guides targeting the essential replication proteins RPA3 and PCNA dropped out up to 100-fold, and guides targeting the validated pan-cancer dependencies Aurora A, Aurora B, and ERCC3 exhibited similar levels of depletion (Fig. 1B). Mutations in Aurora A (19), Aurora B (20), and ERCC3 (21) confer resistance to the cytotoxic agents MLN8054, ZM447439, and triptolide, respectively, thereby providing genetic evidence that they are required for cancer cell growth. In contrast, guide RNAs targeting the non-essential Rosa26 and AAVS1 loci exhibited minimal drop-out over five passages in culture. These GFP competition assays were also capable of identifying cell type-specific dependencies: guides targeting the oncogenic kinase BRAF dropped out in a BRAF-mutant melanoma cell line but not a BRAF-WT colorectal cancer line, whereas guides targeting the gene encoding the estrogen receptor (ESR1) dropped out in an ER-positive breast cancer line but not in a triple-negative breast cancer line (Fig. 1C). We concluded that our CRISPR dropout assay can robustly identify both pan-essential and cancer-specific genetic dependencies.
Figure 1. Cell competition assays to test the essentiality of putative cancer dependencies.
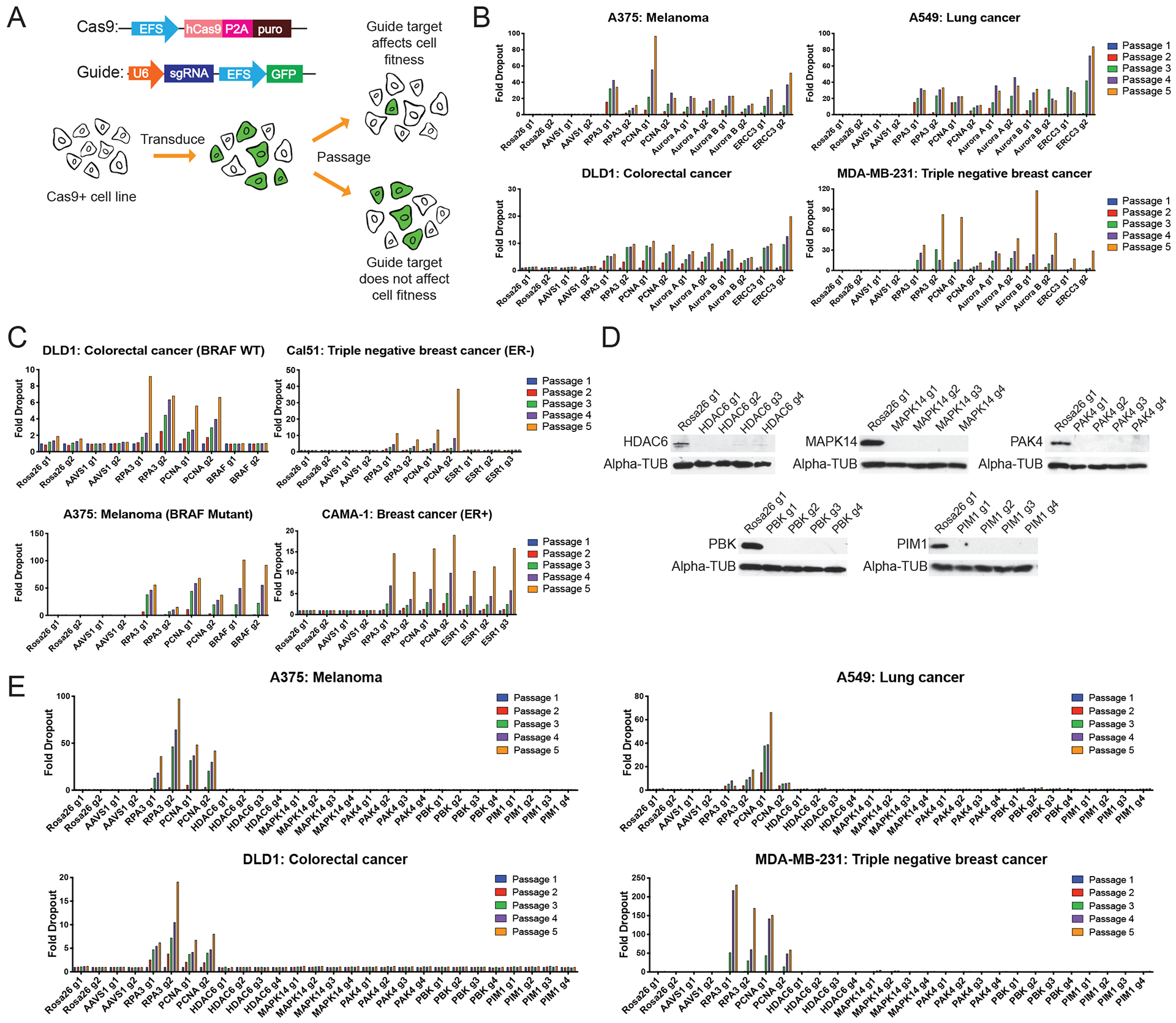
(A) Schematic of the CRISPR-based cell competition assays used in this paper (18).
(B) Cell competition assays comparing guides targeting AAVS1 and ROSA26 (non-essential, negative control genes), RPA3 and PCNA (pan-essential positive control proteins), and Aurora A, Aurora B, and ERCC3 (inhibitor-validated cancer dependencies). Full results from these competition experiments are included in data file S2.
(C) Cell competition assays for the cell type-specific cancer dependencies BRAF and ESR1.
(D) Western blot analysis of A375 populations transduced with the indicated guide RNAs.
(E) Cell competition assays with guide RNAs targeting HDAC6, MAPK14, PAK4, PBK, or PIM1 in four different cancer cell lines.
We next designed guide RNAs against the reported cancer dependencies HDAC6, MAPK14 (p38α), PAK4, PBK, and PIM1. To maximize the likelihood that a CRISPR-induced mutation results in a non-functional allele, guides were designed to target exons that encode key functional domains within a protein (fig. S1A)(18). We used western blotting to verify that each guide resulted in strong protein depletion in four separate cell lines (Fig. 1D and fig. S1B), and we then further confirmed target ablation by performing a second set of western blots with a different antibody that recognizes a distinct protein epitope (fig. S1C). Next, we conducted GFP competition assays in 32 cell lines from 12 different cancer types, which included multiple cell lines in which each gene had previously been reported to be essential (data file S1). In each experiment, four guide RNAs targeting Rosa26 and AAVS1 were used as negative controls, while four guide RNAs targeting PCNA and RPA3 were used as positive controls. These positive control guides dropped out between ~10-fold and ~200-fold over five passages in culture, whereas the negative control guides consistently exhibited <2.5-fold dropout. The variation in positive-control dropout rates likely reflects cellular differences in Cas9 expression, proliferation, and the spectrum of indel mutations produced by the guide RNA. Notably, all guides targeting HDAC6, MAPK14, PAK4, PBK, and PIM1 failed to drop out in every cell line that we tested (Fig. 1E, fig. S2, and data file S2). For instance, HDAC6 has been reported to be a genetic dependency in ARID1A-mutant ovarian cancer (6). However, in ARID1A-mutant ovarian cancer cell lines A2780, OVK18, OVTOKO, and TOV-21G, HDAC6-targeting guides failed to deplete above background levels. Similarly, PIM1 has been reported to be a genetic dependency in triple-negative breast cancer (14, 15), but PIM1-targeting guides were not depleted in any of the seven triple-negative breast cancer cell lines that we tested (data file S2). These results called into question whether these putative drug targets are indeed required for cancer cell growth.
Generation and analysis of CRISPR-derived knockout clones
To further test the essentiality of these genes in cancer, we derived clones harboring CRISPR-induced knockouts in each gene in multiple cancer types. All five genes were knocked out in the triple-negative breast cancer cell line MDA-MB-231 and the melanoma cell line A375. HDAC6, MAPK14, PBK, and PIM1 were knocked out in the colorectal cancer cell line DLD1, whereas PAK4 was knocked out in the colorectal cancer cell line HCT116, because it has previously been reported that PAK4 is not a dependency in DLD1 (11). To minimize the possibility that downstream translational initiation or alternative splicing bypass the effect of a single CRISPR-induced mutation, clones were made by co-transducing cancer cells with guides that targeted two different exons in a gene of interest (Fig. 2A and fig. S1A). Complete target ablation was then verified by western blotting using two antibodies that recognized distinct protein epitopes (Fig. 2B, fig. S3, fig. S4, and fig. S5A). We next compared these knockout clones to control clones transduced with guides targeting Rosa26 or AAVS1. As a positive control, we confirmed that knocking out the verified drug target MEK1 decreased proliferative capacity in A375 clones (fig. S4). However, we found that clones lacking each putative genetic dependency listed in Table 1 proliferated at levels that were indistinguishable from control A375, DLD1, and HCT116 cancer cells (Fig. 2C). For instance, PAK4-KO melanoma cells underwent an average of 20.3 population doublings over the course of 15 days in culture, compared to 19.9 doublings for the Rosa26 guide RNA-transduced clones. To test whether these genes were dispensable for cell division but required for growth in other environments, we also seeded the knockout clones in soft agar and assessed their ability to grow in anchorage-independent conditions. While MEK1-KO clones formed fewer colonies in soft agar (fig. S4E), every HDAC6, MAPK14, PAK4, PBK, and PIM1 knockout exhibited wild-type rates of colony formation, further verifying that these genes are not required for cancer cell fitness (Fig. 2D–E).
Figure 2. Generating and analyzing single cell-derived knockout clones of putative cancer dependencies.
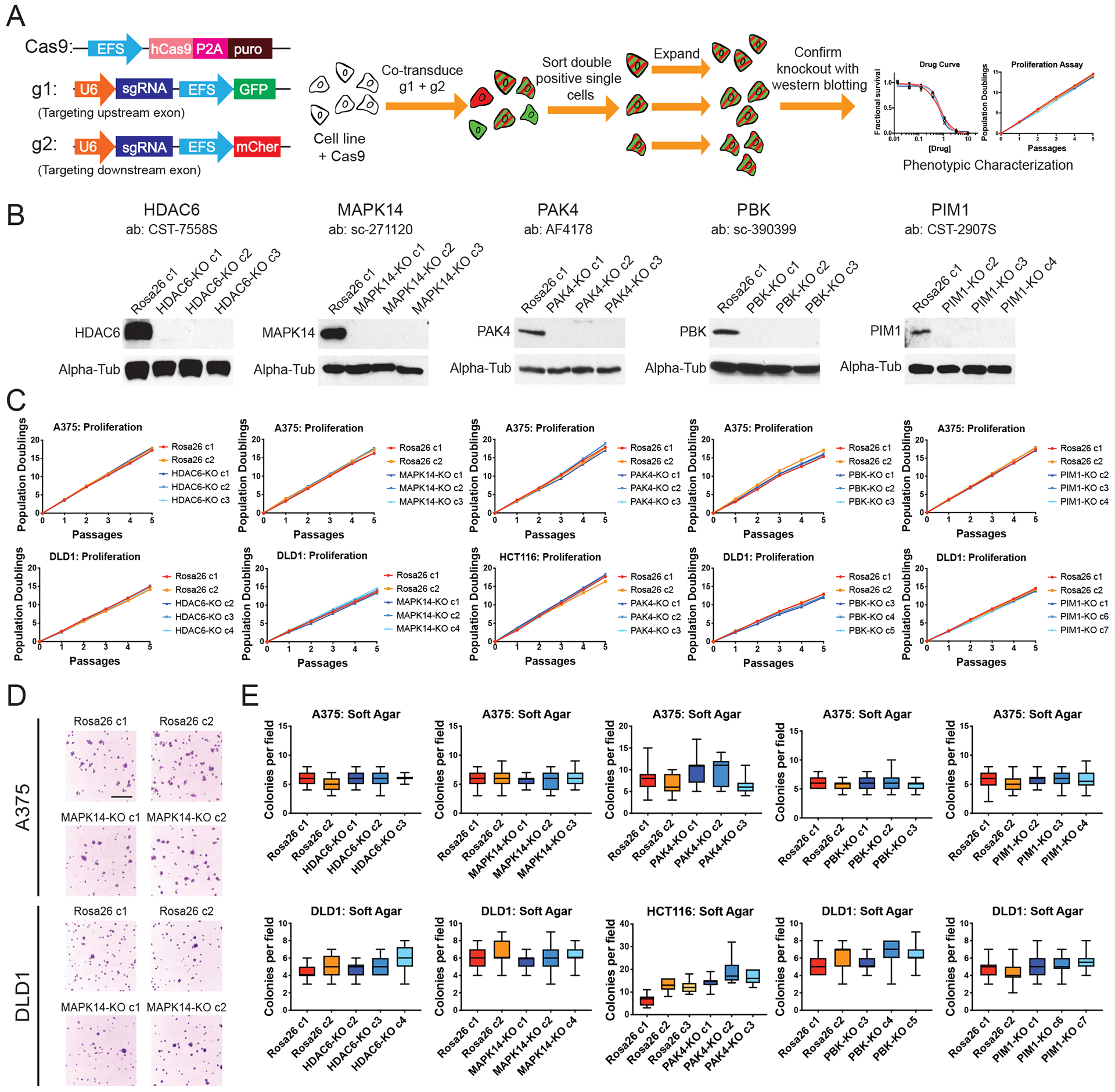
(A) Schematic of the two-guide strategy used to generate clonal knockout cell lines.
(B) Western blot analysis of single-cell derived A375 knockout clones.
(C) Proliferation assays for HDAC6, MAPK14, PAK4, PBK, and PIM1 knockout clones.
(D) Representative images of A375 and DLD1 Rosa26 or MAPK14-KO clones grown in soft agar. Scale bar, 2 mm.
(E) Quantification of colony formation in control or knockout A375, DLD1, and HCT116 clones. Boxes represent the 25th, 50th, and 75th percentiles of colonies per field, and the whiskers represent the 10th and 90th percentiles. For each assay, colonies were counted in at least 15 fields under a 10x objective.
Consistent with previously reported results, Rosa26 and AAVS1 control clones derived from MDA-MB-231 cell populations exhibited some variability in proliferative capacity (3, 4, 22). By analyzing a total of 12 single cell-derived control clones, we established a range of doubling times in which wild-type MDA-MB-231 cells can divide (fig. S5B). Every HDAC6, MAPK14, PAK4, PBK, and PIM1 knockout clone proliferated at a comparable rate to these control clones (fig. S5C). All KO clones were also capable of forming colonies in soft agar at rates comparable to the control clones, further verifying that these putative dependencies are non-essential in breast cancer (fig. S5D).
Lack of homolog upregulation in knockout clones
Null mutations caused by CRISPR may trigger a different cellular response than RNAi-induced gene repression, potentially contributing to the discrepancies between our results and those that had previously been reported. In particular, a recent study suggested that CRISPR-induced nonsense mutations can trigger the up-regulation of the homologs of a targeted gene, potentially compensating for the effects of the lesion (23). We assessed the expression of the closest homologs of HDAC6, MAPK14, PAK4, PBK, and PIM1 in 33 different knockout clones that we generated, but we observed no consistent up-regulation of any target homolog (fig. S6). Additionally, we analyzed RNA-Seq data from 10 published experiments in gene-edited cancer cells from other laboratories, and similarly failed to detect consistent evidence for the up-regulation of target gene homologs (fig. S7). Indeed, in several experiments, we found that the homologs of the targeted gene were down-regulated. These results suggest that homolog up-regulation is not a common consequence of CRISPR mutagenesis in human cancer cells and that compensatory homolog over-expression is unlikely to explain the lack of a detectable growth defect in the CRISPR clones that we have analyzed.
Assessing putative cancer dependencies in whole-genome CRISPR and RNAi screens
Cell lines can exhibit inter-laboratory variability that affects their response to different genetic and chemical perturbations (24). Additionally, although we chose cancer types to study based on the dependency patterns reportedly exhibited by each gene (data file S1), it remained possible that these genes represent dependencies in a cancer lineage not included among the 32 cell lines that we studied. To test this possibility, and to assess whether unique or non-representative features of the cell lines used in our laboratory contributed to our discrepant results, we re-analyzed genetic dependency data from whole-genome CRISPR screens conducted in 485 cancer cell lines (fig. S8A). These screens consistently identified both pan-cancer and cell type-specific genetic dependencies (for example, Aurora B, BRAF, PIK3CA; fig. S8B–C). However, in accordance with our earlier results, these experiments also indicated that our chosen dependencies were fully dispensable for cancer cell fitness (fig. S8A–C). For instance, MAPK14/p38α has previously been reported to be essential in breast cancer (9), but CRISPR screens conducted in 26 different breast cancer cell lines corroborate that its loss is tolerated without a substantial fitness defect (fig. S8D). Strikingly, we also re-analyzed 712 genome-wide shRNA screens, and these knockdown experiments similarly failed to identify HDAC6, MAPK14, PAK4, PBK, or PIM1 as cancer-essential genes (fig. S8E–G). In total, these results indicate that our findings are unlikely to be explained by non-representative features of the cell lines studied in our laboratory, by differences between partial and complete loss-of-function perturbations, or by these genes functioning as genetic dependencies only in certain cancer types. Instead, our data suggest that multiple genes targeted in cancer clinical trials are in fact fully dispensable for cancer cell growth.
Knocking down putative cancer dependencies with CRISPRi
To further investigate whether differences between partial and complete loss-of-function perturbations could explain our discrepant results, we next performed competition experiments using the CRISPRi system. In this approach, catalytically-inactive Cas9 is fused to a transcriptional repressor and targeted to a gene’s promoter, resulting in down-regulation of gene expression without the generation a complete loss-of-function-inducing frameshift mutation (25). We designed three guide RNAs that recognized HDAC6, MAPK14, PAK4, PBK, and PIM1, and verified that these constructs blocked the expression of their targets (fig. S9A). We then conducted competition experiments in four different cell lines, and we found that gRNAs targeting the essential replication protein MCM2 exhibited ~10-fold to ~20-fold dropout, while gRNAs targeting HDAC6, MAPK14, PAK4, PBK, and PIM1 failed to deplete (fig. S9B). These assays further verify that our results cannot be explained by the existence of different cellular responses to partial and complete loss-of-function alterations.
Assessing the sensitivity of target-knockout clones to chemotherapy agents undergoing combination clinical trials
Several of the proteins listed in Table 1 are currently undergoing combination clinical trials using their targeted inhibitors together with other chemotherapy agents. It is conceivable that a protein could be non-essential under normal conditions but that its loss sensitizes cells to specific chemotherapies. For instance, HDAC6 is capable of deacetylating microtubules (26), and HDAC6 inhibition has been reported to render cells vulnerable to drugs that interfere with microtubule dynamics (27). As a result of this preclinical work, two clinical trials are combining HDAC6 inhibitors with the microtubule stabilizer paclitaxel (NCT02632071 and NCT02661815). We therefore tested whether the knockout clones that we had generated were sensitive to various chemotherapy agents (fig. S10A–C). In contrast to previous results, loss of HDAC6 failed to sensitize cells to paclitaxel or to four other anti-cancer drugs (fig. S10A). Similarly, p38α inhibitors have been clinically applied in combination with bortezomib, gemcitabine, carboplatin, and temozolomide (NCT00087867, NCT00095680, NCT01663857, and NCT02364206), but MAPK14/p38α knockout clones in multiple cell lines were as sensitive to these agents as Rosa26 control clones (fig. S10B). These results suggest that, in addition to being non-essential, these putative drug targets do not affect sensitivity to several chemotherapy agents that have been tested in combination trials.
Assessing RNAi promiscuity as a cause of the misidentification of cancer dependencies
If these genes do not drive cancer growth or chemotherapy resistance, then why have inhibitors targeting the proteins that they encode been tested in human cancer patients? A review of the literature indicates that each of these genes has been described to be essential on the basis of RNAi-induced knockdown phenotypes (data file S1). Off-target toxicity has been reported to be a common problem in the design and interpretation of RNAi-based experiments (28–30), though the impact of these issues on the therapeutic development pipeline is not known. We acquired four different RNAi constructs that were used in these prior studies and then tested their effects on the clones that we had generated. While we were able to confirm that each construct decreased the expression of its putative target, we also discovered that these constructs impaired proliferation in both WT clones and clones in which the construct’s target had been knocked out (fig. S11A–C). For example, a recent report found that PAK4-targeting siRNAs blocked cell division in HCT116 colon cancer cells and concluded that PAK4 was a genetic dependency in this cell line (31). However, we found that these same siRNAs induced an equivalent decrease in proliferation in both HCT116 PAK4-KO and HCT116 Rosa26 clones, suggesting that their effects on growth are a consequence of off-target toxicity (fig. S11A). Similarly, while knocking down PIM1 has been reported to block proliferation in the MDA-MB-231 breast cancer cell line (15), this construct had the same effect in MDA-MB-231 PIM1-KO cells (fig. S11B). Our results therefore suggest that these drug targets have advanced to clinical testing due at least in part to promiscuous RNAi constructs.
Assessing the specificity of cancer drugs undergoing clinical trials
Off-target toxicity from small-molecule drugs can cause dangerous side effects and is a major cause of clinical trial failure (32, 33). Our results suggested that the drugs listed in Table 1 were designed to target non-essential cellular proteins, raising the possibility that the anti-cancer effects of these drugs could be due to off-target interactions. We therefore sought to apply CRISPR to differentiate between the on-target and off-target effects of each clinical cancer drug. First, we confirmed that CRISPR could be used to verify the MOA for several genetically-validated therapies. The natural product rapamycin is reported to bind to the prolyl-isomerase FKBP12, and this complex inhibits the essential mTOR kinase (fig. S12A)(34, 35). We knocked out FKBP12 using CRISPR, and we verified that these KO clones exhibited increased resistance to rapamycin treatment (fig. S12B–C). Similarly, knocking out p53 conferred resistance to the experimental p53-activating drug nutlin-3a (fig. S12D–F). Finally, we sought to test whether CRISPR could be used to validate a published resistance-granting point mutation. We used CRISPR-mediated homology-directed repair (HDR) to introduce a missense mutation into the kinase domain of the essential mitotic kinase MPS1, and we verified that this substitution was capable of granting resistance to the small-molecule MPS1 inhibitor AZ3146 (fig. S12G–I)(36). Thus, CRISPR-derived knockout and knock-in cell lines can be used to validate on-target drug activity.
Next, we applied CRISPR to interrogate the MOA of two caspase-3 activating compounds, PAC-1 and 1541B. These drugs are reported to function by catalyzing the conversion of caspase-3 from its inactive, procaspase state to its active, cleaved form, thereby causing cellular apoptosis (fig. S13A)(16, 17). Currently, PAC-1 is undergoing three different clinical trials in cancer patients (NCT02355535, NCT03332355, and NCT03927248). We knocked out the CASP3 gene in four different cell lines and then verified protein ablation using two different antibodies (Fig. 3A and fig. S13). However, these CASP3-KO lines exhibited identical sensitivity to PAC-1 and 1541B compared to Rosa26 controls (Fig. 3B, fig. S13D and S13F). These results suggest that a putative caspase-3 activator undergoing clinical trials actually kills cancer cells in a caspase-3-independent manner.
Figure 3. Target-independent cell killing by multiple anti-cancer drugs.
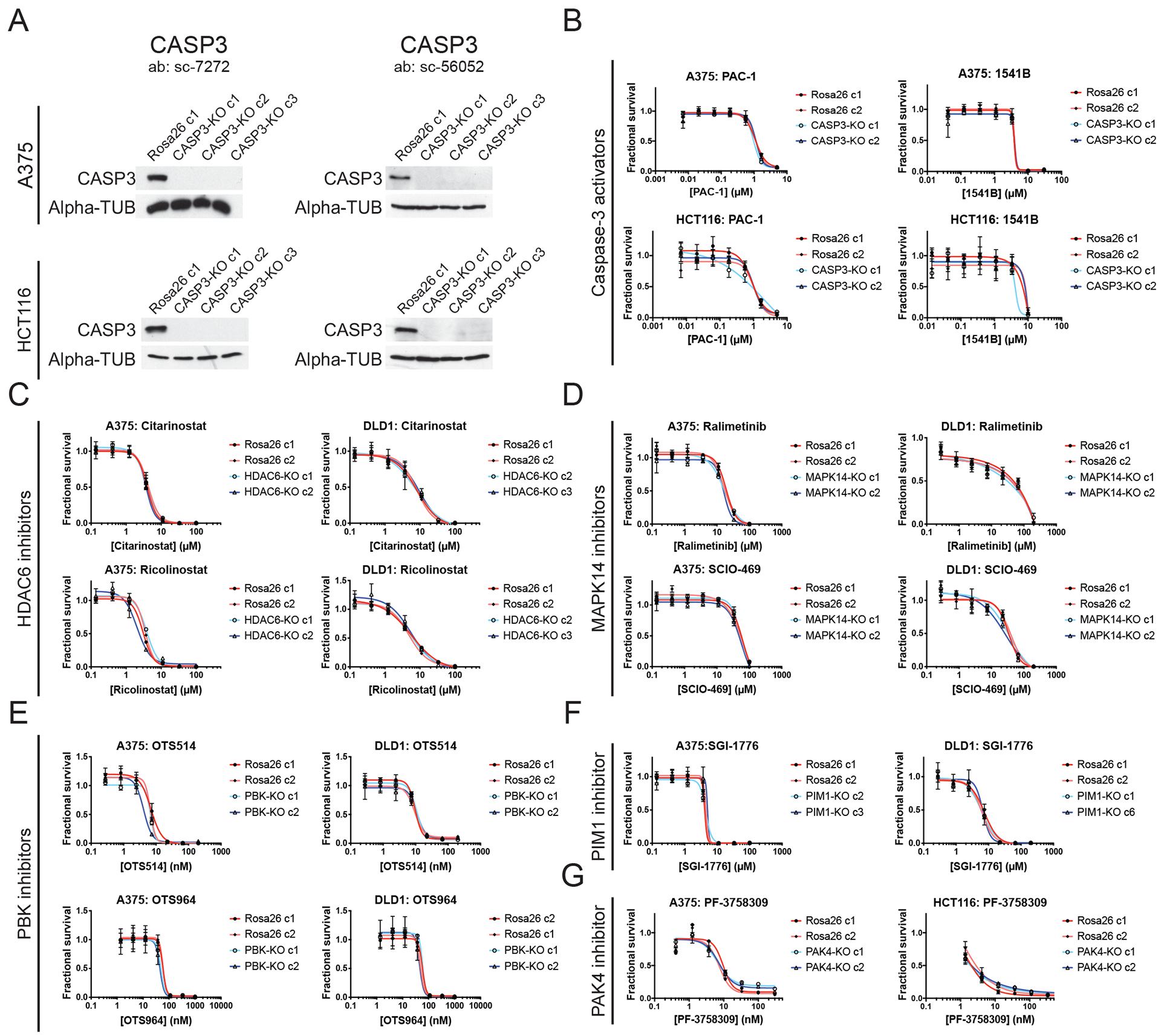
(A) Western blot analysis for caspase-3 in A375 and HCT116 cells.
(B) 7-point dose-response curves of Rosa26 and CASP3-KO A375 and HCT116 cells in the presence of two putative caspase-3 activators, 1541B and PAC-1.
(C) 7-point dose-response curves of Rosa26 and HDAC6-KO A375 and DLD1 cells in the presence of two putative HDAC6 inhibitors, ricolinostat and citarinostat.
(D) 7-point dose-response curves of Rosa26 and MAPK14-KO A375 and DLD1 cells in the presence of two putative MAPK14 inhibitors, ralimetinib and SCIO-469.
(E) 7-point dose-response curves of Rosa26 and PBK-KO A375 and DLD1 cells in the presence of two putative PBK inhibitors, OTS514 and OTS964.
(F) 7-point dose-response curves of Rosa26 and PIM1-KO A375 and DLD1 cells in the presence of a putative PIM1 inhibitor, SGI-1776.
(G) 7-point dose-response curves of Rosa26 and PAK4-KO A375 and HCT116 cells in the presence of a putative PAK4 inhibitor, PF-3758309.
We next tested each putative HDAC6, MAPK14, PAK4, PBK, and PIM1 inhibitor in control and knockout clones. If these drugs act by specifically inhibiting their reported targets, then cancer cells that totally lack the expression of their targets would be expected to be resistant to these drugs’ effects. In contrast, if a drug kills cells in which its reported target has been knocked out, then this drug necessarily kills cells by affecting another protein or proteins. In every instance that we tested, cancer cells in which HDAC6, MAPK14, PAK4, PBK, or PIM1 had been knocked out exhibited wild-type sensitivity to their putative targeted inhibitors (Fig. 3 and fig. S14A–B). For example, we found that the PAK4 inhibitor PF-3758309 blocked the growth of both Rosa26 and PAK4-KO melanoma cells with a GI50 value of ~9 nM (Fig. 3G). Given that this drug is fully capable of killing cells in which its putative target has been deleted, the ability of PF-3758309 to block cancer cell growth must be through an off-target effect. To further interrogate whether the drugs studied in this manuscript could exhibit an on-target MOA in an additional genetic background, we knocked out HDAC6 in TOV-21G cells, an ARID1A-mutant ovarian cancer cell line in which this gene has been reported to be a dependency (fig. S15A)(6). However, TOV-21G HDAC6-KO cells exhibited wild-type fitness in vitro and in soft agar (fig. S15B–C), and these cells remained sensitive to citarinostat and ricolinostat, two putative HDAC6 inhibitors in clinical trials (fig. S15D). In total, all 10 different anti-cancer agents targeting CASP3, HDAC6, MAPK14, PAK4, PBK, or PIM1 exhibited clear evidence of target-independent cell killing in every knockout cell line that we examined.
Finally, we applied these putative inhibitors to investigate several combination chemotherapy trials. As described above, HDAC6 inhibitors are currently undergoing testing in cancer patients along with paclitaxel, and p38α inhibitors have also been combined with several different therapeutic agents. We verified that co-treatment with a targeted inhibitor and a second agent generally caused a greater decrease in cancer cell viability than either agent alone (fig. S14B). However, this synthetic enhancement was observed in both Rosa26 and target-knockout clones, suggesting that these additive effects are also due to an off-target interaction.
Discovering the true target of OTS964
If these clinical anti-cancer therapies do not kill cells by inhibiting their reported targets, then how do they block cancer growth? We note that, although the MOA of each drug has previously been characterized using biochemical and biophysical approaches, there is little genetic evidence linking each drug to its reported target. We hypothesized that an alternative, genetic methodology could shed light on the true target of a therapeutic agent whose MOA was in question.
For this work, we chose to focus on the putative PBK inhibitor OTS964, because it exhibited nanomolar potency in multiple cancer types and our CRISPR experiments had provided clear evidence that PBK was not required for cell proliferation. Moreover, OTS964 has been reported to affect mitotic progression (13), and anti-mitotic drugs have historically proven to be highly successful anti-cancer agents (37). To identify mutations that conferred resistance to OTS964, we used HCT116 colorectal cancer cells, which harbor an increased mutation rate caused by a defect in mismatch repair (38). We cultured HCT116 cells in the presence of a lethal concentration of OTS964 and successfully isolated 12 clones that were capable of growing under these conditions (Fig. 4A). We found that these clones exhibited stable resistance to OTS964, as they failed to revert to OTS964 sensitivity after prolonged growth in normal medium (fig. S16A). Cancer cells commonly acquire chemotherapy resistance by amplifying the P-glycoprotein drug efflux pump (39). However, the OTS964-resistant clones remained sensitive to paclitaxel, a verified P-glycoprotein substrate, suggesting that they had not acquired a multi-drug resistance phenotype (fig. S16B)(40). These experiments indicated that our drug-resistant clones could harbor a mutation or mutations that specifically altered OTS964 sensitivity.
Figure 4. Discovery of CDK11 as the in cellulo target of the mis-characterized anti-cancer drug OTS964.
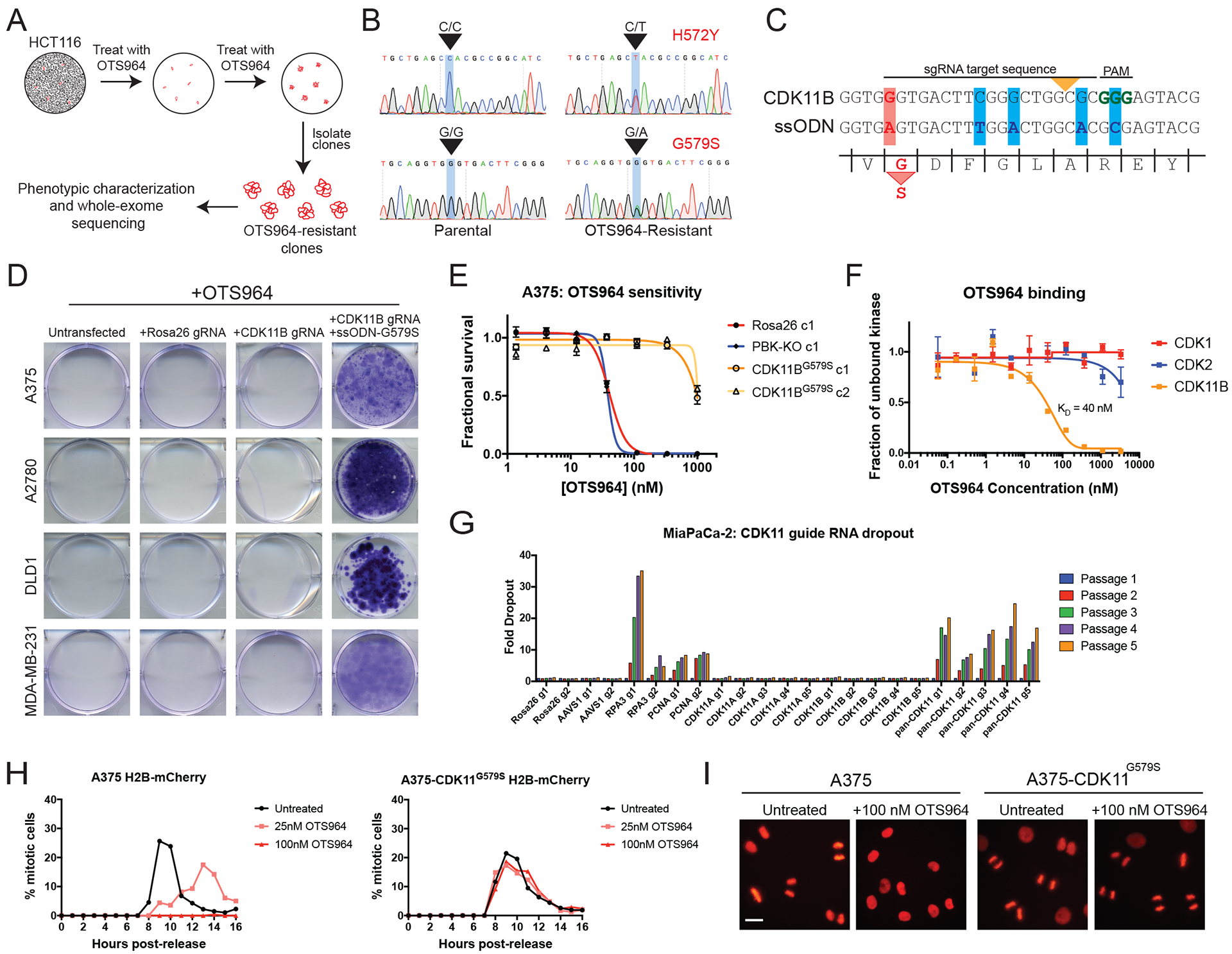
(A) A schematic of the strategy to use the highly mutagenic HCT116 cell line to isolate mutations that confer OTS964 resistance.
(B) Sanger sequencing validation of two heterozygous mutations in the CDK11B kinase domain.
(C) Constructs used to introduce the G579S mutation into CDK11B via CRISPR-mediated HDR. The yellow arrowhead indicates the site of Cas9 cleavage, the red bar indicates the G579S substitution, and the blue bars indicate silent mutations introduced to prevent re-cutting after HDR.
(D) Crystal violet staining of cancer cells transfected with the indicated constructs and then cultured in a lethal concentration of OTS964.
(E) 7-point dose-response curves of Rosa26, PBK-KO, and CDK11BG579S clones grown in varying concentration of OTS964.
(F) Titration experiments reveal that OTS964 binds to CDK11B with a KD of 40 nM.
(G) Pancreatic cancer cell line MiaPaca-2 was transduced with guides specific CDK11A, guides specific for CDK11B, or guides that harbored cut sites in both genes.
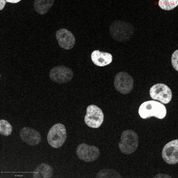
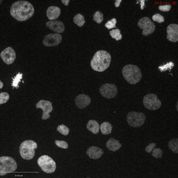
(H) A375 H2B-mCherry cells (left) or A375 H2B-mCherry cells that express CDK11BG579S (right) were arrested at G1/S with a double-thymidine block and then were released into normal medium or medium containing OTS964. The percentage of mitotic cells in each population was scored every hour.
(I) Representative images of the experiments in (H), 9 hours after release from thymidine. Scale bar, 50 μm.
To identify genetic alterations capable of conferring OTS964 resistance, we subjected 10 OTS964-resistant clones, one Rosa26 control clone, and the parental cell population to whole-exome sequencing (WES). Notably, all 10 resistant clones were found to harbor heterozygous missense mutations in the poorly-characterized cyclin-dependent kinase CDK11B (fig. S16C). Eight clones harbored two mutations in this gene, H572Y and G579S, in trans, while two clones harbored only the G579S substitution. No CDK11B mutations were observed in the parental population or in the Rosa26 control clone. Sanger sequencing verified the presence of the CDK11B mutations in two independent drug-resistant clones that were not subjected to WES (Fig. 4B and fig. S16C). These mutations were also absent from additional control clones that were analyzed (fig. S16C) and have not been previously observed in the Catalog of Somatic Mutations in Cancer database (41).
The human genome encodes two CDK11 proteins, CDK11A and CDK11B, that are 97% identical and that arose from an evolutionarily-recent gene duplication event (42). The CDK11 family has been reported to support various cellular processes, including transcription, splicing, and chromosome segregation (43), but its role in cancer is unknown. No drugs have previously been reported to target CDK11, and inhibitors that are specific for single cyclin-dependent kinases (CDKs) are difficult to discover due to the sequence similarity among these kinases (44). We aligned the sequences of the human CDKs, and we noted that 19 out of 20 of these proteins harbored an alanine residue immediately upstream of the magnesium-coordinating DFG motif (fig. S16D). Only CDK11 contained a glycine at this location, and this glycine was mutated to serine in every OTS964-resistant clone that we sequenced (fig. S16C–D). This amino acid position (called “xDFG”) has previously been identified as a key residue that affects kinase inhibitor binding (45), suggesting a potential basis for CDK11-selective inhibition. To test whether the xDFG Gly→Ser mutation was sufficient to confer resistance to OTS964, we designed a strategy to use CRISPR-mediated homology-directed repair to introduce this substitution into the endogenous CDK11B gene (Fig. 4C). These experiments revealed that this point mutation was sufficient to restore viability in A375, A2780, DLD1, and MDA-MB-231 cancer cells grown in a lethal concentration of OTS964 (Fig. 4D–E). To verify that our results were not an off-target effect of CRISPR, we generated a retrovirus to stably express CDK11BG579S cDNA, and we confirmed that this construct was also sufficient to confer OTS964 resistance (fig. S16E). In an HCT116 clone that had spontaneously evolved resistance to OTS964, eliminating mutant CDK11B with CRISPR restored OTS964 sensitivity, demonstrating that this alteration is both necessary and sufficient for drug resistance (fig. S16F). Introducing an alanine substitution into residue 579, so that the CDK11B xDFG motif was identical to the other human CDKs, was also sufficient to decrease the efficacy of OTS964 (fig. S16G–H). Finally, to confirm a direct interaction between OTS964 and CDK11B, we assessed the binding of OTS964 to different CDKs. OTS964 bound to CDK11B with a KD of 40 nM, and it displayed greater than 10-fold selectivity for this kinase compared to several other CDKs (Fig. 4F and fig. S16I). In total, these results indicate that the putative PBK inhibitor OTS964 actually functions by targeting CDK11, and its specificity for this kinase is conferred by CDK11’s distinct xDFG motif.
Discovering the consequences of CDK11 inhibition in cancer
We next determined the cellular effects of OTS964 treatment and CDK11 ablation with CRISPR. In cell competition assays, cancer cells transduced with guide RNAs specific for either CDK11A or CDK11B exhibited minimal dropout. However, guides designed to recognize both isoforms exhibited substantial dropout in every cell line that we tested, including pancreatic cancer and triple-negative breast cancer (Fig. 4G and fig. S17A). Flow cytometry revealed that cells transduced with pan-CDK11 guides accumulated in G2/M with 4C DNA content, suggesting that CDK11 function is required for mitotic progression (fig. S17B). To test whether OTS964 phenocopied the CDK11 guide RNAs, we arrested A375 cells expressing the chromosomal marker H2B-mCherry at G1/S with a double-thymidine block, and then released them into normal medium or medium containing OTS964. Cells treated with a low concentration of OTS964 exhibited delayed nuclear envelope breakdown and progressed slowly through mitosis (Fig. 4H, fig. S17C, and movies S1–2). Cells treated with a lethal concentration of OTS964 arrested in G2, before mitotic entry (movie S3). OTS964 treatment did not perturb DNA replication, as the arrested cells displayed 4C DNA content and did not accumulate 53BP1-foci, a marker of DNA damage (fig. S17D–E). Introducing the G579S substitution into A375 cells rescued normal mitotic entry and progression in the presence of a lethal concentration of OTS964 (fig. 4H–I, fig. S17C, and movie S4). These results establish CDK11 activity as necessary for mitosis in human cancer and suggest that CDK11 is the key in cellulo target of OTS964.
Discussion
It is generally known that small molecules can exhibit off-target effects that may confound the design of specific chemical inhibitors (46). Our data suggest that, rather than simply being the side effect of a drug, these off-target interactions are frequently the mechanism by which small molecules block cancer growth. Every inhibitor tested in this manuscript that lacked a previously-described resistance mutation was found to kill through an off-target effect; these results therefore identify this phenomenon as a common problem that affects cancer clinical trials. As 97% of drug-indication pairs tested in clinical trials fail to receive FDA approval (5), the mis-identification of essential genes in cancer and the mis-characterization of reportedly target-specific inhibitors likely contributes to their exceedingly high failure rate. The adoption of more stringent, genetic target and activity validation studies may alleviate this problem and decrease the failure rate of new cancer drugs.
Each gene that we studied has been reported to be required in a cell-autonomous manner for cancer proliferation by more than 180 publications, and it is this specific claim that our work sought to test (data file S1). Toward this end, we generated knockout clones in at least three cancer cell lines, we performed CRISPR competition assays in 32 cell lines, and we performed CRISPRi-knockdown competition assays in an additional four cell lines, which all consistently demonstrated that HDAC6, MAPK14, PAK4, PBK, and PIM1 are dispensable for cancer cell fitness. While cancer cells harbor the ability to evolve in response to various perturbations, we do not believe that this adaptability is sufficient to explain the robust growth of the CRISPR-modified cells that we have generated. First, in the competition assays that we conducted, cells are analyzed immediately after guide RNA transduction, allowing them no time to adapt to the loss of the targeted gene. For instance, while BRAF-addicted melanoma cells are capable of evolving BRAF-independence over time by acquiring secondary mutations in MEK or NRAS (47), we still observed a strong depletion of BRAF-targeting guides in these dropout assays. Secondly, we knocked out the verified drug target MEK1 and we confirmed that MEK1-KO clones grow substantially more slowly than Rosa26 control clones, demonstrating our ability to validate genetic dependencies in CRISPR-modified clones. Thirdly, while each of the genes that we studied has previously been reported to be essential, our experiments provide a mechanism to explain these discrepant results. In particular, we demonstrate that several RNAi constructs previously used to inhibit these genes exhibit identical anti-proliferative effects in target-WT and target-knockout cancer cells, suggesting that RNAi promiscuity contributed to the mis-identification of these genes as drug targets. Fourthly, while it has been proposed that cells can compensate for CRISPR-induced mutations by up-regulating homologs of the targeted gene (17), we failed to detect any evidence of this in our knockout clones or in genetically-modified cell lines from several independent laboratories. Lastly, high-throughput screens conducted in hundreds of cancer cell lines using both RNAi and CRISPR technologies have also failed to identify these genes as cancer dependencies. We therefore believe that cellular evolution after CRISPR mutagenesis is unlikely to explain the robust growth of the cancer cells lacking the drug targets that we have studied.
The cell lines studied in this paper were chosen based on the literature on each target, but we have not attempted to recapitulate every individual published result with each drug or drug target. Thus, it remains possible these drug targets exhibit a cell type-specific dependency pattern not uncovered in this work. To partially address this concern, we analyzed published whole-genome CRISPR and RNAi screening data from >700 cell lines, which consistently revealed that the genes studied in this work could be eliminated without substantially affecting cell fitness. Due to the breadth of cell lines tested both within our lab and through high-throughput screening, it is unlikely that these genes are genetic dependencies across cancer types or in a common cancer lineage. Nonetheless, we do not rule out the possibility that these genes are essential in a rare cancer type not included among those studied here. Additionally, it remains possible that these genes play a non-cell-autonomous role during tumorigenesis. For instance, while MAPK14/p38α has been reported to be essential for proliferation in breast cancer (9, 48), colon cancer (8), ovarian cancer (49), and several other cancer types, it has also been proposed to mediate inflammatory signaling (50). Thus, while our work provides strong evidence that these proteins are dispensable for cancer proliferation, we do not rule out the possibility that these proteins have some function in other, non-cell-autonomous processes related to tumor development in vivo.
Our results indicate that many cancer drugs in clinical trials kill cells independently of their reported targets. As the application of a predictive biomarker doubles the likelihood that a clinical trial will succeed (5), the inability to decipher a drug’s true target may prevent successful biomarker identification and contribute to trial failure. Moreover, our findings may provide evidence that cancer drug polypharmacology is a common MOA for reportedly target-specific compounds. For example, while ricolinostat has been reported to be a selective HDAC6 inhibitor (51), our work shows that HDAC6 expression is fully dispensable for ricolinostat sensitivity. These results are similar to those reported in (52), which found that ricolinostat continued to kill HDAC6-KO HAP1 cells. The human genome harbors 18 different histone deacetylases (53), and it is possible that this drug kills cells by inhibiting HDAC6 and several synthetically-redundant HDAC family members. Additionally, the invalidation of a drug’s putative target does not necessarily mean that a drug will be ineffective in the clinic, as some broadly non-specific inhibitors have proven efficacious in certain circumstances. In many cases, these successes derive from a thorough understanding of a drug’s MOA. For instance, the multi-targeted kinase inhibitor midostaurin has received FDA approval for use in FLT3+ leukemias because of its demonstrated activity against FLT3 (54). Thus, strong validation of on-target drug activity remains essential.
Alternately, these mis-characterized drugs may kill cells by inhibiting single, specific proteins that are not closely related to their reported targets. For instance, our results demonstrate that the putative PBK inhibitor OTS964 functions by blocking CDK11 activity. Several CDK inhibitors have received FDA approval or are in late-stage trials for various malignancies, underscoring the clinical potential for targeting members of the cyclin-dependent kinase family (55). However, no CDK11-specific inhibitors have been previously described (56). OTS964 and its derivatives could therefore allow us to block cancer growth by inhibiting a previously-undruggable mitotic CDK. Furthermore, our work identifies CDK11’s distinct xDFG motif as a key determinant of drug sensitivity, suggesting a potential structural basis for CDK11-specific inhibition. Although CDK11 has previously been reported to function in chromosome segregation (57), our results demonstrate that its activity is required for entry into mitosis. It will therefore be crucial to investigate whether CDK11 inhibitors are capable of synergizing with PLK1 inhibitors, Aurora A inhibitors, or any other drugs that similarly target mitotic entry (58). Finally, the CDK11 locus on Chromosome 1p has been reported to be deleted or translocated in several cancer types, including melanoma and neuroblastoma (43), raising the exciting possibility that alterations in this gene family could serve as predictive biomarkers for CDK11-inhibitor sensitivity.
More broadly, our results underscore the power of genetic approaches to improve the preclinical characterization of cancer drugs and drug targets. In particular, CRISPR-mediated gene editing is a powerful methodology for interrogating the effects of loss-of-function alterations in disease-relevant genes, and head-to-head comparisons have verified that CRISPR is less susceptible to off-target effects than RNA interference (59, 60). Although biochemical and biophysical approaches can demonstrate target engagement by a potential therapeutic molecule, these assays alone are insufficient to demonstrate the relevance of this interaction in cellulo. Mutagenesis experiments, using either spontaneous or CRISPR-directed approaches, can complement these assays to verify or discover a drug’s true MOA and indicate potential biomarkers for sensitivity and resistance. We suggest that the adoption of stringent genetic characterization assays in the preclinical setting will decrease the number of drugs used in human cancer patients that fail to provide any clinical benefit.
Materials and methods
Study design
In this work, we sought to determine whether several drug targets were truly essential for cancer cell fitness. After discovering that many of these drug targets were non-essential, we investigated whether the drugs used to target them killed cells through an off-target effect. The cell lines used in this study were selected based on an analysis of the literature on these drug targets (summarized in data file S1). No predetermined sample sizes were used for this analysis. No randomization or blinding was performed. Cell competition experiments represent single biological replicates. Proliferation assays represent two to three biological replicates. Drug-sensitivity curves were generated with three to six technical replicates. Soft agar experiments represent three technical replicates with at least 15 independent fields counted for each experiment. Raw data for the cell competition experiments are included in data file S2.
Selecting drug targets to study
Our lab previously investigated the role of MELK in cancer (3, 4). We found that, contrary to previous results obtained with RNAi, cancer cells tolerated CRISPR-induced ablation of MELK with no loss in cell fitness. Additionally, we discovered that OTS167, a small-molecule inhibitor of MELK in clinical trials, killed cells in a MELK-independent manner. These findings led us to investigate whether MELK and OTS167 were aberrations, or whether other drugs and drug targets had been similarly mischaracterized.
To begin this project, we generated a list of drugs and drug targets to study. We constructed this list using a few criteria, informed in part by our experience studying MELK. First, we sought to identify cancer genes that reportedly played a cell-autonomous role in cancer growth, so that we could study the most relevant phenotypes that resulted from their ablation in cell culture. Thus, we did not consider drugs that primarily target angiogenesis, the immune checkpoints, or related in vivo processes. (Importantly, our work does not rule out in vivo roles for the genes studied in this paper). Secondly, we only considered drugs that were reported to act by targeting single, specific proteins. If a drug was believed to act by inhibiting multiple proteins, then this would confound our CRISPR experiments, as the genetic ablation of a single gene would not be expected to phenocopy the effects of the inhibitor. Thirdly, we focused on genes that were reported to have broad dependency patterns, allowing us to study the consequences of their inhibition in a wide range of cell lines. Fourthly, we focused on genes that had been largely characterized using RNA interference, though we did not exclude genes that had been previously studied using CRISPR, transgenic mice, dominant-negative alleles, or other approaches. Fifthly, we chose drugs that were in advanced preclinical or clinical testing. Sixthly, we posited that the gold standard for showing on-target drug activity was the identification of a mutation that confers resistance to a targeted inhibitor, and we sought to study drugs that lacked known resistance-granting mutations.
Using these criteria, we searched PubMed, the database of American clinical trials (https://clinicaltrials.gov/), and other related resources (61) for drugs and drug targets that fit these criteria. We did not aim to comprehensively identify every drug that met the above criteria, but instead chose to limit ourselves to a small number of targets such that we could deeply characterize each one. Using these approaches, we chose to study CASP3, HDAC6, MAPK14, PAK4, PBK, and PIM1. We first became aware of the putative PBK inhibitor OTS964 because it was developed by the same company that created the MELK inhibitor that we previously studied. A press release on this drug reported that clinical trials would soon be initiated, though to our knowledge these clinical trials have not yet begun (62). Additionally, our list of drugs initially included the putative PAK4 inhibitor KPT-9274. However, while performing the research described in this paper, a report was published that identified mutations in NAMPT that granted resistance to KPT-9274, and we therefore did not further pursue this compound (63). After the initial submission of this manuscript, a second group independently demonstrated that the putative HDAC6 inhibitor ricolinostat kills HDAC6-KO HAP1 cancer cells, in accordance with our results (52).
We also chose to study two drugs, PAC-1 and 1541B, that reportedly function by activating the apoptosis protein caspase-3. While caspase-3 is not considered to be a “cancer dependency”, its activation by small molecules has been reported to trigger cancer cell apoptosis (64, 65). The first activator, PAC-1, was introduced in a publication that used in vitro methods to demonstrate the ability of PAC-1 to induce pro-caspase 3 cleavage and activation. Furthermore, the group implied specificity of PAC-1 for caspase-3 by showing that it exhibited a higher IC50 value in MCF7 cells, a caspase 3-deficient breast cancer cell line, compared to caspase 3-expressing cell lines (64). After this study, however, concerns were raised over whether caspase-3 activation was the true MOA of PAC-1. In a letter to Nature Chemical Biology, another laboratory reportedly failed to see significant activation of pro-caspase 3 in in vitro studies of PAC-1 (66). Additionally, the group stated that the caspase 3-deficient cell line, MCF7, displayed similar sensitivities to the drug as two caspase 3-expressing cell lines. Although this letter raised questions as to the true MOA of PAC-1, the original developers of this compound disputed these concerns, arguing that the different in vitro results were a consequence of different pro-caspase concentrations and buffer conditions used in the assays (67). In support of this claim, in later publications the group detailed the in vitro MOA of PAC-1 as a zinc chelator, describing a mechanism wherein zinc prevents procaspase-3 activation; therefore, the in vitro efficacy of PAC-1 is highly dependent on the concentration of zinc in the buffer (68). This group also argued that the activity of PAC-1 against the MCF7 cell line only occurs under conditions of low cell density and high drug concentration, and that the mechanism of death seems to resemble necrosis more than caspase-mediated apoptosis (67). Indeed, a number of other publications using PAC-1 and second-generation caspase-3 activators reported a similar resistance of MCF7 cells to caspase-3-activating compounds (65, 69). On the basis of this evidence, many in the field have continued to use PAC-1, not only in biological investigations but also in a number of clinical trials, under its listing as a caspase-3 activator (Table 1 and data file S1). In contrast to PAC-1, later caspase-3 activators, namely 1541, were reported to have direct interactions with caspase-3. 1541, the parental compound to the 1541B inhibitor used in our study, was not only able to induce caspase-3 activation in in vitro conditions where PAC-1 exhibited no effect, but 1541-resistance mutations in the CASP3 gene were also described (65). Because caspase-3 deficiencies have been linked with decreased sensitivity to a wide range of chemotherapeutic agents, we considered it possible that this putative resistance could be caused by an indirect effect on apoptosis (70, 71). Thus, due to the controversy and conflicting data concerning the MOA of different caspase 3-activators, we decided to include these compounds in our study.
Cell culture
The sources of each cell line are listed in data file S3. The identities of all human cell lines used in this study were confirmed using STR profiling (University of Arizona Genetics Core). A375, A549, A673, Cal51, Cama1, DLD1, HCT116, HEK293T, MDA-MB-231, PC3, SK-MEL-28, and U87 cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Thermo Fisher Scientific; Cat. No. 11995–073) supplemented with 10% Fetal Bovine Serum (FBS) (Sigma-Aldrich; Cat. No. F2442), 2 mM glutamine (Lonza; Cat. No. 17–605F), and 100 U/ml penicillin and streptomycin (Life Technologies; Cat. No. 15140122). A2780, DU-145, HCC1143, HCC38, HT29, K562, LNCaP, MDA-MB-453, MDA-MB-468, NCI-H82, OVK18, OVTOKO, SUIT2, SW480, and TOV-21G cell lines were cultured in Roswell Park Memorial Institute (RPMI)-1640 medium (Lonza; Cat. No. 12–115F/12) supplemented with 10% FBS, 2 mM glutamine, and 100 U/ml penicillin and streptomycin. MiaPaCa-2 cells were cultured in DMEM medium supplemented 10% FBS, 2.5% horse serum (Thermo Fisher Scientific; Cat. No. 26050088), 2 mM glutamine, and 100 U/ml penicillin and streptomycin. MCF7 cells were cultured in DMEM supplemented with 10% FBS, 0.01 mg/ml insulin (Thermo Fisher Scientific; Cat. No. 12585–014), 2 mM glutamine, and 100 U/ml penicillin and streptomycin. T24 cells were cultured in McCoy’s 5A medium (Life Technologies; Cat. No. 16600–108) supplemented with 10% FBS, 2 mM glutamine, and 100 U/ml penicillin and streptomycin. HepG2 cells were cultured in Eagle’s Minimum Essential medium (ATCC; Cat. No. 30–2003) supplemented with 10% FBS, 2 mM glutamine, and 100 U/ml penicillin and streptomycin. RPE1 cells were cultured in DMEM/F12 (Thermo Fisher Scientific; Cat. No. 11320–033) supplemented with 10% FBS, 2 mM glutamine, and 100 U/ml penicillin and streptomycin. Sum149 cells were cultured in Ham’s F12 medium (Lonza; Cat. No. 12–615F) supplemented with 5% FBS, 0.01 mg/ml insulin, 500 ng/ml hydrocortisone (STEMCELL Technologies; Cat. No. 07926), 2 mM glutamine, and 100 U/ml penicillin and streptomycin.
Additional details on the conduct of these studies are included in the Supplementary Materials and Methods section.
Statistical analysis
For box plots, the boxes represent the 25th, 50th, and 75th percentiles of the colonies per field, while the whiskers represent the 10th and 90th percentiles. In fig. S4, a Student’s t test (two-sided) was used to compare control and MEK1-KO colony formation efficiency.
Supplementary Material
Figure S1. Drug target ablation with CRISPR/Cas9.
Figure S2. Cell competition assays in multiple cancer types.
Figure S3. Verification of drug target knockouts.
Figure S4. Knocking out the verified genetic dependency MEK1 in A375 melanoma cells.
Figure S5. MDA-MB-231 clonal analysis.
Figure S6. Analysis of homolog gene expression in CRISPR-KO clones.
Figure S7. Analysis of homolog gene expression in published RNA-Seq experiments.
Figure S8. Assessing putative cancer dependencies in whole-genome CRISPR and RNAi screens.
Figure S9. Targeting several putative cancer dependencies with CRISPRi.
Figure S10. Knocking out putative cancer dependencies fails to sensitize cells to several clinical chemotherapy agents.
Figure S11. Target-independent toxicity of RNAi reagents previously used to investigate several putative cancer dependencies.
Figure S12. Using CRISPR to validate the MOA of several anti-cancer drugs.
Figure S13. Off-target toxicity of two caspase 3-activating compounds in CASP3-KO clones.
Figure S14. Target-independent cancer cell killing in single-agent and combination-therapy experiments.
Figure S15. Off-target toxicity of two putative HDAC6-inhibiting compounds in HDAC6-KO ovarian cancer clones.
Figure S16. OTS964-resistant clones harbor a mutation in the xDFG residue of CDK11B.
Figure S17. CDK11 activity is required for progression through mitosis.
Data file S1. Literature supporting the designation of HDAC6, MAPK14, PAK4, PBK, and PIM1 as cancer genetic dependencies and CASP3 as a drug target.
Data file S2. Cell competition assay results.
Data file S3. Sources of the cell lines used in this manuscript.
Data file S4. CRISPR gRNA sequences.
Data file S5. CRISPRi gRNA sequences.
Data file S6. qPCR primers.
Data file S7. Antibody sources and concentrations.
Data file S8. Drugs and drug sources.
Movie S1. A375 cells expressing H2B-mCherry released from a double-thymidine block into normal medium.
{kind=link}
Movie S2. A375 cells expressing H2B-mCherry released from a double-thymidine block into medium with 25 nM OTS964.
{kind=link}
Movie S3. A375 cells expressing H2B-mCherry released from a double-thymidine block into medium with 100 nM OTS964.
{kind=link}
Movie S4. A375CDK11B-G579S cells expressing H2B-mCherry released from a double-thymidine block into medium with 100 nM OTS964.
{kind=link}
Acknowledgments.
We thank the following individuals for sharing plasmids and cell lines used in this work: Chris Vakoc (CSHL), Camila dos Santos (CSHL), Ken Chang (CSHL), Lloyd Trotman (CSHL), Marco Jost and Jonathan Weissman (UCSF), Jeroen van den Berg and René Medema (Netherlands Cancer Institute), Hani Goodarzi (UCSF), David Solomon (UCSF), David Livingston (Harvard), Julian Lum (University of Victoria), Hensin Tsao and Keith Flaherty (MGH), Selvendiran Karuppaiyah (Ohio State University), and Susan Murphy (Duke).
Funding. Research in the Sheltzer Lab is supported by an NIH Early Independence Award (1DP5OD021385), a Breast Cancer Alliance Young Investigator Award, a Damon Runyon-Rachleff Innovation Award, a Gates Foundation Innovative Technology Solutions grant, and a CSHL-Northwell Translational Cancer Research Grant. This work was performed with assistance from the CSHL Flow Cytometry Shared Resource, which is supported by the CSHL Cancer Center Support Grant 5P30CA045508. A.L. and C.J.G. are supported by NSF Graduate Research Fellowships.
Footnotes
Competing interests. The authors declare that they have no competing interests.
Data and materials availability. All data associated with this study are present in the paper or the Supplementary Materials.
References and notes
- 1.Luo J, Solimini NL, Elledge SJ, Principles of Cancer Therapy: Oncogene and Non-oncogene Addiction, Cell 136, 823–837 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharma SV, Settleman J, Oncogene addiction: setting the stage for molecularly targeted cancer therapy, Genes Dev. 21, 3214–3231 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Lin A, Giuliano CJ, Sayles NM, Sheltzer JM, CRISPR/Cas9 mutagenesis invalidates a putative cancer dependency targeted in on-going clinical trials, eLife 6, e24179 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giuliano CJ, Lin A, Smith JC, Palladino AC, Sheltzer JM, MELK expression correlates with tumor mitotic activity but is not required for cancer growth, eLife 7, e32838 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wong CH, Siah KW, Lo AW, Estimation of clinical trial success rates and related parameters, Biostatistics (2018), doi: 10.1093/biostatistics/kxx069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bitler BG, Wu S, Park PH, Hai Y, Aird KM, Wang Y, Zhai Y, Kossenkov AV, Vara-Ailor A, Iii FJR, Zou W, Speicher DW, Huntsman DG, Conejo-Garcia JR, Cho KR, Christianson DW, Zhang R, ARID1A-mutated ovarian cancers depend on HDAC6 activity, Nat. Cell Biol 19, 962–973 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Putcha P, Yu J, Rodriguez-Barrueco R, Saucedo-Cuevas L, Villagrasa P, Murga-Penas E, Quayle SN, Yang M, Castro V, Llobet-Navas D, Birnbaum D, Finetti P, Woodward WA, Bertucci F, Alpaugh ML, Califano A, Silva J, HDAC6 activity is a non-oncogene addiction hub for inflammatory breast cancers, Breast Cancer Res. BCR 17 (2015), doi: 10.1186/s13058-015-0658-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gupta J, del Barco Barrantes I, Igea A, Sakellariou S, Pateras IS, Gorgoulis VG, Nebreda AR, Dual Function of p38α MAPK in Colon Cancer: Suppression of Colitis-Associated Tumor Initiation but Requirement for Cancer Cell Survival, Cancer Cell 25, 484–500 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Cánovas B, Igea A, Sartori AA, Gomis RR, Paull TT, Isoda M, Pérez-Montoyo H, Serra V, González-Suárez E, Stracker TH, Nebreda AR, Targeting p38α Increases DNA Damage, Chromosome Instability, and the Anti-tumoral Response to Taxanes in Breast Cancer Cells, Cancer Cell 33, 1094–1110.e8 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Siu MKY, Chan HY, Kong DSH, Wong ESY, Wong OGW, Ngan HYS, Tam KF, Zhang H, Li Z, Chan QKY, Tsao SW, Strömblad S, Cheung ANY, p21-activated kinase 4 regulates ovarian cancer cell proliferation, migration, and invasion and contributes to poor prognosis in patients, Proc. Natl. Acad. Sci 107, 18622–18627 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murray BW, Guo C, Piraino J, Westwick JK, Zhang C, Lamerdin J, Dagostino E, Knighton D, Loi C-M, Zager M, Kraynov E, Popoff I, Christensen JG, Martinez R, Kephart SE, Marakovits J, Karlicek S, Bergqvist S, Smeal T, Small-molecule p21-activated kinase inhibitor PF-3758309 is a potent inhibitor of oncogenic signaling and tumor growth, Proc. Natl. Acad. Sci 107, 9446–9451 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park J-H, Lin M-L, Nishidate T, Nakamura Y, Katagiri T, PDZ-Binding Kinase/T-LAK Cell-Originated Protein Kinase, a Putative Cancer/Testis Antigen with an Oncogenic Activity in Breast Cancer, Cancer Res. 66, 9186–9195 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Matsuo Y, Park J-H, Miyamoto T, Yamamoto S, Hisada S, Alachkar H, Nakamura Y, TOPK inhibitor induces complete tumor regression in xenograft models of human cancer through inhibition of cytokinesis, Sci. Transl. Med 6, 259ra145–259ra145 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Horiuchi D, Camarda R, Zhou AY, Yau C, Momcilovic O, Balakrishnan S, Corella AN, Eyob H, Kessenbrock K, Lawson DA, Marsh LA, Anderton BN, Rohrberg J, Kunder R, Bazarov AV, Yaswen P, McManus MT, Rugo HS, Werb Z, Goga A, PIM1 kinase inhibition as a targeted therapy against triple-negative breast tumors with elevated MYC expression, Nat. Med 22, 1321–1329 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brasó-Maristany F, Filosto S, Catchpole S, Marlow R, Quist J, Francesch-Domenech E, Plumb DA, Zakka L, Gazinska P, Liccardi G, Meier P, Gris-Oliver A, Cheang MCU, Perdrix-Rosell A, Shafat M, Noël E, Patel N, McEachern K, Scaltriti M, Castel P, Noor F, Buus R, Mathew S, Watkins J, Serra V, Marra P, Grigoriadis A, Tutt AN, PIM1 kinase regulates cell death, tumor growth and chemotherapy response in triple-negative breast cancer, Nat. Med 22, 1303–1313 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Putt KS, Chen GW, Pearson JM, Sandhorst JS, Hoagland MS, Kwon J-T, Hwang S-K, Jin H, Churchwell MI, Cho M-H, Doerge DR, Helferich WG, Hergenrother PJ, Small-molecule activation of procaspase-3 to caspase-3 as a personalized anticancer strategy, Nat. Chem. Biol 2, 543–550 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Wolan DW, Zorn JA, Gray DC, Wells JA, Small-Molecule Activators of a Proenzyme, Science 326, 853–858 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, Vakoc CR, Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains, Nat. Biotechnol 33, 661–667 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sloane DA, Trikic MZ, Chu MLH, Lamers MBAC, Mason CS, Mueller I, Savory WJ, Williams DH, Eyers PA, Drug-Resistant Aurora A Mutants for Cellular Target Validation of the Small Molecule Kinase Inhibitors MLN8054 and MLN8237, ACS Chem. Biol 5, 563–576 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Girdler F, Sessa F, Patercoli S, Villa F, Musacchio A, Taylor S, Molecular Basis of Drug Resistance in Aurora Kinases, Chem. Biol 15, 552–562 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Smurnyy Y, Cai M, Wu H, McWhinnie E, Tallarico JA, Yang Y, Feng Y, DNA sequencing and CRISPR-Cas9 gene editing for target validation in mammalian cells, Nat. Chem. Biol 10, 623–625 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Khan GN, Kim EJ, Shin TS, Lee SH, Heterogeneous Cell Types in Single-cell-derived Clones of MCF7 and MDA-MB-231 Cells, Anticancer Res. 37, 2343–2354 (2017). [DOI] [PubMed] [Google Scholar]
- 23.El-Brolosy MA, Kontarakis Z, Rossi A, Kuenne C, Günther S, Fukuda N, Kikhi K, Boezio GLM, Takacs CM, Lai S-L, Fukuda R, Gerri C, Giraldez AJ, Stainier DYR, Genetic compensation triggered by mutant mRNA degradation, Nature 568, 193 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ben-David U, Siranosian B, Ha G, Tang H, Oren Y, Hinohara K, Strathdee CA, Dempster J, Lyons NJ, Burns R, Nag A, Kugener G, Cimini B, Tsvetkov P, Maruvka YE, O’Rourke R, Garrity A, Tubelli AA, Bandopadhayay P, Tsherniak A, Vazquez F, Wong B, Birger C, Ghandi M, Thorner AR, Bittker JA, Meyerson M, Getz G, Beroukhim R, Golub TR, Genetic and transcriptional evolution alters cancer cell line drug response, Nature , 1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, Qi LS, Kampmann M, Weissman JS, Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation, Cell 159, 647–661 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang X-F, Yao T-P, HDAC6 is a microtubule-associated deacetylase, Nature 417, 455–458 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Huang P, Almeciga-Pinto I, Jarpe M, van Duzer JH, Mazitschek R, Yang M, Jones SS, Quayle SN, Selective HDAC inhibition by ACY-241 enhances the activity of paclitaxel in solid tumor models, Oncotarget 8, 2694–2707 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M, Li B, Cavet G, Linsley PS, Expression profiling reveals off-target gene regulation by RNAi, Nat. Biotechnol 21, 635–637 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Birmingham A, Anderson EM, Reynolds A, Ilsley-Tyree D, Leake D, Fedorov Y, Baskerville S, Maksimova E, Robinson K, Karpilow J, Marshall WS, Khvorova A, 3’ UTR seed matches, but not overall identity, are associated with RNAi off-targets, Nat. Methods 3, 199–204 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Putzbach W, Gao QQ, Patel M, van Dongen S, Haluck-Kangas A, Sarshad AA, Bartom ET, Kim K-YA, Scholtens DM, Hafner M, Zhao JC, Murmann AE, Peter ME, Many si/shRNAs can kill cancer cells by targeting multiple survival genes through an off-target mechanism, eLife 6, e29702 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tabusa H, Brooks T, Massey AJ, Knockdown of PAK4 or PAK1 Inhibits the Proliferation of Mutant KRAS Colon Cancer Cells Independently of RAF/MEK/ERK and PI3K/AKT Signaling, Mol. Cancer Res (2012), doi: 10.1158/1541-7786.MCR-12-0466. [DOI] [PubMed] [Google Scholar]
- 32.Harrison RK, Phase II and phase III failures: 2013–2015 Nat. Rev. Drug Discov (2016), doi: 10.1038/nrd.2016.184. [DOI] [PubMed] [Google Scholar]
- 33.Force T, Kolaja KL, Cardiotoxicity of kinase inhibitors: the prediction and translation of preclinical models to clinical outcomes, Nat. Rev. Drug Discov 10, 111–126 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Heitman J, Movva NR, Hall MN, Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast, Science 253, 905–909 (1991). [DOI] [PubMed] [Google Scholar]
- 35.Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH, RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs, Cell 78, 35–43 (1994). [DOI] [PubMed] [Google Scholar]
- 36.Gurden MD, Westwood IM, Faisal A, Naud S, Cheung K-MJ, McAndrew C, Wood A, Schmitt J, Boxall K, Mak G, Workman P, Burke R, Hoelder S, Blagg J, Van Montfort RLM, Linardopoulos S, Naturally Occurring Mutations in the MPS1 Gene Predispose Cells to Kinase Inhibitor Drug Resistance, Cancer Res. 75, 3340–3354 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Janssen A, Medema RH, Mitosis as an anti-cancer target, Oncogene 30, 2799–2809 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Glaab WE, Tindall KR, Mutation rate at the hprt locus in human cancer cell lines with specific mismatch repair-gene defects., Carcinogenesis 18, 1–8 (1997). [DOI] [PubMed] [Google Scholar]
- 39.Nielsen D, Skovsgaard T, P-glycoprotein as multidrug transporter: a critical review of current multidrug resistant cell lines, Biochim. Biophys. Acta 1139, 169–183 (1992). [DOI] [PubMed] [Google Scholar]
- 40.Kim RB, Drugs as P-glycoprotein substrates, inhibitors, and inducers, Drug Metab. Rev 34, 47–54 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, Kok CY, Jia M, De T, Teague JW, Stratton MR, McDermott U, Campbell PJ, COSMIC: exploring the world’s knowledge of somatic mutations in human cancer, Nucleic Acids Res. 43, D805–D811 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gururajan R, Lahti JM, Grenet J, Easton J, Gruber I, Ambros PF, Kidd VJ, Duplication of a Genomic Region Containing the Cdc2L1–2 and MMP21–22 Genes on Human Chromosome 1p36.3 and their Linkage to D1Z2, Genome Res. 8, 929–939 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dos Santos Paparidis NF, Canduri F, The Emerging Picture of CDK11: Genetic, Functional and Medicinal Aspects, Curr. Med. Chem 25, 880–888 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Ferguson FM, Gray NS, Kinase inhibitors: the road ahead, Nat. Rev. Drug Discov 17, 353–377 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Treiber DK, Shah NP, Ins and Outs of Kinase DFG Motifs, Chem. Biol 20, 745–746 (2013). [DOI] [PubMed] [Google Scholar]
- 46.Klaeger S, Heinzlmeir S, Wilhelm M, Polzer H, Vick B, Koenig P-A, Reinecke M, Ruprecht B, Petzoldt S, Meng C, Zecha J, Reiter K, Qiao H, Helm D, Koch H, Schoof M, Canevari G, Casale E, Depaolini SR, Feuchtinger A, Wu Z, Schmidt T, Rueckert L, Becker W, Huenges J, Garz A-K, Gohlke B-O, Zolg DP, Kayser G, Vooder T, Preissner R, Hahne H, Tõnisson N, Kramer K, Götze K, Bassermann F, Schlegl J, Ehrlich H-C, Aiche S, Walch A, Greif PA, Schneider S, Felder ER, Ruland J, Médard G, Jeremias I, Spiekermann K, Kuster B, The target landscape of clinical kinase drugs, Science 358, eaan4368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rizos H, Menzies AM, Pupo GM, Carlino MS, Fung C, Hyman J, Haydu LE, Mijatov B, Becker TM, Boyd SC, Howle J, Saw R, Thompson JF, Kefford RF, Scolyer RA, Long GV, BRAF Inhibitor Resistance Mechanisms in Metastatic Melanoma: Spectrum and Clinical Impact, Clin. Cancer Res 20, 1965–1977 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Chen L, Mayer JA, Krisko TI, Speers CW, Wang T, Hilsenbeck SG, Brown PH, Inhibition of the p38 Kinase Suppresses the Proliferation of Human ER-Negative Breast Cancer Cells, Cancer Res. 69, 8853–8861 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matrone A, Grossi V, Chiacchiera F, Fina E, Cappellari M, Caringella AM, Di Naro E, Loverro G, Simone C, p38α Is Required for Ovarian Cancer Cell Metabolism and Survival, Int. J. Gynecol. Cancer 20, 203 (2010). [DOI] [PubMed] [Google Scholar]
- 50.Kim C, Sano Y, Todorova K, Carlson BA, Arpa L, Celada A, Lawrence T, Otsu K, Brissette JL, Arthur JSC, Park JM, p38α MAP kinase serves cell type-specific inflammatory functions in skin injury and coordinates pro- and anti-inflammatory gene expression, Nat. Immunol 9, 1019–1027 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vogl DT, Raje NS, Jagannath S, Richardson PG, Hari P, Orlowski RZ, Supko JG, Tamang D, Yang M, Jones SS, Wheeler C, Markelewicz RJ, Lonial S, Ricolinostat, the first selective histone deacetylase 6 inhibitor, in combination with bortezomib and dexamethasone for relapsed or refractory multiple myeloma, Clin. Cancer Res , clincanres.2526.2016 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Depetter Y, Geurs S, De Vreese R, Goethals S, Vandoorn E, Laevens A, Steenbrugge J, Meyer E, de Tullio P, Bracke M, D’hooghe M, De Wever O, Selective pharmacological inhibitors of HDAC6 reveal biochemical activity but functional tolerance in cancer models, Int. J. Cancer (2019), doi: 10.1002/ijc.32169. [DOI] [PubMed] [Google Scholar]
- 53.Seto E, Yoshida M, Erasers of Histone Acetylation: The Histone Deacetylase Enzymes, Cold Spring Harb. Perspect. Biol 6 (2014), doi: 10.1101/cshperspect.a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Levis M, Midostaurin approved for FLT3-mutated AML, Blood 129, 3403–3406 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Whittaker SR, Mallinger A, Workman P, Clarke PA, Inhibitors of cyclin-dependent kinases as cancer therapeutics, Pharmacol. Ther 173, 83–105 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou Y, Shen JK, Hornicek FJ, Kan Q, Duan Z, The emerging roles and therapeutic potential of cyclin-dependent kinase 11 (CDK11) in human cancer, Oncotarget 7, 40846–40859 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hu D, Valentine M, Kidd VJ, Lahti JM, CDK11(p58) is required for the maintenance of sister chromatid cohesion, J. Cell Sci 120, 2424–2434 (2007). [DOI] [PubMed] [Google Scholar]
- 58.Manchado E, Guillamot M, Malumbres M, Killing cells by targeting mitosis, Cell Death Differ. 19, 369–377 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smith I, Greenside PG, Natoli T, Lahr DL, Wadden D, Tirosh I, Narayan R, Root DE, Golub TR, Subramanian A, Doench JG, Evaluation of RNAi and CRISPR technologies by large-scale gene expression profiling in the Connectivity Map, PLOS Biol. 15, e2003213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Evers B, Jastrzebski K, Heijmans JPM, Grernrum W, Beijersbergen RL, Bernards R, CRISPR knockout screening outperforms shRNA and CRISPRi in identifying essential genes, Nat. Biotechnol 34, 631–633 (2016). [DOI] [PubMed] [Google Scholar]
- 61.Xu J, Wang P, Yang H, Zhou J, Li Y, Li X, Xue W, Yu C, Tian Y, Zhu F, Comparison of FDA Approved Kinase Targets to Clinical Trial Ones: Insights from Their System Profiles and Drug-Target Interaction Networks, BioMed Res. Int. 2016 (2016), doi: 10.1155/2016/2509385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Highly effective new anti-cancer drug shows few side effects—in mice Univ. Chic. News (available at https://news.uchicago.edu/story/highly-effective-new-anti-cancer-drug-shows-few-side-effects-mice). [Google Scholar]
- 63.Neggers JE, Kwanten B, Dierckx T, Noguchi H, Voet A, Bral L, Minner K, Massant B, Kint N, Delforge M, Vercruysse T, Baloglu E, Senapedis W, Jacquemyn M, Daelemans D, Target identification of small molecules using large-scale CRISPR-Cas mutagenesis scanning of essential genes, Nat. Commun 9, 502 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Putt KS, Chen GW, Pearson JM, Sandhorst JS, Hoagland MS, Kwon J-T, Hwang S-K, Jin H, Churchwell MI, Cho M-H, Doerge DR, Helferich WG, Hergenrother PJ, Small-molecule activation of procaspase-3 to caspase-3 as a personalized anticancer strategy, Nat. Chem. Biol 2, 543 (2006). [DOI] [PubMed] [Google Scholar]
- 65.Wolan DW, Zorn JA, Gray DC, Wells JA, Small-Molecule Activators of a Proenzyme, Science 326, 853–858 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Denault J-B, Drag M, Salvesen GS, Alves J, Heidt AB, Deveraux Q, Harris JL, Small molecules not direct activators of caspases, Nat. Chem. Biol 3, 519 (2007). [DOI] [PubMed] [Google Scholar]
- 67.Reply to “Small molecules not direct activators of caspases,” Nat. Chem. Biol 3, 520 (2007). [DOI] [PubMed] [Google Scholar]
- 68.Peterson QP, Goode DR, West DC, Ramsey KN, Lee JJY, Hergenrother PJ, PAC-1 activates procaspase-3 in vitro through relief of zinc-mediated inhibition, J. Mol. Biol 388, 144–158 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang F, Wang L, Zhao Y, Li Y, Ping G, Xiao S, Chen K, Zhu W, Gong P, Yang J, Wu C, A novel small-molecule activator of procaspase-3 induces apoptosis in cancer cells and reduces tumor growth in human breast, liver and gallbladder cancer xenografts, Mol. Oncol 8, 1640–1652 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang X-H, Sladek TL, Liu X, Butler BR, Froelich CJ, Thor AD, Reconstitution of Caspase 3 Sensitizes MCF-7 Breast Cancer Cells to Doxorubicin- and Etoposide-induced Apoptosis, Cancer Res 61, 348–354 (2001). [PubMed] [Google Scholar]
- 71.Yang X, Zheng F, Xing H, Gao Q, Wei W, Lu Y, Wang S, Zhou J, Hu W, Ma D, Resistance to chemotherapy-induced apoptosis via decreased caspase-3 activity and overexpression of antiapoptotic proteins in ovarian cancer, J. Cancer Res. Clin. Oncol 130, 423–428 (2004). [DOI] [PubMed] [Google Scholar]
- 72.Giuliano CJ, Lin A, Girish V, Sheltzer JM, Generating Single Cell–Derived Knockout Clones in Mammalian Cells with CRISPR/Cas9, Curr. Protoc. Mol. Biol 128, e100 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tarumoto Y, Lu B, Somerville TDD, Huang Y-H, Milazzo JP, Wu XS, Klingbeil O, Demerdash OE, Shi J, Vakoc CR, LKB1, Salt-Inducible Kinases, and MEF2C Are Linked Dependencies in Acute Myeloid Leukemia, Mol. Cell 69, 1017–1027.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F, Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells, Science 343, 84–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Horlbeck MA, Gilbert LA, Villalta JE, Adamson B, Pak RA, Chen Y, Fields AP, Park CY, Corn JE, Kampmann M, Weissman JS, Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation, eLife 5, e19760 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Smale ST, Calcium Phosphate Transfection of 3T3 Fibroblasts, Cold Spring Harb. Protoc 2010, pdb.prot5372 (2010). [DOI] [PubMed] [Google Scholar]
- 77.van den Berg J, Manjón AG, Kielbassa K, Feringa FM, Freire R, Medema RH, A limited number of double-strand DNA breaks is sufficient to delay cell cycle progression, Nucleic Acids Res. 46, 10132–10144 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Edgar R, Domrachev M, Lash AE, Gene Expression Omnibus: NCBI gene expression and hybridization array data repository, Nucleic Acids Res. 30, 207–210 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mou H, Moore J, Malonia SK, Li Y, Ozata DM, Hough S, Song C-Q, Smith JL, Fischer A, Weng Z, Green MR, Xue W, Genetic disruption of oncogenic Kras sensitizes lung cancer cells to Fas receptor-mediated apoptosis, Proc. Natl. Acad. Sci 114, 3648–3653 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Boratyn GM, Camacho C, Cooper PS, Coulouris G, Fong A, Ma N, Madden TL, Matten WT, McGinnis SD, Merezhuk Y, Raytselis Y, Sayers EW, Tao T, Ye J, Zaretskaya I, BLAST: a more efficient report with usability improvements, Nucleic Acids Res. 41, W29–W33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.DepMap B, DepMap Achilles 18Q3 public (2018), doi: 10.6084/m9.figshare.6931364.v1. [DOI] [Google Scholar]
- 82.Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, Goodale A, Lee Y, Ali LD, Jiang G, Lubonja R, Harrington WF, Strickland M, Wu T, Hawes DC, Zhivich VA, Wyatt MR, Kalani Z, Chang JJ, Okamoto M, Stegmaier K, Golub TR, Boehm JS, Vazquez F, Root DE, Hahn WC, Tsherniak A, Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells, Nat. Genet 49, 1779–1784 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.DEMETER2 data (2018), doi: 10.6084/m9.figshare.6025238.v2. [DOI] [Google Scholar]
- 84.McFarland JM, Ho ZV, Kugener G, Dempster JM, Montgomery PG, Bryan JG, Krill-Burger JM, Green TM, Vazquez F, Boehm JS, Golub TR, Hahn WC, Root DE, Tsherniak A, Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration, bioRxiv , 305656 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sadelain M, Papapetrou EP, Bushman FD, Safe harbours for the integration of new DNA in the human genome, Nat. Rev. Cancer 12, 51–58 (2012). [DOI] [PubMed] [Google Scholar]
- 86.Cerbini T, Funahashi R, Luo Y, Liu C, Park K, Rao M, Malik N, Zou J, Transcription activator-like effector nuclease (TALEN)-mediated CLYBL targeting enables enhanced transgene expression and one-step generation of dual reporter human induced pluripotent stem cell (iPSC) and neural stem cell (NSC) lines, PloS One 10, e0116032 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP, Comprehensive analysis of kinase inhibitor selectivity, Nat. Biotechnol 29, 1046–1051 (2011). [DOI] [PubMed] [Google Scholar]
- 88.Ai J, Wang Y, Dar JA, Liu J, Liu L, Nelson JB, Wang Z, HDAC6 regulates androgen receptor hypersensitivity and nuclear localization via modulating Hsp90 acetylation in castration-resistant prostate cancer, Mol. Endocrinol. Baltim. Md 23, 1963–1972 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rolando Alberto R-F, Yudibeth S-L, Jonathan F-VM, Raul F-M, Laura Cristina C-P, Ismael V-M, Martha Cecilia R-H, Martiniano B, M M-A, Jose Guadalupe T-F, Elvia B-M, Jose C-B, Design, Synthesis and Biological Evaluation of a Phenyl Butyric Acid Derivative, N-(4-chlorophenyl)-4-phenylbutanamide: A HDAC6 Inhibitor with Anti-proliferative Activity on Cervix Cancer and Leukemia Cells (2017), doi:info:doi/ 10.2174/1871520617666170103092851. [DOI] [Google Scholar]
- 90.Aldana-Masangkay GI, Rodriguez-Gonzalez A, Lin T, Ikeda AK, Hsieh Y-T, Kim Y-M, Lomenick B, Okemoto K, Landaw EM, Wang D, Mazitschek R, Bradner JE, Sakamoto KM, Tubacin suppresses proliferation and induces apoptosis of acute lymphoblastic leukemia cells, Leuk. Lymphoma 52, 1544–1555 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Amengual JE, Johannet P, Lombardo M, Zullo K, Hoehn D, Bhagat G, Scotto L, Jirau-Serrano X, Radeski D, Heinen J, Jiang H, Cremers S, Zhang Y, Jones S, O’Connor OA, Dual Targeting of Protein Degradation Pathways with the Selective HDAC6 Inhibitor ACY-1215 and Bortezomib Is Synergistic in Lymphoma, Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res 21, 4663–4675 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bai J, Lei Y, An G, He L, Down-Regulation of Deacetylase HDAC6 Inhibits the Melanoma Cell Line A375.S2 Growth through ROS-Dependent Mitochondrial Pathway, PLOS ONE 10, e0121247 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Balliu M, Guandalini L, Romanelli MN, D’Amico M, Paoletti F, HDAC-inhibitor (S)-8 disrupts HDAC6-PP1 complex prompting A375 melanoma cell growth arrest and apoptosis, J. Cell. Mol. Med 19, 143–154 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bergman JA, Woan K, Perez-Villarroel P, Villagra A, Sotomayor EM, Kozikowski AP, Selective histone deacetylase 6 inhibitors bearing substituted urea linkers inhibit melanoma cell growth, J. Med. Chem 55, 9891–9899 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cao J, Lv W, Wang L, Xu J, Yuan P, Huang S, He Z, Hu J, Ricolinostat (ACY-1215) suppresses proliferation and promotes apoptosis in esophageal squamous cell carcinoma via miR-30d/PI3K/AKT/mTOR and ERK pathways, Cell Death Dis. 9 (2018), doi: 10.1038/s41419-018-0788-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chuang M-J, Wu S-T, Tang S-H, Lai X-M, Lai H-C, Hsu K-H, Sun K-H, Sun G-H, Chang S-Y, Yu D-S, Hsiao P-W, Huang S-M, Cha T-L, The HDAC Inhibitor LBH589 Induces ERK-Dependent Prometaphase Arrest in Prostate Cancer via HDAC6 Inactivation and Down-Regulation, PLOS ONE 8, e73401 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cosenza M, Civallero M, Marcheselli L, Sacchi S, Pozzi S, Ricolinostat, a selective HDAC6 inhibitor, shows anti-lymphoma cell activity alone and in combination with bendamustine, Apoptosis Int. J. Program. Cell Death 22, 827–840 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ding G, Liu H-D, Huang Q, Liang H-X, Ding Z-H, Liao Z-J, Huang G, HDAC6 promotes hepatocellular carcinoma progression by inhibiting P53 transcriptional activity, FEBS Lett. 587, 880–886 (2013). [DOI] [PubMed] [Google Scholar]
- 99.Dong J, Zheng N, Wang X, Tang C, Yan P, Zhou H-B, Huang J, A novel HDAC6 inhibitor exerts an anti-cancer effect by triggering cell cycle arrest and apoptosis in gastric cancer, Eur. J. Pharmacol 828, 67–79 (2018). [DOI] [PubMed] [Google Scholar]
- 100.Fu X-H, Zhang X, Yang H, Xu X-W, Hu Z-L, Yan J, Zheng X-L, Wei R-R, Zhang Z-Q, Tang S-R, Geng M-Y, Huang X, CUDC-907 displays potent antitumor activity against human pancreatic adenocarcinoma in vitro and in vivo through inhibition of HDAC6 to downregulate c-Myc expression, Acta Pharmacol. Sin 40, 677–688 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gradilone SA, Radtke BN, Bogert PS, Huang BQ, Gajdos GB, LaRusso NF, HDAC6 inhibition restores ciliary expression and decreases tumor growth, Cancer Res. 73, 2259–2270 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hackanson B, Rimmele L, Benkißer M, Abdelkarim M, Fliegauf M, Jung M, Lübbert M, HDAC6 as a target for antileukemic drugs in acute myeloid leukemia, Leuk. Res 36, 1055–1062 (2012). [DOI] [PubMed] [Google Scholar]
- 103.Hideshima T, Qi J, Paranal RM, Tang W, Greenberg E, West N, Colling ME, Estiu G, Mazitschek R, Perry JA, Ohguchi H, Cottini F, Mimura N, Görgün G, Tai Y-T, Richardson PG, Carrasco RD, Wiest O, Schreiber SL, Anderson KC, Bradner JE, Discovery of selective small-molecule HDAC6 inhibitor for overcoming proteasome inhibitor resistance in multiple myeloma, Proc. Natl. Acad. Sci. U. S. A 113, 13162–13167 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Inks ES, Josey BJ, Jesinkey SR, Chou CJ, A novel class of small molecule inhibitors of HDAC6, ACS Chem. Biol 7, 331–339 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kaliszczak M, Trousil S, Åberg O, Perumal M, Nguyen Q-D, Aboagye EO, A novel small molecule hydroxamate preferentially inhibits HDAC6 activity and tumour growth, Br. J. Cancer 108, 342–350 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kaliszczak M, van Hechanova E, Li Y, Alsadah H, Parzych K, Auner HW, Aboagye EO, The HDAC6 inhibitor C1A modulates autophagy substrates in diverse cancer cells and induces cell death, Br. J. Cancer 119, 1278 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lee H-Y, Tsai A-C, Chen M-C, Shen P-J, Cheng Y-C, Kuo C-C, Pan S-L, Liu Y-M, Liu J-F, Yeh T-K, Wang J-C, Chang C-Y, Chang J-Y, Liou J-P, Azaindolylsulfonamides, with a More Selective Inhibitory Effect on Histone Deacetylase 6 Activity, Exhibit Antitumor Activity in Colorectal Cancer HCT116 Cells, J. Med. Chem 57, 4009–4022 (2014). [DOI] [PubMed] [Google Scholar]
- 108.Lee J-H, Mahendran A, Yao Y, Ngo L, Venta-Perez G, Choy ML, Kim N, Ham W-S, Breslow R, Marks PA, Development of a histone deacetylase 6 inhibitor and its biological effects, Proc. Natl. Acad. Sci 110, 15704–15709 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lee Y-S, Lim K-H, Guo X, Kawaguchi Y, Gao Y, Barrientos T, Ordentlich P, Wang X-F, Counter CM, Yao T-P, The cytoplasmic deacetylase HDAC6 is required for efficient oncogenic tumorigenesis, Cancer Res. 68, 7561–7569 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li N, Tie X-J, Liu P-J, Zhang Y, Ren H-Z, Gao X, Xu Z-Q, Effects of down-regulation of HDAC6 expression on proliferation, cell cycling and migration of esophageal squamous cell carcinoma cells and related molecular mechanisms, Asian Pac. J. Cancer Prev. APJCP 14, 685–689 (2013). [DOI] [PubMed] [Google Scholar]
- 111.Li S, Liu X, Chen X, Zhang L, Wang X, Histone deacetylase 6 promotes growth of glioblastoma through inhibition of SMAD2 signaling, Tumor Biol. 36, 9661–9665 (2015). [DOI] [PubMed] [Google Scholar]
- 112.Lienlaf M, Perez-Villarroel P, Knox T, Pabon M, Sahakian E, Powers J, Woan KV, Lee C, Cheng F, Deng S, Smalley KSM, Montecino M, Kozikowski A, Pinilla-Ibarz J, Sarnaik A, Seto E, Weber J, Sotomayor EM, Villagra A, Essential role of HDAC6 in the regulation of PD‐L1 in melanoma, Mol. Oncol 10, 735–750 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Liu J, Gu J, Feng Z, Yang Y, Zhu N, Lu W, Qi F, Both HDAC5 and HDAC6 are required for the proliferation and metastasis of melanoma cells, J. Transl. Med 14 (2016), doi: 10.1186/s12967-015-0753-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liu Y-M, Lee H-Y, Lai M-J, Pan S-L, Huang H-L, Kuo F-C, Chen M-C, Liou J-P, Pyrimidinedione-mediated selective histone deacetylase 6 inhibitors with antitumor activity in colorectal cancer HCT116 cells, Org. Biomol. Chem 13, 10226–10235 (2015). [DOI] [PubMed] [Google Scholar]
- 115.Lwin T, Zhao X, Cheng F, Zhang X, Huang A, Shah B, Zhang Y, Moscinski LC, Choi YS, Kozikowski AP, Bradner JE, Dalton WS, Sotomayor E, Tao J, A microenvironment-mediated c-Myc/miR-548m/HDAC6 amplification loop in non-Hodgkin B cell lymphomas, J. Clin. Invest 123, 4612–4626 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mak AB, Nixon AML, Kittanakom S, Stewart JM, Chen GI, Curak J, Gingras A-C, Mazitschek R, Neel BG, Stagljar I, Moffat J, Regulation of CD133 by HDAC6 Promotes β-Catenin Signaling to Suppress Cancer Cell Differentiation, Cell Rep 2, 951–963 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Medler TR, Craig JM, Fiorillo AA, Feeney YB, Harrell JC, Clevenger CV, HDAC6 Deacetylates HMGN2 to Regulate Stat5a Activity and Breast Cancer Growth, Mol. Cancer Res 14, 994–1008 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pai J-T, Hsu C-Y, Hua K-T, Yu S-Y, Huang C-Y, Chen C-N, Liao C-H, Weng M-S, NBM-T-BBX-OS01, Semisynthesized from Osthole, Induced G1 Growth Arrest through HDAC6 Inhibition in Lung Cancer Cells, Molecules 20, 8000–8019 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Park SJ, Kim JK, Bae HJ, Eun JW, Shen Q, Kim HS, Shin WC, Yang HD, Lee EK, You JS, Park WS, Lee JY, Nam SW, HDAC6 sustains growth stimulation by prolonging the activation of EGF receptor through the inhibition of rabaptin-5-mediated early endosome fusion in gastric cancer, Cancer Lett. 354, 97–106 (2014). [DOI] [PubMed] [Google Scholar]
- 120.Putcha P, Yu J, Rodriguez-Barrueco R, Saucedo-Cuevas L, Villagrasa P, Murga-Penas E, Quayle SN, Yang M, Castro V, Llobet-Navas D, Birnbaum D, Finetti P, Woodward WA, Bertucci F, Alpaugh ML, Califano A, Silva J, HDAC6 activity is a non-oncogene addiction hub for inflammatory breast cancers, Breast Cancer Res. 17, 149 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ryu H-W, Lee D-H, Shin D-H, Kim SH, Kwon SH, Aceroside VIII is a New Natural Selective HDAC6 Inhibitor that Synergistically Enhances the Anticancer Activity of HDAC Inhibitor in HT29 Cells, Planta Med. 81, 222–227 (2015). [DOI] [PubMed] [Google Scholar]
- 122.Santo L, Hideshima T, Kung AL, Tseng J-C, Tamang D, Yang M, Jarpe M, van Duzer JH, Mazitschek R, Ogier WC, Cirstea D, Rodig S, Eda H, Scullen T, Canavese M, Bradner J, Anderson KC, Jones SS, Raje N, Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma, Blood 119, 2579–2589 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Seidel C, Schnekenburger M, Mazumder A, Teiten M-H, Kirsch G, Dicato M, Diederich M, 4-Hydroxybenzoic acid derivatives as HDAC6-specific inhibitors modulating microtubular structure and HSP90α chaperone activity against prostate cancer, Biochem. Pharmacol 99, 31–52 (2016). [DOI] [PubMed] [Google Scholar]
- 124.Song YW, Lim Y, Cho SK, 2,4‑Di‑tert‑butylphenol, a potential HDAC6 inhibitor, induces senescence and mitotic catastrophe in human gastric adenocarcinoma AGS cells, Biochim. Biophys. Acta Mol. Cell Res 1865, 675–683 (2018). [DOI] [PubMed] [Google Scholar]
- 125.Subramanian C, Jarzembowski JA, Opipari AW, Castle VP, Kwok RPS, HDAC6 deacetylates Ku70 and regulates Ku70-Bax binding in neuroblastoma, Neoplasia N. Y. N 13, 726–734 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tan Y, Zhang S, Zhu H, Chu Y, Zhou H, Liu D, Huo J, Histone deacetylase 6 selective inhibitor ACY1215 inhibits cell proliferation and enhances the chemotherapeutic effect of 5-fluorouracil in HCT116 cells, Ann. Transl. Med 7 (2019), doi: 10.21037/atm.2018.11.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Valente S, Trisciuoglio D, Tardugno M, Benedetti R, Labella D, Secci D, Mercurio C, Boggio R, Tomassi S, Di Maro S, Novellino E, Altucci L, Del Bufalo D, Mai A, Cosconati S, tert-Butylcarbamate-containing histone deacetylase inhibitors: apoptosis induction, cytodifferentiation, and antiproliferative activities in cancer cells, ChemMedChem 8, 800–811 (2013). [DOI] [PubMed] [Google Scholar]
- 128.Wang F, Zhong B-W, Zhao Z-R, ACY 1215, a histone deacetylase 6 inhibitor, inhibits cancer cell growth in melanoma, J. Biol. Regul. Homeost. Agents 32, 851–858 (2018). [PubMed] [Google Scholar]
- 129.Wang L, Kofler M, Brosch G, Melesina J, Sippl W, Martinez ED, Easmon J, 2-Benzazolyl-4-Piperazin-1-Ylsulfonylbenzenecarbohydroxamic Acids as Novel Selective Histone Deacetylase-6 Inhibitors with Antiproliferative Activity, PLoS ONE 10 (2015), doi: 10.1371/journal.pone.0134556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wang L, Xiang S, Williams KA, Dong H, Bai W, Nicosia SV, Khochbin S, Bepler G, Zhang X, Depletion of HDAC6 Enhances Cisplatin-Induced DNA Damage and Apoptosis in Non-Small Cell Lung Cancer Cells, PLOS ONE 7, e44265 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wang X, Ma Y, Meng P, Han J, Yu H, Bi L, miR-433 inhibits oral squamous cell carcinoma (OSCC) cell growth and metastasis by targeting HDAC6, Oral Oncol 51, 674–682 (2015). [DOI] [PubMed] [Google Scholar]
- 132.Wang Z, Hu P, Tang F, Lian H, Chen X, Zhang Y, He X, Liu W, Xie C, HDAC6 promotes cell proliferation and confers resistance to temozolomide in glioblastoma, Cancer Lett. 379, 134–142 (2016). [DOI] [PubMed] [Google Scholar]
- 133.Wang Z, Tang F, Hu P, Wang Y, Gong J, Sun S, Xie C, HDAC6 promotes cell proliferation and confers resistance to gefitinib in lung adenocarcinoma, Oncol. Rep 36, 589–597 (2016). [DOI] [PubMed] [Google Scholar]
- 134.Wei L-H, Torng P-L, Hsiao S-M, Jeng Y-M, Chen M-W, Chen C-A, Histone deacetylase 6 regulates estrogen receptor alpha in uterine leiomyoma, Reprod. Sci. Thousand Oaks Calif 18, 755–762 (2011). [DOI] [PubMed] [Google Scholar]
- 135.Woan KV, Lienlaf M, Perez-Villaroel P, Lee C, Cheng F, Knox T, Woods DM, Barrios K, Powers J, Sahakian E, Wang HW, Canales J, Marante D, Smalley KSM, Bergman J, Seto E, Kozikowski A, Pinilla-Ibarz J, Sarnaik A, Celis E, Weber J, Sotomayor EM, Villagra A, Targeting histone deacetylase 6 mediates a dual anti-melanoma effect: Enhanced antitumor immunity and impaired cell proliferation, Mol. Oncol 9, 1447–1457 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Xiang W, Guo F, Cheng W, Zhang J, Huang J, Wang R, Ma Z, Xu K, HDAC6 inhibition suppresses chondrosarcoma by restoring the expression of primary cilia, Oncol. Rep 38, 229–236 (2017). [DOI] [PubMed] [Google Scholar]
- 137.Yang F, Wang F, Liu Y, Wang S, Li X, Huang Y, Xia Y, Cao C, Sulforaphane induces autophagy by inhibition of HDAC6-mediated PTEN activation in triple negative breast cancer cells, Life Sci. 213, 149–157 (2018). [DOI] [PubMed] [Google Scholar]
- 138.Yang MH, Laurent G, Bause AS, Spang R, German N, Haigis MC, Haigis KM, HDAC6 and SIRT2 regulate the acetylation state and oncogenic activity of mutant K-RAS, Mol. Cancer Res. MCR 11, 1072–1077 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yang Z, Wang T, Wang F, Niu T, Liu Z, Chen X, Long C, Tang M, Cao D, Wang X, Xiang W, Yi Y, Ma L, You J, Chen L, Discovery of Selective Histone Deacetylase 6 Inhibitors Using the Quinazoline as the Cap for the Treatment of Cancer, J. Med. Chem 59, 1455–1470 (2016). [DOI] [PubMed] [Google Scholar]
- 140.Yuan L, Lou W, Sang J, [Effect of HDAC6 down-regulation on the growth of xenografted human laryngeal carcinoma cell line Hep-2 in nude mice and underlying mechanism], Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi 47, 481–486 (2012). [PubMed] [Google Scholar]
- 141.Zhang I, Beus M, Stochaj U, Le PU, Zorc B, Rajić Z, Petrecca K, Maysinger D, Inhibition of glioblastoma cell proliferation, invasion, and mechanism of action of a novel hydroxamic acid hybrid molecule, Cell Death Discov. 4 (2018), doi: 10.1038/s41420-018-0103-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Abdelhafez OM, Ahmed EY, Abdel Latif NA, Arafa RK, Abd Elmageed ZY, Ali HI, Design and molecular modeling of novel P38α MAPK inhibitors targeting breast cancer, synthesized from oxygen heterocyclic natural compounds, Bioorg. Med. Chem 27, 1308–1319 (2019). [DOI] [PubMed] [Google Scholar]
- 143.Antoon JW, Bratton MR, Guillot LM, Wadsworth S, Salvo VA, Burow ME, Inhibition of p38-MAPK alters SRC coactivation and estrogen receptor phosphorylation, Cancer Biol. Ther 13, 1026–1033 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Campbell RM, Anderson BD, Brooks NA, Brooks HB, Chan EM, Dios AD, Gilmour R, Graff JR, Jambrina E, Mader M, McCann D, Na S, Parsons SH, Pratt SE, Shih C, Stancato LF, Starling JJ, Tate C, Velasco JA, Wang Y, Ye XS, Characterization of LY2228820 Dimesylate, a Potent and Selective Inhibitor of p38 MAPK with Antitumor Activity, Mol. Cancer Ther 13, 364–374 (2014). [DOI] [PubMed] [Google Scholar]
- 145.Chiacchiera F, Matrone A, Ferrari E, Ingravallo G, Lo Sasso G, Murzilli S, Petruzzelli M, Salvatore L, Moschetta A, Simone C, p38alpha blockade inhibits colorectal cancer growth in vivo by inducing a switch from HIF1alpha- to FoxO-dependent transcription, Cell Death Differ. 16, 1203–1214 (2009). [DOI] [PubMed] [Google Scholar]
- 146.Comes F, Matrone A, Lastella P, Nico B, Susca FC, Bagnulo R, Ingravallo G, Modica S, Lo Sasso G, Moschetta A, Guanti G, Simone C, A novel cell type-specific role of p38alpha in the control of autophagy and cell death in colorectal cancer cells, Cell Death Differ. 14, 693–702 (2007). [DOI] [PubMed] [Google Scholar]
- 147.Del Barco Barrantes I, Stephan-Otto Attolini C, Slobodnyuk K, Igea A, Gregorio S, Gawrzak S, Gomis RR, Nebreda AR, Regulation of Mammary Luminal Cell Fate and Tumorigenesis by p38α, Stem Cell Rep 10, 257–271 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Gill K, Nigam L, Singh R, Kumar S, Subbarao N, Chauhan SS, Dey S, The Rational Design of Specific Peptide Inhibitor against p38α MAPK at Allosteric-Site: A Therapeutic Modality for HNSCC, PLOS ONE 9, e101525 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 149.Leelahavanichkul K, Amornphimoltham P, Molinolo AA, Basile JR, Koontongkaew S, Gutkind JS, A role for p38 MAPK in head and neck cancer cell growth and tumor-induced angiogenesis and lymphangiogenesis, Mol. Oncol 8, 105–118 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Limoge M, Safina A, Truskinovsky AM, Aljahdali I, Zonneville J, Gruevski A, Arteaga CL, Bakin AV, Tumor p38MAPK signaling enhances breast carcinoma vascularization and growth by promoting expression and deposition of pro-tumorigenic factors, Oncotarget 8, 61969–61981 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Matrone A, Grossi V, Chiacchiera F, Fina E, Cappellari M, Caringella AM, Di Naro E, Loverro G, Simone C, p38alpha is required for ovarian cancer cell metabolism and survival, Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc 20, 203–211 (2010). [DOI] [PubMed] [Google Scholar]
- 152.Medicherla S, Reddy M, Ying J, Navas TA, Li L, Nguyen AN, Kerr I, Hanjarappa N, Protter AA, Higgins LS, p38alpha-selective MAP kinase inhibitor reduces tumor growth in mouse xenograft models of multiple myeloma, Anticancer Res. 28, 3827–3833 (2008). [PubMed] [Google Scholar]
- 153.Nguyen AN, Stebbins EG, Henson M, O’Young G, Choi SJ, Quon D, Damm D, Reddy M, Ma JY, Haghnazari E, Kapoun AM, Medicherla S, Protter A, Schreiner GF, Kurihara N, Anderson J, Roodman GD, Navas TA, Higgins LS, Normalizing the bone marrow microenvironment with p38 inhibitor reduces multiple myeloma cell proliferation and adhesion and suppresses osteoclast formation, Exp. Cell Res 312, 1909–1923 (2006). [DOI] [PubMed] [Google Scholar]
- 154.Planchard D, Camara-Clayette V, Dorvault N, Soria J-C, Fouret P, p38 Mitogen-activated protein kinase signaling, ERCC1 expression, and viability of lung cancer cells from never or light smoker patients, Cancer 118, 5015–5025 (2012). [DOI] [PubMed] [Google Scholar]
- 155.Song W-J, Dong Y, Luo C, Chen Y-Y, p38MAPK family isoform p38α and activating transcription factor 2 are associated with the malignant phenotypes and poor prognosis of patients with ovarian adenocarcinoma, Pathol. Res. Pract 213, 1282–1288 (2017). [DOI] [PubMed] [Google Scholar]
- 156.Vanderkerken K, Medicherla S, Coulton L, De Raeve H, Willems A, Lawson M, Van Camp B, Protter AA, Higgins LS, Menu E, Croucher PI, Inhibition of p38alpha mitogen-activated protein kinase prevents the development of osteolytic bone disease, reduces tumor burden, and increases survival in murine models of multiple myeloma, Cancer Res. 67, 4572–4577 (2007). [DOI] [PubMed] [Google Scholar]
- 157.Zarredar H, Pashapour S, Farajnia S, Ansarin K, Baradaran B, Ahmadzadeh V, Safari F, Targeting the KRAS, p38α, and NF-κB in lung adenocarcinoma cancer cells: The effect of combining RNA interferences with a chemical inhibitor, J. Cell. Biochem 120, 10670–10677 (2019). [DOI] [PubMed] [Google Scholar]
- 158.Zhang Y, Wang X, Qin X, Wang X, Liu F, White E, Zheng XFS, PP2AC Level Determines Differential Programming of p38-TSC-mTOR Signaling and Therapeutic Response to p38-Targeted Therapy in Colorectal Cancer, EBioMedicine 2, 1944–1956 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Aboukameel A, Muqbil I, Senapedis W, Baloglu E, Landesman Y, Shacham S, Kauffman M, Philip PA, Mohammad RM, Azmi AS, Novel p21-Activated Kinase 4 (PAK4) Allosteric Modulators Overcome Drug Resistance and Stemness in Pancreatic Ductal Adenocarcinoma, Mol. Cancer Ther 16, 76–87 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Callow MG, Clairvoyant F, Zhu S, Schryver B, Whyte DB, Bischoff JR, Jallal B, Smeal T, Requirement for PAK4 in the anchorage-independent growth of human cancer cell lines, J. Biol. Chem 277, 550–558 (2002). [DOI] [PubMed] [Google Scholar]
- 161.Castillo J, Knol JC, Korver CM, Piersma SR, Pham TV, de Goeij-de Haas RR, van Pelt AMM, Jimenez CR, Jansen BJH, Human Testis Phosphoproteome Reveals Kinases as Potential Targets in Spermatogenesis and Testicular Cancer, Mol. Cell. Proteomics MCP 18, S132–S144 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Cosset É, Ilmjärv S, Dutoit V, Elliott K, von Schalscha T, Camargo MF, Reiss A, Moroishi T, Seguin L, Gomez G, Moo J-S, Preynat-Seauve O, Krause K-H, Chneiweiss H, Sarkaria JN, Guan K-L, Dietrich P-Y, Weis SM, Mischel PS, Cheresh DA, Glut3 Addiction Is a Druggable Vulnerability for a Molecularly Defined Subpopulation of Glioblastoma, Cancer Cell 32, 856–868.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Fu X, Feng J, Zeng D, Ding Y, Yu C, Yang B, PAK4 confers cisplatin resistance in gastric cancer cells via PI3K/Akt- and MEK/ERK-dependent pathways, Biosci. Rep 34 (2014), doi: 10.1042/BSR20130102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.He L-F, Xu H-W, Chen M, Xian Z-R, Wen X-F, Chen M-N, Du C-W, Huang W-H, Wu J-D, Zhang G-J, Activated-PAK4 predicts worse prognosis in breast cancer and promotes tumorigenesis through activation of PI3K/AKT signaling, Oncotarget 8, 17573–17585 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kimmelman AC, Hezel AF, Aguirre AJ, Zheng H, Paik J-H, Ying H, Chu GC, Zhang JX, Sahin E, Yeo G, Ponugoti A, Nabioullin R, Deroo S, Yang S, Wang X, McGrath JP, Protopopova M, Ivanova E, Zhang J, Feng B, Tsao MS, Redston M, Protopopov A, Xiao Y, Futreal PA, Hahn WC, Klimstra DS, Chin L, DePinho RA, Genomic alterations link Rho family of GTPases to the highly invasive phenotype of pancreas cancer, Proc. Natl. Acad. Sci. U. S. A 105, 19372–19377 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.King H, Thillai K, Whale A, Arumugam P, Eldaly H, Kocher HM, Wells CM, PAK4 interacts with p85 alpha: implications for pancreatic cancer cell migration, Sci. Rep 7, 42575 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Li N, Lopez MA, Linares M, Kumar S, Oliva S, Martinez-Lopez J, Xu L, Xu Y, Perini T, Senapedis W, Baloglu E, Shammas MA, Hunter Z, Anderson KC, Treon SP, Munshi NC, Fulciniti M, Dual PAK4-NAMPT Inhibition Impacts Growth and Survival, and Increases Sensitivity to DNA-Damaging Agents in Waldenström Macroglobulinemia, Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res 25, 369–377 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Li Q, Zhang X, Wei N, Liu S, Ling Y, Wang H, p21-activated kinase 4 as a switch between caspase-8 apoptosis and NF-κB survival signals in response to TNF-α in hepatocarcinoma cells, Biochem. Biophys. Res. Commun 503, 3003–3010 (2018). [DOI] [PubMed] [Google Scholar]
- 169.Li S-Q, Wang Z-H, Mi X-G, Liu L, Tan Y, MiR-199a/b-3p suppresses migration and invasion of breast cancer cells by downregulating PAK4/MEK/ERK signaling pathway, IUBMB Life 67, 768–777 (2015). [DOI] [PubMed] [Google Scholar]
- 170.Li Z, Li X, Xu L, Tao Y, Yang C, Chen X, Fang F, Wu Y, Ding X, Zhao H, Li M, Qian G, Xu Y, Ren J, Du W, Wang J, Lu J, Hu S, Pan J, Inhibition of neuroblastoma proliferation by PF-3758309, a small-molecule inhibitor that targets p21-activated kinase 4, Oncol. Rep 38, 2705–2716 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lu H, Liu S, Zhang G, Wu null Bin, Zhu Y, Frederick DT, Hu Y, Zhong W, Randell S, Sadek N, Zhang W, Chen G, Cheng C, Zeng J, Wu LW, Zhang J, Liu X, Xu W, Krepler C, Sproesser K, Xiao M, Miao B, Liu J, Song CD, Liu JY, Karakousis GC, Schuchter LM, Lu Y, Mills G, Cong Y, Chernoff J, Guo J, Boland GM, Sullivan RJ, Wei Z, Field J, Amaravadi RK, Flaherty KT, Herlyn M, Xu X, Guo W, PAK signalling drives acquired drug resistance to MAPK inhibitors in BRAF-mutant melanomas, Nature 550, 133–136 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lu S-X, Zhang CZ, Luo R-Z, Wang C-H, Liu L-L, Fu J, Zhang L, Wang H, Xie D, Yun J-P, Zic2 promotes tumor growth and metastasis via PAK4 in hepatocellular carcinoma, Cancer Lett. 402, 71–80 (2017). [DOI] [PubMed] [Google Scholar]
- 173.Park M-H, Lee H-S, Lee C-S, You ST, Kim D-J, Park B-H, Kang MJ, Heo WD, Shin E-Y, Schwartz MA, Kim E-G, p21-Activated kinase 4 promotes prostate cancer progression through CREB, Oncogene 32, 2475–2482 (2013). [DOI] [PubMed] [Google Scholar]
- 174.Siu MKY, Chan HY, Kong DSH, Wong ESY, Wong OGW, Ngan HYS, Tam KF, Zhang H, Li Z, Chan QKY, Tsao SW, Strömblad S, Cheung ANY, p21-activated kinase 4 regulates ovarian cancer cell proliferation, migration, and invasion and contributes to poor prognosis in patients, Proc. Natl. Acad. Sci. U. S. A 107, 18622–18627 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Tyagi N, Bhardwaj A, Singh AP, McClellan S, Carter JE, Singh S, p-21 activated kinase 4 promotes proliferation and survival of pancreatic cancer cells through AKT- and ERK-dependent activation of NF-κB pathway, Oncotarget 5, 8778–8789 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Wong LE, Chen N, Karantza V, Minden A, The Pak4 protein kinase is required for oncogenic transformation of MDA-MB-231 breast cancer cells, Oncogenesis 2, e50 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wu T, Pang Y, Guo J, Yin W, Zhu M, Hao C, Wang K, Wang J, Zhao D, Cheng M, Discovery of 2-(4-Substituted-piperidin/piperazine-1-yl)-N-(5-cyclopropyl-1H-pyrazol-3-yl)-quinazoline-2,4-diamines as PAK4 Inhibitors with Potent A549 Cell Proliferation, Migration, and Invasion Inhibition Activity, Molecules 23 (2018), doi: 10.3390/molecules23020417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Zeng B, Shi W, Tan G, MiR-199a/b-3p inhibits gastric cancer cell proliferation via down-regulating PAK4/MEK/ERK signaling pathway, BMC Cancer 18, 34 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Zhang H-Y, Zhang J, Hao C-Z, Zhou Y, Wang J, Cheng M-S, Zhao D-M, Li F, LC-0882 targets PAK4 and inhibits PAK4-related signaling pathways to suppress the proliferation and invasion of gastric cancer cells, Am. J. Transl. Res 9, 2736–2747 (2017). [PMC free article] [PubMed] [Google Scholar]
- 180.Zhang J, Wang J, Guo Q, Wang Y, Zhou Y, Peng H, Cheng M, Zhao D, Li F, LCH-7749944, a novel and potent p21-activated kinase 4 inhibitor, suppresses proliferation and invasion in human gastric cancer cells, Cancer Lett. 317, 24–32 (2012). [DOI] [PubMed] [Google Scholar]
- 181.Alachkar H, Mutonga M, Malnassy G, Park J-H, Fulton N, Woods A, Meng L, Kline J, Raca G, Odenike O, Takamatsu N, Miyamoto T, Matsuo Y, Stock W, Nakamura Y, T-LAK cell-originated protein kinase presents a novel therapeutic target in FLT3-ITD mutated acute myeloid leukemia, Oncotarget 6, 33410–33425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Ayllón V, O’connor R, PBK/TOPK promotes tumour cell proliferation through p38 MAPK activity and regulation of the DNA damage response, Oncogene 26, 3451–3461 (2007). [DOI] [PubMed] [Google Scholar]
- 183.Chai Y, Xue H, Wu Y, Du X, Zhang Z, Zhang Y, Zhang L, Zhang S, Zhang Z, Xue Z, MicroRNA-216b-3p inhibits lung adenocarcinoma cell growth via regulating PDZ binding kinase/T-LAK-cell-originated protein kinase, Exp. Ther. Med 15, 4822–4828 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Chen F, Li R, Wang C, Cao L, Wang Y, Yu L, T-LAK cell-originated protein kinase is essential for the proliferation of hepatocellular carcinoma SMMC-7721 cells, Cell Biochem. Funct 31, 736–742 (2013). [DOI] [PubMed] [Google Scholar]
- 185.Chen J-H, Liang Y-X, He H-C, Chen J-Y, Lu J-M, Chen G, Lin Z-Y, Fu X, Ling X-H, Han Z-D, Jiang F-N, Zhong W-D, Overexpression of PDZ-binding kinase confers malignant phenotype in prostate cancer via the regulation of E2F1, Int. J. Biol. Macromol 81, 615–623 (2015). [DOI] [PubMed] [Google Scholar]
- 186.Dou X, Wei J, Sun A, Shao G, Childress C, Yang W, Lin Q, PBK/TOPK mediates geranylgeranylation signaling for breast cancer cell proliferation, Cancer Cell Int 15, 27 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Gao G, Zhang T, Wang Q, Reddy K, Chen H, Yao K, Wang K, Roh E, Zykova T, Ma W, Ryu J, Curiel-Lewandrowski C, Alberts D, Dickinson SE, Bode AM, Xing Y, Dong Z, ADA-07 Suppresses Solar Ultraviolet-Induced Skin Carcinogenesis by Directly Inhibiting TOPK, Mol. Cancer Ther 16, 1843–1854 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Gao T, Hu Q, Hu X, Lei Q, Feng Z, Yu X, Peng C, Song X, He H, Xu Y, Zuo W, Zeng J, Liu Z, Yu L, Novel selective TOPK inhibitor SKLB-C05 inhibits colorectal carcinoma growth and metastasis, Cancer Lett. 445, 11–23 (2019). [DOI] [PubMed] [Google Scholar]
- 189.Herrero-Martín D, Osuna D, Ordóñez JL, Sevillano V, Martins AS, Mackintosh C, Campos M, Madoz-Gúrpide J, Otero-Motta AP, Caballero G, Amaral AT, Wai DH, Braun Y, Eisenacher M, Schaefer K-L, Poremba C, de Alava E, Stable interference of EWS-FLI1 in an Ewing sarcoma cell line impairs IGF-1/IGF-1R signalling and reveals TOPK as a new target, Br. J. Cancer 101, 80–90 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hu F, Gartenhaus RB, Eichberg D, Liu Z, Fang H-B, Rapoport AP, PBK/TOPK interacts with the DBD domain of tumor suppressor p53 and modulates expression of transcriptional targets including p21, Oncogene 29, 5464–5474 (2010). [DOI] [PubMed] [Google Scholar]
- 191.Hu F, Gartenhaus RB, Zhao XF, Fang H-B, Minkove S, Poss DE, Rapoport AP, c-Myc and E2F1 drive PBK/TOPK expression in high-grade malignant lymphomas, Leuk. Res 37, 447–454 (2013). [DOI] [PubMed] [Google Scholar]
- 192.Ikeda Y, Park J-H, Miyamoto T, Takamatsu N, Kato T, Iwasa A, Okabe S, Imai Y, Fujiwara K, Nakamura Y, Hasegawa K, T-LAK Cell-Originated Protein Kinase (TOPK) as a Prognostic Factor and a Potential Therapeutic Target in Ovarian Cancer, Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res 22, 6110–6117 (2016). [DOI] [PubMed] [Google Scholar]
- 193.Joel M, Mughal AA, Grieg Z, Murrell W, Palmero S, Mikkelsen B, Fjerdingstad HB, Sandberg CJ, Behnan J, Glover JC, Langmoen IA, Stangeland B, Targeting PBK/TOPK decreases growth and survival of glioma initiating cells in vitro and attenuates tumor growth in vivo, Mol. Cancer 14, 121 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kato T, Inoue H, Imoto S, Tamada Y, Miyamoto T, Matsuo Y, Nakamura Y, Park J-H, Oncogenic roles of TOPK and MELK, and effective growth suppression by small molecular inhibitors in kidney cancer cells, Oncotarget 7, 17652–17664 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kim DJ, Li Y, Reddy K, Lee M-H, Kim MO, Cho Y-Y, Lee S-Y, Kim J-E, Bode AM, Dong Z, Novel TOPK inhibitor HI-TOPK-032 effectively suppresses colon cancer growth, Cancer Res. 72, 3060–3068 (2012). [DOI] [PubMed] [Google Scholar]
- 196.Lei B, Qi W, Zhao Y, Li Y, Liu S, Xu X, Zhi C, Wan L, Shen H, PBK/TOPK expression correlates with mutant p53 and affects patients’ prognosis and cell proliferation and viability in lung adenocarcinoma, Hum. Pathol 46, 217–224 (2015). [DOI] [PubMed] [Google Scholar]
- 197.Liu Y, Liu H, Cao H, Song B, Zhang W, Zhang W, PBK/TOPK mediates promyelocyte proliferation via Nrf2-regulated cell cycle progression and apoptosis, Oncol. Rep 34, 3288–3296 (2015). [DOI] [PubMed] [Google Scholar]
- 198.Ohashi T, Komatsu S, Ichikawa D, Miyamae M, Okajima W, Imamura T, Kiuchi J, Nishibeppu K, Kosuga T, Konishi H, Shiozaki A, Fujiwara H, Okamoto K, Tsuda H, Otsuji E, Overexpression of PBK/TOPK Contributes to Tumor Development and Poor Outcome of Esophageal Squamous Cell Carcinoma, Anticancer Res. 36, 6457–6466 (2016). [DOI] [PubMed] [Google Scholar]
- 199.Ohashi T, Komatsu S, Ichikawa D, Miyamae M, Okajima W, Imamura T, Kiuchi J, Kosuga T, Konishi H, Shiozaki A, Fujiwara H, Okamoto K, Tsuda H, Otsuji E, Overexpression of PBK/TOPK relates to tumour malignant potential and poor outcome of gastric carcinoma, Br. J. Cancer 116, 218–226 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Park J-H, Inoue H, Kato T, Zewde M, Miyamoto T, Matsuo Y, Salgia R, Nakamura Y, TOPK (T-LAK cell-originated protein kinase) inhibitor exhibits growth suppressive effect on small cell lung cancer, Cancer Sci. 108, 488–496 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Park J-H, Lin M-L, Nishidate T, Nakamura Y, Katagiri T, PDZ-binding kinase/T-LAK cell-originated protein kinase, a putative cancer/testis antigen with an oncogenic activity in breast cancer, Cancer Res. 66, 9186–9195 (2006). [DOI] [PubMed] [Google Scholar]
- 202.Song X, Yin S, Zhang E, Fan L, Ye M, Zhang Y, Hu H, Glycycoumarin exerts anti-liver cancer activity by directly targeting T-LAK cell-originated protein kinase, Oncotarget 7, 65732–65743 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Stauffer S, Zeng Y, Zhou J, Chen X, Chen Y, Dong J, CDK1-mediated mitotic phosphorylation of PBK is involved in cytokinesis and inhibits its oncogenic activity, Cell. Signal 39, 74–83 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Sugimori M, Hayakawa Y, Koh M, Hayashi T, Tamura R, Kuroda S, Targeting the T-Lak cell originated protein kinase by OTS964 shrinks the size of power-law coded heterogeneous glioma stem cell populations, Oncotarget 9, 3043–3059 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Uchida E, Suwa S, Yoshimoto R, Watanabe K, Kasama T, Miura O, Fukuda T, TOPK is regulated by PP2A and BCR/ABL in leukemia and enhances cell proliferation, Int. J. Oncol 54, 1785–1796 (2019). [DOI] [PubMed] [Google Scholar]
- 206.Vishchuk OS, Sun H, Wang Z, Ermakova SP, Xiao J, Lu T, Xue P, Zvyagintseva TN, Xiong H, Shao C, Yan W, Duan Q, Zhu F, PDZ-binding kinase/T-LAK cell-originated protein kinase is a target of the fucoidan from brown alga Fucus evanescens in the prevention of EGF-induced neoplastic cell transformation and colon cancer growth, Oncotarget 7, 18763–18773 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Wang M-Y, Lin Z-R, Cao Y, Zheng L-S, Peng L-X, Sun R, Meng D-F, Xie P, Yang J-P, Cao L, Xu L, Huang B-J, Qian C-N, PDZ binding kinase (PBK) is a theranostic target for nasopharyngeal carcinoma: driving tumor growth via ROS signaling and correlating with patient survival, Oncotarget 7, 26604–26616 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Xiao J, Duan Q, Wang Z, Yan W, Sun H, Xue P, Fan X, Zeng X, Chen J, Shao C, Zhu F, Phosphorylation of TOPK at Y74, Y272 by Src increases the stability of TOPK and promotes tumorigenesis of colon, Oncotarget 7, 24483–24494 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Yang J, Yuan D, Xing T, Su H, Zhang S, Wen J, Bai Q, Dang D, Ginsenoside Rh2 inhibiting HCT116 colon cancer cell proliferation through blocking PDZ-binding kinase/T-LAK cell-originated protein kinase, J. Ginseng Res 40, 400–408 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Zheng M, Luan S, Gao S, Cheng L, Hao B, Li J, Chen Y, Hou X, Chen L, Li H, Proton pump inhibitor ilaprazole suppresses cancer growth by targeting T-cell-originated protein kinase, Oncotarget 8, 39143–39153 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Zhu F, Zykova TA, Kang BS, Wang Z, Ebeling MC, Abe Y, Ma W-Y, Bode AM, Dong Z, Bidirectional signals transduced by TOPK-ERK interaction increase tumorigenesis of HCT116 colorectal cancer cells, Gastroenterology 133, 219–231 (2007). [DOI] [PubMed] [Google Scholar]
- 212.Bellon M, Lu L, Nicot C, Constitutive activation of Pim1 kinase is a therapeutic target for adult T-cell leukemia, Blood 127, 2439–2450 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Blanco-Aparicio C, Collazo AMG, Oyarzabal J, Leal JF, Albarán MI, Lima FR, Pequeño B, Ajenjo N, Becerra M, Alfonso P, Reymundo MI, Palacios I, Mateos G, Quiñones H, Corrionero A, Carnero A, Pevarello P, Lopez AR, Fominaya J, Pastor J, Bischoff JR, Pim 1 kinase inhibitor ETP-45299 suppresses cellular proliferation and synergizes with PI3K inhibition, Cancer Lett. 300, 145–153 (2011). [DOI] [PubMed] [Google Scholar]
- 214.Chen J, Kobayashi M, Darmanin S, Qiao Y, Gully C, Zhao R, Kondo S, Wang H, Wang H, Yeung S-CJ, Lee M-H, Hypoxia-mediated up-regulation of Pim-1 contributes to solid tumor formation, Am. J. Pathol 175, 400–411 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Deng D, Wang L, Chen Y, Li B, Xue L, Shao N, Wang Q, Xia X, Yang Y, Zhi F, MicroRNA-124–3p regulates cell proliferation, invasion, apoptosis, and bioenergetics by targeting PIM1 in astrocytoma, Cancer Sci. 107, 899–907 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Fan R-F, Lu Y, Fang Z-G, Guo X-Y, Chen Y-X, Xu Y-C, Lei Y-M, Liu K-F, Lin D-J, Liu L-L, Liu X-F, PIM-1 kinase inhibitor SMI-4a exerts antitumor effects in chronic myeloid leukemia cells by enhancing the activity of glycogen synthase kinase 3β, Mol. Med. Rep 16, 4603–4612 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Herzog S, Fink MA, Weitmann K, Friedel C, Hadlich S, Langner S, Kindermann K, Holm T, Böhm A, Eskilsson E, Miletic H, Hildner M, Fritsch M, Vogelgesang S, Havemann C, Ritter CA, Meyer zu Schwabedissen HE, Rauch B, Hoffmann W, Kroemer HK, Schroeder H, Bien-Möller S, Pim1 kinase is upregulated in glioblastoma multiforme and mediates tumor cell survival, Neuro-Oncol. 17, 223–242 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Hou X, Yu Y, Feng J, Wang J, Zheng C, Ling Z, Ge M, Zhu X, Biochemical changes of salivary gland adenoid cystic carcinoma cells induced by SGI-1776, Exp. Cell Res 352, 403–411 (2017). [DOI] [PubMed] [Google Scholar]
- 219.Jie W, He Q-Y, Luo B-T, Zheng S-J, Kong Y-Q, Jiang H-G, Li R-J, Guo J-L, Shen Z-H, Inhibition of Pim-1 attenuates the proliferation and migration in nasopharyngeal carcinoma cells, Asian Pac. J. Trop. Med 5, 645–650 (2012). [DOI] [PubMed] [Google Scholar]
- 220.Karatas OF, Antiproliferative potential of miR-33a in laryngeal cancer Hep-2 cells via targeting PIM1, Head Neck 40, 2455–2461 (2018). [DOI] [PubMed] [Google Scholar]
- 221.Kim J-E, Son JE, Jeong H, Joon Kim D, Seo SK, Lee E, Lim TG, Kim JR, Chen H, Bode AM, Lee KW, Dong Z, A Novel Cinnamon-Related Natural Product with Pim-1 Inhibitory Activity Inhibits Leukemia and Skin Cancer, Cancer Res. 75, 2716–2728 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Kirschner AN, Wang J, van der Meer R, Anderson PD, Franco-Coronel OE, Kushner MH, Everett JH, Hameed O, Keeton EK, Ahdesmaki M, Grosskurth SE, Huszar D, Abdulkadir SA, PIM kinase inhibitor AZD1208 for treatment of MYC-driven prostate cancer, J. Natl. Cancer Inst 107 (2015), doi: 10.1093/jnci/dju407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Leung CO, Wong CC, Fan DN, Kai AK, Tung EK, Xu IM, Ng IO, Lo RC, PIM1 regulates glycolysis and promotes tumor progression in hepatocellular carcinoma, Oncotarget 6, 10880–10892 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Li J-Q, Yang X, Zhou X-M, PIM1 gene silencing inhibits proliferation and promotes apoptosis of human esophageal cancer cell line Eca-109, Cancer Biomark. Sect. Dis. Markers 18, 149–154 (2017). [DOI] [PubMed] [Google Scholar]
- 225.Li J, Hu XF, Loveland BE, Xing PX, Pim-1 expression and monoclonal antibody targeting in human leukemia cell lines, Exp. Hematol 37, 1284–1294 (2009). [DOI] [PubMed] [Google Scholar]
- 226.Li K, Li Y, Zhou D, Fan Y, Guo H, Ma T, Wen J, Liu D, Zhao L, Synthesis and biological evaluation of quinoline derivatives as potential anti-prostate cancer agents and Pim-1 kinase inhibitors, Bioorg. Med. Chem 24, 1889–1897 (2016). [DOI] [PubMed] [Google Scholar]
- 227.Liao Y, Feng Y, Shen J, Gao Y, Cote G, Choy E, Harmon D, Mankin H, Hornicek F, Duan Z, Clinical and biological significance of PIM1 kinase in osteosarcoma, J. Orthop. Res. Off. Publ. Orthop. Res. Soc 34, 1185–1194 (2016). [DOI] [PubMed] [Google Scholar]
- 228.Liu K, Gao H, Wang Q, Wang L, Zhang B, Han Z, Chen X, Han M, Gao M, Hispidulin suppresses cell growth and metastasis by targeting PIM1 through JAK2/STAT3 signaling in colorectal cancer, Cancer Sci. 109, 1369–1381 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 229.Mawas AS, Amatya VJ, Suzuki R, Kushitani K, Mohi El-Din MM, Takeshima Y, PIM1 knockdown inhibits cell proliferation and invasion of mesothelioma cells, Int. J. Oncol 50, 1029–1034 (2017). [DOI] [PubMed] [Google Scholar]
- 230.Morishita D, Takami M, Yoshikawa S, Katayama R, Sato S, Kukimoto-Niino M, Umehara T, Shirouzu M, Sekimizu K, Yokoyama S, Fujita N, Cell-permeable carboxyl-terminal p27(Kip1) peptide exhibits anti-tumor activity by inhibiting Pim-1 kinase, J. Biol. Chem 286, 2681–2688 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Mou S, Wang G, Ding D, Yu D, Pei Y, Teng S, Fu Q, Expression and function of PIM kinases in osteosarcoma, Int. J. Oncol 49, 2116–2126 (2016). [DOI] [PubMed] [Google Scholar]
- 232.Nakano H, Saito N, Parker L, Tada Y, Abe M, Tsuganezawa K, Yokoyama S, Tanaka A, Kojima H, Okabe T, Nagano T, Rational evolution of a novel type of potent and selective proviral integration site in Moloney murine leukemia virus kinase 1 (PIM1) inhibitor from a screening-hit compound, J. Med. Chem 55, 5151–5164 (2012). [DOI] [PubMed] [Google Scholar]
- 233.Schroeder RL, Goyal N, Bratton M, Townley I, Pham NA, Tram P, Stone T, Geathers J, Nguyen K, Sridhar J, Identification of quinones as novel PIM1 kinase inhibitors, Bioorg. Med. Chem. Lett 26, 3187–3191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Shannan B, Watters A, Chen Q, Mollin S, Dörr M, Meggers E, Xu X, Gimotty PA, Perego M, Li L, Benci J, Krepler C, Brafford P, Zhang J, Wei Z, Zhang G, Liu Q, Yin X, Nathanson KL, Herlyn M, Vultur A, PIM kinases as therapeutic targets against advanced melanoma, Oncotarget 7, 54897–54912 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Smedt RD, Peirs S, Morscio J, Matthijssens F, Roels J, Reunes L, Lintermans B, Goossens S, Lammens T, Roy NV, Touzart A, Jenni S, Tsai Y-C, Lovisa F, Mussolin L, Serafin V, Nieuwerburgh FV, Deforce D, Uyttebroeck A, Tousseyn T, Burkhardt B, Klapper W, Moerloose BD, Benoit Y, Macintyre E, Bourquin J-P, Basso G, Accordi B, Bornhauser B, Meijerink J, Vandenberghe P, Vlierberghe PV, Preclinical evaluation of second generation PIM inhibitors for the treatment of T-cell acute lymphoblastic leukemia and lymphoma, Haematologica , haematol.2018 199257 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Thomas M, Lange-Grünweller K, Weirauch U, Gutsch D, Aigner A, Grünweller A, Hartmann RK, The proto-oncogene Pim-1 is a target of miR-33a, Oncogene 31, 918–928 (2012). [DOI] [PubMed] [Google Scholar]
- 237.Wang J, Anderson PD, Luo W, Gius D, Roh M, Abdulkadir SA, Pim1 kinase is required to maintain tumorigenicity in MYC-expressing prostate cancer cells, Oncogene 31, 1794–1803 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Wang Q, Jiang Y, Guo R, Lv R, Liu T, Wei H, Ming H, Tian X, Physcion 8-O-β-glucopyranoside suppresses tumor growth of Hepatocellular carcinoma by downregulating PIM1, Biomed. Pharmacother. Biomedecine Pharmacother 92, 451–458 (2017). [DOI] [PubMed] [Google Scholar]
- 239.Weirauch U, Beckmann N, Thomas M, Grünweller A, Huber K, Bracher F, Hartmann RK, Aigner A, Functional Role and Therapeutic Potential of the Pim-1 Kinase in Colon Carcinoma, Neoplasia N. Y. N 15, 783–794 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Wu Y, Deng Y, Zhu J, Duan Y, Weng W, Wu X, Pim1 promotes cell proliferation and regulates glycolysis via interaction with MYC in ovarian cancer, OncoTargets Ther. 11, 6647–6656 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Xie J, Bai J, SGI-1776, an imidazo pyridazine compound, inhibits the proliferation of ovarian cancer cells by inactivating Pim-1, Zhong Nan Da Xue Xue Bao Yi Xue Ban 39, 649–657 (2014). [DOI] [PubMed] [Google Scholar]
- 242.Xu D, Allsop SA, Witherspoon SM, Snider JL, Yeh JJ, Fiordalisi JJ, White CD, Williams D, Cox AD, Baines AT, The oncogenic kinase Pim-1 is modulated by K-Ras signaling and mediates transformed growth and radioresistance in human pancreatic ductal adenocarcinoma cells, Carcinogenesis 32, 488–495 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Yan B, Yau EX, Samanta S, Ong CW, Yong KJ, Ng LK, Bhattacharya B, Lim KH, Soong R, Yeoh KG, Deng N, Tan P, Lam Y, Salto-Tellez M, Singapore Gastric Cancer Consortium, Clinical and therapeutic relevance of PIM1 kinase in gastric cancer, Gastric Cancer Off. J. Int. Gastric Cancer Assoc. Jpn. Gastric Cancer Assoc 15, 188–197 (2012). [DOI] [PubMed] [Google Scholar]
- 244.Ye C, Zhang C, Huang H, Yang B, Xiao G, Kong D, Tian Q, Song Q, Song Y, Tan H, Wang Y, Zhou T, Zi X, Sun Y, The Natural Compound Myricetin Effectively Represses the Malignant Progression of Prostate Cancer by Inhibiting PIM1 and Disrupting the PIM1/CXCR4 Interaction, Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol 48, 1230–1244 (2018). [DOI] [PubMed] [Google Scholar]
- 245.Zhang C, Qie Y, Yang T, Wang L, Du E, Liu Y, Xu Y, Qiao B, Zhang Z, Kinase PIM1 promotes prostate cancer cell growth via c-Myc-RPS7-driven ribosomal stress, Carcinogenesis 40, 202 (2019). [DOI] [PubMed] [Google Scholar]
- 246.Zhang G, Liu Z, Cui G, Wang X, Yang Z, MicroRNA-486–5p targeting PIM-1 suppresses cell proliferation in breast cancer cells, Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med 35, 11137–11145 (2014). [DOI] [PubMed] [Google Scholar]
- 247.Zhang M, Liu T, Sun H, Weng W, Zhang Q, Liu C, Han Y, Sheng W, Pim1 supports human colorectal cancer growth during glucose deprivation by enhancing the Warburg effect, Cancer Sci. 109, 1468–1479 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Zhao B, Liu L, Mao J, Zhang Z, Wang Q, Li Q, PIM1 mediates epithelial-mesenchymal transition by targeting Smads and c-Myc in the nucleus and potentiates clear-cell renal-cell carcinoma oncogenesis, Cell Death Dis. 9, 307 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Zhu X, Xu J, Hu S, Feng J, Jiang L, Hou X, Cao J, Han J, Ling Z, Ge M, Pim-1 acts as an oncogene in human salivary gland adenoid cystic carcinoma, J. Exp. Clin. Cancer Res. CR 33, 114 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Botham RC, Fan TM, Im I, Borst LB, Dirikolu L, Hergenrother PJ, Dual small-molecule targeting of procaspase-3 dramatically enhances zymogen activation and anticancer activity, J. Am. Chem. Soc 136, 1312–1319 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Joshi AD, Botham RC, Schlein LJ, Roth HS, Mangraviti A, Borodovsky A, Tyler B, Joslyn S, Looper JS, Podell M, Fan TM, Hergenrother PJ, Riggins GJ, Synergistic and targeted therapy with a procaspase-3 activator and temozolomide extends survival in glioma rodent models and is feasible for the treatment of canine malignant glioma patients, Oncotarget 8, 80124–80138 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Lucas PW, Schmit JM, Peterson QP, West DC, Hsu DC, Novotny CJ, Dirikolu L, Churchwell MI, Doerge DR, Garrett LD, Hergenrother PJ, Fan TM, Pharmacokinetics and derivation of an anticancer dosing regimen for PAC-1, a preferential small molecule activator of procaspase-3, in healthy dogs, Invest. New Drugs 29, 901–911 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Ma J, Chen D, Lu K, Wang L, Han X, Zhao Y, Gong P, Design, synthesis, and structure-activity relationships of novel benzothiazole derivatives bearing the ortho-hydroxy N-carbamoylhydrazone moiety as potent antitumor agents, Eur. J. Med. Chem 86, 257–269 (2014). [DOI] [PubMed] [Google Scholar]
- 254.Patel V, Balakrishnan K, Keating MJ, Wierda WG, Gandhi V, Expression of executioner procaspases and their activation by a procaspase-activating compound in chronic lymphocytic leukemia cells, Blood 125, 1126–1136 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Peh J, Fan TM, Wycislo KL, Roth HS, Hergenrother PJ, The combination of vemurafenib and procaspase-3 activation is synergistic in mutant BRAF melanomas, Mol. Cancer Ther 15, 1859–1869 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Peterson QP, Hsu DC, Novotny CJ, West DC, Kim D, Schmit JM, Dirikolu L, Hergenrother PJ, Fan TM, Discovery and canine preclinical assessment of a nontoxic procaspase-3-activating compound, Cancer Res. 70, 7232–7241 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Roth HS, Hergenrother PJ, Derivatives of Procaspase-Activating Compound 1 (PAC-1) and their Anticancer Activities, Curr. Med. Chem 23, 201–241 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Wang F, Liu Y, Wang L, Yang J, Zhao Y, Wang N, Cao Q, Gong P, Wu C, Targeting procaspase-3 with WF-208, a novel PAC-1 derivative, causes selective cancer cell apoptosis, J. Cell. Mol. Med 19, 1916–1928 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Wang F, Wang L, Li Y, Wang N, Wang Y, Cao Q, Gong P, Yang J, Wu C, PAC-1 and its derivative WF-210 Inhibit Angiogenesis by inhibiting VEGF/VEGFR pathway, Eur. J. Pharmacol 821, 29–38 (2018). [DOI] [PubMed] [Google Scholar]
- 260.West DC, Qin Y, Peterson QP, Thomas DL, Palchaudhuri R, Morrison KC, Lucas PW, Palmer AE, Fan TM, Hergenrother PJ, Differential effects of procaspase-3 activating compounds in the induction of cancer cell death, Mol. Pharm 9, 1425–1434 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Zaman S, Wang R, Gandhi V, Targeting executioner procaspase-3 with the procaspase-activating compound B-PAC-1 induces apoptosis in multiple myeloma cells, Exp. Hematol 43, 951–962.e3 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Zhai X, Huang Q, Jiang N, Wu D, Zhou H, Gong P, Discovery of hybrid dual N-acylhydrazone and diaryl urea derivatives as potent antitumor agents: design, synthesis and cytotoxicity evaluation, Molecules 18, 2904–2923 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Drug target ablation with CRISPR/Cas9.
Figure S2. Cell competition assays in multiple cancer types.
Figure S3. Verification of drug target knockouts.
Figure S4. Knocking out the verified genetic dependency MEK1 in A375 melanoma cells.
Figure S5. MDA-MB-231 clonal analysis.
Figure S6. Analysis of homolog gene expression in CRISPR-KO clones.
Figure S7. Analysis of homolog gene expression in published RNA-Seq experiments.
Figure S8. Assessing putative cancer dependencies in whole-genome CRISPR and RNAi screens.
Figure S9. Targeting several putative cancer dependencies with CRISPRi.
Figure S10. Knocking out putative cancer dependencies fails to sensitize cells to several clinical chemotherapy agents.
Figure S11. Target-independent toxicity of RNAi reagents previously used to investigate several putative cancer dependencies.
Figure S12. Using CRISPR to validate the MOA of several anti-cancer drugs.
Figure S13. Off-target toxicity of two caspase 3-activating compounds in CASP3-KO clones.
Figure S14. Target-independent cancer cell killing in single-agent and combination-therapy experiments.
Figure S15. Off-target toxicity of two putative HDAC6-inhibiting compounds in HDAC6-KO ovarian cancer clones.
Figure S16. OTS964-resistant clones harbor a mutation in the xDFG residue of CDK11B.
Figure S17. CDK11 activity is required for progression through mitosis.
Data file S1. Literature supporting the designation of HDAC6, MAPK14, PAK4, PBK, and PIM1 as cancer genetic dependencies and CASP3 as a drug target.
Data file S2. Cell competition assay results.
Data file S3. Sources of the cell lines used in this manuscript.
Data file S4. CRISPR gRNA sequences.
Data file S5. CRISPRi gRNA sequences.
Data file S6. qPCR primers.
Data file S7. Antibody sources and concentrations.
Data file S8. Drugs and drug sources.
Movie S1. A375 cells expressing H2B-mCherry released from a double-thymidine block into normal medium.
Movie S2. A375 cells expressing H2B-mCherry released from a double-thymidine block into medium with 25 nM OTS964.
Movie S3. A375 cells expressing H2B-mCherry released from a double-thymidine block into medium with 100 nM OTS964.
Movie S4. A375CDK11B-G579S cells expressing H2B-mCherry released from a double-thymidine block into medium with 100 nM OTS964.