

Abstract
Activation of Ca2+/calmodulin kinase II (CaMKII) and the N-Methyl D-aspartate receptor (NMDAR), particularly its GluN2B subunit, contribute to the central sensitization of nociceptive pathways and persistent pain. Using mutant mice wherein the activity-driven binding of CaMKII to S1303 in GluN2B is abrogated (GluN2BKI), this study investigated the importance of this interaction for acute and persistent inflammatory nociception. GluN2BKI, wild type and heterozygote mice did not differ in responses to acute noxious heat stimuli as measured with tail flick, paw flick, or hot plate assays, nor did they differ in their responses to mechanical stimulation with von Frey filaments. Surprisingly, the three genotypes exhibited similar spontaneous pain behaviors and hypersensitivity to heat or mechanical stimuli induced by intraplantar injection of capsaicin; however, GluN2BKI mice did not immediately attend to the paw. WT and GluN2BKI mice also did not differ in the nociceptive behaviors elicited by intraplantar injection of formalin, even though MK801 greatly reduced these behaviors in both genotypes concordant with NMDAR dependence. CaMKII binding to GluN2B at S1303 therefore does not appear to be critical for the development of inflammatory nociception. Finally, intrathecal KN93 reduced formalin-induced nociceptive behaviors in GluN2BKI mice. KN93 does not inhibit CaKMII, but rather binds Ca2+/calmodulin. It has multiple other targets including Ca2+-, Na+- and K+-channels, as well as various kinases. Therefore, the use of GluN2BKI mice provided genetic specificity in assessing the role of CaMKII in inflammatory pain signaling cascades. These results challenge current thinking on the involvement of the CaMKII-NMDAR interaction in inflammatory pain.
Keywords: Nociception, NMDA receptor, Capsaicin, GluN2B, CaMKII
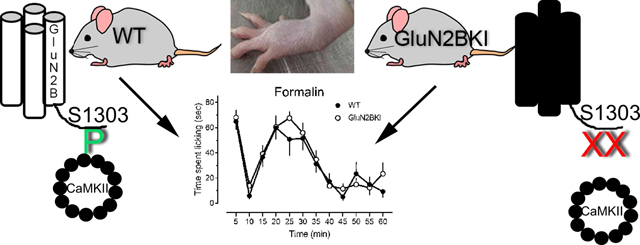
Graphical Abstract
1. Introduction
The role of the NMDA receptor (NMDAR) has been extensively studied and documented in both inflammatory and neuropathic models of nociception (Bourinet et al., 2014; Fisher et al., 2000; Petrenko et al., 2003). In response to noxious peripheral stimuli, the NMDAR mediates changes in neuronal protein expression and function in both the peripheral and central nervous systems. These changes alter responsiveness to subsequent noxious stimuli long after the inciting stimuli have lapsed; this process is thought to underlie the development of central and peripheral sensitization and chronic pain states (Ji et al., 2003; Woolf, 2011). Indeed, several drugs that antagonize NMDARs, such as ketamine, memantine and dextromethorphan, are now used clinically to treat chronic pain (Hewitt, 2000; Kreutzwiser and Tawfic, 2019; Schroeder and Schroeder, 2019).
NMDARs assemble as tetramers composed of two GluN1 subunits and two GluN2 or GluN3 subunits. NMDARs in nociceptive pathways of the dorsal horn include GluN2B (MacDermott, 2014; Momiyama, 2000), an isoform of GluN2 that binds Ca2+/calmodulin (CaM) kinase II (CaMKII). At excitatory synapses, NMDAR-mediated increases in postsynaptic Ca2+ concentration lead to the Ca2+/CaM-dependent activation of CaMKII, and its subsequent translocation to postsynaptic sites, where it can associate with the GluN1 or GluN2B subunits of the NMDAR (Leonard et al., 1999; Strack and Colbran, 1998). Binding of Ca2+/CaM to CaMKII induces a conformational change that exposes the catalytic site of CaMKII, enabling autophosphorylation that allows CaMKII to remain activated independent of intracellular Ca2+ levels (Bayer et al., 2001; Strack and Colbran, 1998). Mice with the point mutation in CaMKII that prevents α-CaMKII autophosphorylation (T286A) are deficient in NMDAR-dependent LTP (Glazewski et al., 2000), and show reduced pain behaviors in the second phase of the formalin test (Zeitz et al., 2004). Pain behaviors in the second phase of the formalin test, as well as sensitization of neurons in the spinal cord following intradermal injection of capsaicin are also blocked by KN93 (Choi et al., 2006; Fang et al., 2002). KN93 binds to Ca2+/CaM (Pellicena and Schulman, 2014; Wong et al., 2019), preventing its binding to the regulatory site and accompanying exposure of CaMKII’s catalytic site, thereby inhibiting only stimulated but not ongoing autonomous CaMKII activity (Barcomb et al., 2015; Vest et al., 2010). Inhibition of CaMKII either pharmacologically or with siRNA also reduces bone cancer pain, chemotherapy-induced neuropathic pain, and visceral pain (Halt et al., 2012; Liu et al., 2014; Shirahama et al., 2012). Interestingly, in each of these models, activity of CaMKII was linked with GluN2B, which is abundantly expressed at synapses in the dorsal horn of the spinal cord (Hildebrand et al., 2014). Of note, specific antagonists of GluN2B reverse spontaneous pain related behaviors evoked by intraplantar injection of capsaicin (Taniguchi et al., 1997) or formalin (Borza et al., 2007). Further, GluN2B antagonism reduces mechanical allodynia without the deficits in motor coordination seen with complete NMDAR antagonism (Boyce et al., 1999).
Given that postsynaptic CaMKII activity is linked to NMDAR activation, especially via its binding to the GluN2B subunit, specific disruption of this interaction could be a means to selectively abrogate NMDAR-dependent sensitization in response to noxious peripheral stimuli without complete inhibition of the NMDAR. To investigate this idea, we have used mice with point mutations (L1298A and R1300Q) on the C-terminal portion of GluN2B that specifically prevent activity-driven CaMKII binding (Halt et al., 2012). The goal of the present study was to characterize the pain phenotype of these mice. We hypothesized that (1) wildtype (WT) and GluN2BKI (KI) mice would not differ in responsiveness to acute noxious stimuli as these stimuli are largely NMDAR-independent; and (2) hypersensitivity would be reduced in GluN2BKI mice compared to WT mice in models of inflammatory pain. To that end, we tested responsiveness to acute mechanical and thermal stimuli as well as development of pain behaviors in the capsaicin and formalin models of inflammatory pain in these KI mice and their heterozygous (HET) and WT littermates.
2. Results
2.1. Abrogation of CaMKII binding to S1303 in GluN2B does not alter acute response to heat or mechanical stimuli
GluN2BKI mice did not differ in their responsiveness to acute noxious heat stimuli. No differences in response latency were observed among WT, HET or GluN2BKI mice at any stimulus intensity in either the tail flick or the hot plate test. All three genotypes exhibited an intensity-dependent decrease in response latency as the stimulus intensity increased (Fig. 1A and B). GluN2BKI mice also did not differ from WT or HET mice in their latency to withdraw the hind paw from a noxious heat stimulus (Fig. 1C). Furthermore, WT, HET and GluN2BKI mice did not differ with respect to their basal mechanical withdrawal threshold (Fig. 1D and Table 1). Collectively, these data suggest that binding of CaMKII to the GluN2B subunit of NMDAR does not mediate sensitivity to acute noxious heat or innocuous mechanical stimuli.
Fig. 1.

Response latencies to acute noxious mechanical and heat stimuli in wildtype (WT), heterozygous (HET), and GluN2B knock-in (GluN2BKI) mice. A) Latency to tail withdrawal from low and high intensity heat. [Two-way repeated measures ANOVA Genotype: F(2,25) = 0.29, P = 0.0818; Intensity F(1,25) = 18.38, P = 0.0002; Genotype × Intensity: F(2,25) = 0.42, P = 0.6643] B) Response latency on hot plate to different temperatures. [Two-way repeated measures ANOVA Genotype: F(2,25) = 0.29, P = 0.7532; Intensity F(2,50) = 130.48, P < 0.0001; Genotype × Intensity: F(4,50) = 0.21, P = 0.9343]. C) Latency to paw withdrawal to heat before (Baseline) and 20 min after injection of 0.1% capsaicin (CAP; 10 μl) in the dorsal surface of the left hind paw. [Two-way repeated measures ANOVA Genotype: F(2,24) = 1.57, P = 0.2286; Time: F(1,24) = 130.50, P < 0.0001; Genotype × Time: F(2,24) =3.61, P = 0.0425]. For panels A-C: WT (open bars, circles), HET (solid bars, squares), and GluN2BKI mice (KI, hatched bars, triangles). D) Percent response to application of von Frey applications of incremental force before (Baseline, open symbols) and 20 min after injection of 0.1% capsaicin (10 μl, solid symbols) in the dorsal surface of the right hind paw. Abscissa indicates log mN of force. See also Table 1. For panel D: WT (circle), HET (square), GluN2BKI (triangle) mice. Error bars are omitted for clarity. Values are the mean ± S.E.M of determinations in 7 −11 mice in each genotype. Panels A-C: **P < 0.01 compared to lowest intensity or baseline value;
Table 1.
Response Thresholds for Withdrawal from von Frey Filaments Before and 20 min After Intraplantar Injection of 0.1% Capsaicin in the Hind paw.
| Baseline | After Capsaicin | |||
|---|---|---|---|---|
| Ipsilateral | Contralateral | Ipsilateral | Contralateral | |
| WT | 4.1 a (4.0–4.1) | 4.1 a (4.1–4.2) | 2.7 ** (2.3–3.0) | 4.1 a (4.0 – 4.2) |
| MET | 4.1 a (3.9–4.1) | 4.1 a (4.1–4.2) | 2.5 ** (2.4–2.7) | 3.9 ** (3.6 – 4.1) |
| GIuN2BKl | 4.1 a (4.1–4.2) | 4.1 a (4.1 –4.2) | 2.6 ** (2.3–2.7) | 4.1 a (4.0 – 4.2) |
Values are the EF50 in mN and 95% comfidence limits. a indicates that the EF50 values and slopes do not differ.
p <0.01 compared to baseline values for the corresponding hind paw. WT: F(2,40) = 26.47 p < 0.0001; HET: F (2,56) = 28.97 < 0.0001; KI: F(2,44) =31.01 p < 0.0001.
2.2. Abrogation of CaMKII binding to S1303 GluN2B increases time to onset, but not duration of capsaicin-induced spontaneous pain behaviors or hypersensitivity to heat or mechanical stimuli
Spontaneous licking and biting behaviors were measured in the initial 15 min after injection of capsaicin. Substantial variability was observed within each genotype. GluN2BKI mice did not differ from HET or WT mice in their response to capsaicin injection into either the left or right hind paw. Moreover, the duration of licking and biting evoked by capsaicin did not differ between the left or right hind paw for any of the genotypes. (Fig. 2A). During the first assessment of licking and biting behaviors after injection into the left hind paw, the blinded observer noted that a subset of the mice did not immediately initiate licking and biting of the hind paw. The onset of licking and biting of the hind paw was therefore also recorded after the injection of capsaicin into the right paw the following week. On breaking the blind, it was determined that WT and HET mice initiated licking and biting within 39.8 and 42.1 sec, respectively, of capsaicin injection. However, GluN2BKI mice did not initiate spontaneous behaviors on average until 135.3 seconds after capsaicin injection (Fig. 2B). Interestingly, this delay appeared to arise because of licking and biting misdirected to the perianal region, a behavior that was not observed in WT or HET mice. Capsaicin induced equivalent heat hypersensitivity in GluN2BKI, HET and WT mice (Fig. 1C). It also shifted the force-response curve for tactile stimulation with von Frey filaments to the left to an equivalent extent in all three genotypes. Taken together, these findings suggest that binding of CaMKII to the GluN2B subunit is not essential for the immediate development of hypersensitivity to mechanical or heat stimuli.
Fig. 2.

Capsaicin-evoked pain behaviors. A) Duration of licking and biting behaviors during the 15 min after intraplantar injection of capsaicin in the left (first exposure) or the right (second exposure) hind paw of WT (open bar, circle), HET (solid bar, square), and GluN2BKI (hatched bar, triangle) mice. [Two-way repeated measures ANOVA followed by Holm-Sidak test. Genotype: F(2,23) = 0.416, P = 0.665; Paw: F(1,23) = 7.032, P = 0.014; Genotype × Paw: F(2,25) = 0.247, P = 0.783] B) Time to onset of licking and biting of the hind paw; symbols as in A. ANOVA followed by Holm-Sidak test. F(2, 24) = 9.298, P = 0.001, Holm-Sidak for WT vs. KI adjusted and for KI vs HET P = 0.0027. All values are the mean ± S.E.M of determinations in 7 – 10 mice in each genotype.
2.3. Abrogation of CaMKII binding to S1303 in GluN2B does not alter formalin-induced pain behaviors
The finding that GluN2BKI mice did not differ in the magnitude of capsaicin-induced behaviors was unexpected, particularly in light of multiple reports that KN93 and the CaMKII autophosphorylation mutation, T286A, attenuate both capsaicin and formalin-induced behaviors (Choi et al., 2006; Fang et al., 2002; Zeitz et al., 2004). Therefore, a second model of inflammatory pain, intraplantar injection of formalin, was used to assess spontaneous pain behaviors in GluN2BKI mice and WT littermates; HET littermates were not tested. Both WT and GluN2BKI mice exhibited the characteristic first and second phase of formalin evoked behaviors (Fig. 3A and C). They did not differ in the magnitude or time course of behaviors [compare open symbols, Fig. 3 panels A and C; Two-way repeated measures ANOVA, Genotype: F(1,19) = 0.066 P = 0.209; Time: F(11,209) = 189.639 P < 0.001; Genotype × Time: F (11,209) = 0.604 P = 0.824]. This finding suggests that formalin-evoked pain behaviors are independent of CaMKII binding to NMDAR that contain GluN2B subunits. Unlike the capsaicin test, GluN2BKI mice did not exhibit misdirected licking or biting, or a delayed response to formalin. To confirm that the second phase of the formalin-induced behaviors was dependent on activation of the NMDAR, WT and GluN2BKI mice were pretreated with saline or the noncompetitive NMDAR antagonist MK801 (0.08 mg/kg s.c.). This dose was based on its in vivo binding to the NMDAR (Price et al., 1988) and ability to block NMDA-induced convulsions (Parsons et al., 1995). Pretreatment with MK801 did not diminish the magnitude of the first phase in either genotype. However, it decreased the magnitude of the second phase in both WT and GluN2BKI mice. The latter finding confirms that phase two behaviors are dependent on NMDAR activation in both genotypes (Fig. 3A and B).
Fig. 3.

Subcutaneous pretreatment with 0.08 mg/kg MK801 attenuates formalin-evoked pain behaviors in paw in both WT littermates (A,B) and GluN2BKI (C,D) mice. Panels A and C illustrate the time course of the effect. Arrow indicates time of MK801 administration. Arrowhead indicates time of formalin injection. MK801 treatment significantly decreased the duration of licking and biting in both WT (panel A) and GluN2BKI (panel C) mice. Panels B and D depict total time spent licking and biting during the second phase (10–40 min). Open symbols: saline control. Solid symbols: MK801. All values are mean ± S.E.M of determinations of 7–11 mice per genotype. Panels A and C: *P < 0.05, **P < 0.01. Two-way repeated measures ANOVA followed by Holm-Sidak test [WT - Treatment: F(1,15) = 26.237 P < 0.001 Time: F(11,165) = 9.760, P < 0.001; Treatment × Time: F(11,165) = 1.831 P =0.053. GluN2BKI - Treatment: F(1,16) = 10.805 P = 0.005; Time: F(11,176) = 13.683 P < 0.001) Treatment × Time F(11,176) = 2.177, P 0.018]. Panels B and D: **, P < 0.01; ***P < 0.01 Student’s t-test C: t(15) = 4.376,P= 0.0005 D: (t(16) = 4.276, P =0.0006.
2.4. Inhibition of Ca2+/CaM with KN93 suppresses formalin-evoked behaviors in GluN2BKI mice
A final set of experiments determined whether KN93, now understood to bind to Ca2+/CaM and interfere with its binding to CaMKII (Wong et al., 2019), could inhibit formalin-evoked behaviors in WT and GluN2BKI mice. WT and GluN2BKI mice were intrathecally injected with 20 μg (33.3 nmol) KN93 or its DMSO-based vehicle 20 min before intraplantar injection of formalin. This dose was selected on the basis of previous reports, which demonstrated that it prevented injury-induced phosphorylation of CaMKII at T286 in the dorsal horn of rats (Chen et al., 2009; Dai et al., 2005). Intrathecal administration of KN93 did not suppress the first phase formalin behavior in WT mice. Although KN93 appeared to suppress the first phase response in KI mice, this effect did not achieve statistical significance compared to the vehicle-treated group (Holm-Sidak test, P = 0.102). For reasons that are unclear, the DMSO vehicle suppressed second-phase formalin responses in the WT mice (compare Fig. 4A with Fig. 3A), which hampered interpretation of the effects of KN93 in these mice (Fig. 4A, B). This was not the case for KI mice, and the ability of KN93 to suppress phase 2 behaviors was evident (Fig. 4C, D).
Fig. 4.

Intrathecal pretreatment with 20 μg (33 nmol) KN93 differentially attenuates formalin-evoked pain behaviors. (A,B) WT littermates (C,D) GluN2BKI mice. Panels A and C illustrate the time course of the effect. Arrow indicates time of KN93 administration. Arrowhead indicates time of formalin injection. KN93 treatment did not significantly decrease the duration of licking and biting in WT (panel A) but did so in GluN2BKI (panel C) mice. Panels B and D depict total time spent licking and biting during the second phase (15–45 min). Open symbols: DMSO control. Solid symbols: KN93. All values are mean ± S.E.M of determinations of 6–8 mice per genotype. Panels A and C: *P< 0.05, **P< 0.01. Two-way repeated measures ANOVA followed by Holm-Sidak test [WT - Treatment: F(1,11) = 4.182, P = 0.066 Time: F(11,121) = 8.281, P < 0.001; Treatment × Time F: (11,121) = 1.052, P = 0.406. GluN2BKI - Treatment: F(1,12) = 7.356, P = 0.019; Time F(11,132) = 9.178, P < 0.001) Treatment × Time F(11,132) = 2.093, P = 0.025]. Panels B and D: *P < 0.05; Student’s t test B: t(11) = 2..055, P = 0.065 D: t(12) = 2.236, P = 0.045
3. Discussion
This study used GluN2BKI mice to investigate the importance of activity-driven CaMKII binding to the GluN2B subunit of the NMDAR in several models of acute pain as well as the capsaicin and formalin models of inflammatory pain. The GluN2B subunit contains two binding sites for CaMKII (S1303 and a more proximal region between residues 839 and 1140)(Leonard et al., 1999). The point mutations in GluN2BKI mice abrogate activity-driven binding of CaMKII to S1303, but do not alter the amounts of GluN2B or the other NMDAR subunits, as well as CaMKII and other synaptic proteins in forebrain (Halt et al., 2012). In GluN2BKI mice, NMDA does not increase CaMKII binding to GluN2B nor does it increase clustering of CaMKII at postsynaptic sites. In fact, CaMKII binding to GluN2B under activated conditions in GluN2BKI mice is less than half that of basal conditions in WT mice (Halt et al., 2012). The loss of an activity-driven interaction of CaMKII and NMDAR results in a 50% reduction of hippocampal LTP. GluN2BKI mice exhibit mild spatial learning deficits, as well as impaired recall following learning Morris water maze specific tasks (Halt et al., 2012; Stein et al., 2014) although they do not exhibit deficits in learning associated with Barnes maze and contextual fear conditioning (Stein et al., 2014). Finally, activity-driven spine outgrowth, which requires binding of CaMKII to GluN2B, is also blocked in these mice (Hamilton et al., 2012). Thus, GluN2BKI mice were used to understand the requirement for activity‐driven binding of CaMKII to GluN2B-containing NMDAR in models of acute and persistent inflammatory nociception.
3.1. Acute Nociception
Impairing the activity-driven binding of CaMKII binding to GluN2B was of no consequence in the tail flick, hot plate, and paw flick tests, capsaicin-induced licking/biting behaviors, or the first phase of the formalin test. This was an expected result. First, at the molecular level, GluN2BKI mice have intact basal neurotransmission at least in the hippocampal CA1 area (Halt et al., 2012). Second, antagonism or knock-down of NMDAR does not affect responsiveness to acute noxious thermal, mechanical or electrical stimuli (McRoberts et al., 2011; Petrenko et al., 2006) Third, these pain models are short-lived in nature, predominantly dependent on the activation of nociceptors, and do not entail either peripheral or central sensitization (Le Bars et al., 2001).
3.2. Persistent Nociception
We expected that the binding of CaMKII to GluN2B would be critical in models of persistent inflammatory pain given the well-chronicled role of NMDAR coupled with strong evidence for the involvement of CaMKII in both the capsaicin and formalin models. With respect to the role of NMDAR, administration of NMDAR antagonists such as APV, CPP, MK801 and memantine attenuates capsaicin-induced pain behaviors as well as mechanical and heat hypersensitivity (Sakurada et al., 1998; Soliman et al., 2005; Taniguchi et al., 1997). Pharmacologic inhibition of NMDARs in either the spinal cord or hippocampus, or selective knockdown in primary afferent neurons decreases the magnitude of the second phase of formalin-induced pain behaviors (Chaplan et al., 1997; Haley and Dickenson, 2016; McKenna and Melzack, 2001; McRoberts et al., 2011; Nazarian et al., 2008). Moreover, spontaneous behaviors induced by capsaicin (Sakurada et al., 1998; Taniguchi et al., 1997) and the second phase of formalin-induced pain behaviors are diminished by specific GluN2B subunit inhibitors, ifenprodil and CP101–606 (Borza et al., 2007). Knock-down of GluN2B using siRNA, or preventing GluN2B association with postsynaptic density protein 95, are able to block pain behaviors associated with the second phase of formalin (D’Mello et al., 2011; Zhang et al., 2013) and attenuate hyperalgesia in diabetic rats (Zhu et al., 2020). Evidence for the role of CaMKII includes findings that the T286A mutation that blocks autophosphorylation of CaMKII attenuates the second phase of the formalin test (Choi et al., 2006; Dai et al., 2005; Zeitz et al., 2004). Finally, capsaicin increases phosphorylation of CaMKII in the dorsal horn, and increases CaMKII-dependent phosphorylation of the ionotropic AMPA receptor subunit, GluA1, and its translocation from the cytoplasm to the cell membrane in vivo and in vitro (Fang et al., 2002; Galan et al., 2004; Price et al., 2005). Additionally, CaMKII phosphorylation of TPRV1, the capsaicin receptor, is required for TRPV1 activation (Jung et al., 2004).
Given the importance of CaMKII as a downstream effector following NMDAR activation in both models and the demonstrated deficits in LTP in GluN2BKI mice (Halt et al., 2012), we hypothesized that capsaicin and formalin-induced nociception would be reduced in GluN2BKI mice compared to litter-matched WT and HET mice. Instead, apart from a delay in onset of capsaicin-induced licking and biting, GluN2BKI mice did not differ from either WT or HET mice with respect to duration of spontaneous pain behaviors or heat and mechanical hypersensitivity. Similarly, GluN2BKI mice did not differ from WT mice in their response to formalin. Collectively, these data suggest that activity-driven binding of CaMKII at S1303 of the GluN2B subunit of NMDAR is not necessary for inflammatory nociception as modeled by spontaneous behaviors or the mechanical and thermal hypersensitivity produced by capsaicin, or second phase of the formalin test.
3.3. Neuronal Pathfinding
The misdirection of licking and biting behavior to the perianal region, and the accompanying delay in onset to licking the hind paw observed in the GluN2BKI mice is intriguing. The NMDAR is involved in developmental neuronal pathfinding (McKinney et al., 1999), and knock-down of the GluN2B subunit in the ventricular zone impairs accurate migration of cortical neurons (Jiang et al., 2015), indicating a role for NMDARs in guiding the developing pre-synaptic neuron (Fedder and Sabo, 2015). The misdirection observed in the capsaicin test was not observed in the formalin test. Whether abrogation of CaMKII binding to GluN2B results in a developmental defect in TRPV1 expressing sensory afferents or the cortical representation of sensory information carried by these afferents remains to be determined.
3.4. Revisiting conclusions from administration of KN93
KN93 has been used as a CaMKII inhibitor for nearly two decades, despite an inability to demonstrate its binding to CaMKII. Critically, a role of CaMKII in inflammatory nociception has been inferred from the ability of KN93 to attenuate those behaviors. At the time these experiments were conducted, it was expected that KN93 would be unable to diminish formalin-evoked behaviors in GluN2BKI mice because the interaction with CaMKII was already abrogated. The finding that it did so was unexpected. Very recently, new approaches have revealed that KN93 actually binds to Ca2+/CaM (Wong et al., 2019). Ca2+/CaM interacts with multiple other targets including Ca2+-, Na+- and K+-channels, as well as various kinases that can also be disrupted by KN93. Therefore, although findings that KN93 blocks capsaicin- and formalin-evoked pain behaviors are an intriguing correlation, they do not demonstrate causation by CaMKII activity. The inhibition of formalin-induced pain behaviors by KN93 in GluN2BKI mice implicates other targets and mechanisms that merit investigation.
3.5. Limitations and Possible Mechanisms
This study specifically investigated and excluded a role for activity-driven CaMKII phosphorylation of S1303 in GluN2B in acute and persistent inflammatory pain. These negative findings contrast with a wealth of data supporting the important role of CAMKII binding to GluN2B in neuropathic pain (Zhou et al., 2017), some of which is based on the use of KN-93. Despite the limitations of this study, including the small number of mice and their reuse, the negative findings do not fully exclude a role of CaMKII in inflammatory pain. Several possibilities can be entertained. First, CaMKII can bind to a second site (a.a. 839–1120) on GluN2B. Unfortunately, this region has not been extensively studied, most likely because the region containing S1303 is essential for activity-driven CaMKII binding to GluN2B. Second, CaMKII binding to other postsynaptic proteins such as GluN1 and GluN2A (Gardoni et al., 1998; Gardoni et al., 2001; Leonard et al., 1999) may be sufficient to mediate hypersensitivity following inflammatory injury although we note that CaMKII binds to GluN2B with the highest affinity and efficiency (She et al., 2012). Third, these findings do not rule out a role for presynaptic CaMKII activity, independent of its binding to GluN2B, where it is necessary for synaptic plasticity (Ninan and Arancio, 2004), possibly by regulating neurotransmitter release (Liu et al., 2007; Luo et al., 2008). Fourth, in addition to its role as a kinase, CaMKII also has a kinase-independent structural role at NMDAR that does not require its binding to the NMDAR to maintain basal NMDAR function (Incontro et al., 2018). This report raises the intriguing possibility that sensitization and persistent nociception in nociceptive circuits depend on the kinase-independent, structural function of the CaMKII protein. Finally, it is also possible that phosphorylation of NMDAR initiated by PSD95 binding may play an important role (Zhu et al., 2020).
3.6. Summary
The GluN2BKI mice used in this study provide genetic specificity in assessing the role of CaMKII binding to S1303 of GluN2B in NMDAR-mediated inflammatory pain signaling cascades. It was surprising that loss of this interaction did not decrease formalin-evoked pain behaviors or capsaicin-induced hypersensitivity, suggesting that activity-driven binding of CaMKII to GluN2B is not a crucial component of peripheral or central sensitization, or of inflammatory nociception. Of note, binding of CaMKII to GluN2B requires phosphorylation of T1472 by the src family tyrosine kinase Fyn (Nakazawa et al., 2006). It is perhaps not unexpected then that Fyn-mediated phosphorylation of T1472 in GluN2B also does not contribute to inflammatory pain (Matsumura et al., 2010). In contrast, Fyn-mediated phosphorylation of T1472 in GluN2B plays a critical role in neuropathic pain (Hildebrand et al., 2016; Matsumura et al., 2010). Whether activity-driven CaMKII binding to GluN2B at S1303 contributes to neuropathic pain remains to be determined. To this end, it would be interesting to further test the role of activity-drive binding of CaMKII in inflammatory and neuropathic pain using mice with a K42M or K42R mutation in CaMKII, in which glutamate-induced synaptic translocation of CaMKII and binding to GluN2B are significantly reduced (Tullis et al., 2020).
The alternatives and limitations notwithstanding, the negative results obtained with this pharmacogenetic approach and new insights into the mechanism of action of KN93 challenge current thinking on the involvement of the GluN2B-NMDAR-CaMKII interaction in persistent inflammatory nociception. These results further suggest that drugs targeting this interaction may not be effective for treating inflammatory pain conditions.
4. Materials and Methods
4.1. Animals
These experiments were approved by the University of Iowa Animal Care and Use Committee and were conducted in accordance with the guidelines of the International Association for the Study of Pain and the National Resource Council Guide for the Care and Use of Laboratory Animals. All mice used were bred and housed at the University of Iowa Animal Care Facility in temperature- and humidity-controlled rooms with 12 hour light and dark cycles. Mice had free access to food and water. GluN2BKI, WT and HET mice were obtained as detailed by Halt et al. Briefly, GluN2BKI+/− (heterozygous) mice were bred to obtain litters containing homozygous KI (GluN2BKI), WT and HET mice. Mouse genotypes were determined through diagnostic PCR followed by restriction enzyme digest using BssHII, which cleaves the PCR product in GluN2BKI but not WT mice. All three genotypes were of the C57BL/6 background and were physically indistinguishable. In all experiments, control WT and HET mice were littermates of the GluN2BKI mice. Adult mice (~8 weeks of age) of both genders were used. As the numbers of male and female mice were insufficient to enable meaningful analysis of gender differences, the genders were pooled for analysis.
4.2. General Experimental Design
The first set of experiments evaluated responsiveness to different intensities of acute heat using the tail flick and hot plate tests; these tests were conducted using the same mice. Using a different cohort of mice, the second set of experiments examined whether the genotypes differed in the magnitude of capsaicin-induced licking and biting of the hind paw or in capsaicin-induced heat or tactile hypersensitivity. Due to limited availability of mice, capsaicin-induced hypersensitivity and licking and biting behaviors were assessed in the same mice using alternate hind paws. After assessment of baseline heat threshold, using the paw flick test, the left hind paw was injected with capsaicin. The duration of time spent licking and biting the hind paw was recorded for 15 min followed by reassessment of paw flick latency. On week later, after assessment of baseline mechanical threshold using von Frey filaments, the right hind paw was injected with capsaicin. The duration of time spent licking and biting the right hind paw was recorded for 15 min followed by reassessment of mechanical threshold. Using a third cohort of mice, the final set of experiments examined whether the genotypes differed in the magnitude of formalin-induced pain behaviors and whether these behaviors were differentially sensitive to antagonism by the NMDAR antagonist, MK-801, or the Ca2+/CaM inhibitor, KN93.
4.3. Measurements of Nociception
All mice were acclimated to the test environment and equipment for three days prior to each experiment. The experimenter was blind to the genotype of the mice. Hot plate latency was defined as the time elapsed to a lick of the hind paw or a display of escape behavior after the mouse was placed on a copper surface heated to 48, 52 or 55°C. One measure of hot plate response latency was made at each temperature for each mouse. Cut-off latencies of 60, 50 and 40 sec (respective of set of increasing temperatures) were imposed to prevent tissue damage.
For the tail flick assay, the tail of each mouse was blackened with a black permanent marker to control for potential differences in pigmentation that may alter heat absorbance. A high intensity light beam (IITC Tail Flick Apparatus Analgesia Meter; Woodland Hills, CA) was directed at the blackened portion dorsal surface of the mouse’s tail. Tail flick latency was defined as the time elapsed to remove the tail from the light beam as measured by an automatic timer. Two measures of tail flick latency were made and averaged to yield a single value at each intensity for each mouse. Cut-off latencies of 20 and 14 sec were used at the low and high intensity to prevent tissue damage.
Paw flick latency was measured as previously reported by our group (Hurley and Hammond, 2000). Measurements were obtained by focusing a high intensity light beam on the hind paw of the mouse. An automatic timer recorded when the mouse removed its paw from the path of the light beam. A cut-off latency of 20 sec was imposed to prevent tissue damage. Mice that didn’t respond within 20 seconds were assigned that value. One measure was made on each hind paw per animal.
Mechanical threshold was determined using von Frey filaments (Smith and Nephew Roylan Inc.) ranging in force from 1.65 to 4.31 log milli-Newtons (mN). Each filament was applied five times to each hind paw, starting with the finest filament until a force was reached at which the mouse responded to each of the five filament applications by raising its paw (100% response).
4.4. Models of Inflammatory Pain
For the capsaicin model of inflammatory pain, the dorsal aspect of the hind paw was injected with 10 μL capsaicin (0.1% in saline, Sigma). The time spent exhibiting spontaneous behaviors (licking and biting of the injected paw) was recorded over a 15 min period. Twenty min after capsaicin injection, when spontaneous behaviors had subsided, thresholds for heat or mechanical stimuli were reassessed on both the ipsilateral and contralateral hind paws.
The formalin test was conducted by injecting 20 μl of 2% formalin in the plantar surface of one hind paw. Time spend exhibiting spontaneous behaviors (licking and biting of the injected paw) was measured in five min intervals over the course of one hour.
4.5. Drugs and Administration
MK801 and KN93 were obtained from Sigma-Aldrich (St. Louis, MO). MK801 was dissolved in saline, which served as its vehicle, and was injected subcutaneously in the scruff of the neck 20 min before formalin. KN93 was first dissolved in DMSO and then diluted with saline. The final concentration of DMSO was 25% (v/v), and this served as the vehicle for intrathecal injections. Mice were lightly anesthetized with isoflurane before intrathecal injection of vehicle or KN93 in a volume of 5 μL as previously described (Hylden and Wilcox, 1980).
4.6. Data Analysis
All data are presented as mean ± S.E.M. Tail flick and hot plate latencies were compared among the genotypes by a two-way repeated measures analysis of variance (ANOVA) in which genotype was one factor and stimulus intensity was the repeated measure. For analysis of capsaicin-induced heat hyperalgesia, paw flick latencies were compared among the genotypes by using a two-way repeated measures ANOVA in which genotype was one factor and time was the repeated measure. For comparison of of capsaicin-evoked pain behaviors between hind paws among the genotypes, a two-way repeated measures ANOVA in which genotype was one factor and hind paw injected was the repeated factor. Time to onset of licking after capsaicin was compared among the genotypes by one-way ANOVA followed by Holm-Sidak post hoc test. Responses to von Frey filaments were fit by non-linear regression, and EF50 and 95% confidence limits determined using GraphPad Prism (San Diego, CA). The extra sum of squares test in GraphPad Prism was then used to compare slopes and EF50 of the stimulus-response functions for the different genotypes before and after capsaicin. Duration of licking after formalin was compared by a two-way repeated measures ANOVA with genotype as one factor and time as the repeated factor. Mean duration of licking during the second phase of the formalin test was compared by Student’s t-test. For all comparisons, a P < 0.05 was considered significant.
Highlights.
Acute and inflammatory pain were tested in GluN2BKI mice impaired in CaMKII binding
Loss of activity-driven CaMKII binding to GluN2B did not diminish acute pain
Loss of activity-driven CaMKII binding to GluN2B did not obtund inflammatory pain
KN93 inhibited formalin pain in GluN2BKI mice implicating other targets of Ca2+/CaM
CaMKII binding to S1303 in GluN2B is not critical for inflammatory pain signaling
Acknowledgements
We thank Dr. Robert Dallapiazza for stimulating discussions.
Funding
This publication was made possible by Grant Number UL1RR024979 from the National Center for Research Resources (NCRR), a part of the National Institutes of Health (NIH). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the CTSA or NIH. Work on the GluN2B KI mouse was funded by NIH R01 NS046450 (JWH).
Abbreviations
- ANOVA
analysis of variance
- Ca2+
calcium
- CaM
calmodulin
- CaMKII
Ca2+/CaM kinase II
- CAP
capsaicin
- DMSO
dimethyl sulfoxide
- EF50
50% of the effective force
- GluN1, 2A, 2B
NMDAR subunits
- HET
heterozygote
- K+
potassium
- KI
knock in
- KN93
N-[2-[[[3-(4-Chlorophenyl)-2-propenyl]methylamino]methyl]phenyl]-N-(2hydroxyethyl)-4-methoxybenzenesulphonamide
- MK801
(5R,10S)-(−)-5-Methyl-10,11-dihydro-5H-dibenzo[a,d]cylcohepten-5,10-imine maleate
- mN
milli-Newtons
- Na+
sodium
- NMDAR
N-Methyl D-aspartate receptor
- PCR
polymerase chain reaction
- S.E.M.
standard error of the mean
- s.c.
subcutaneous
- siRNA
small interfering RNA
- WT
wild type
Footnotes
Declaration of interests
The authors declare that they do not have any competing interests
Data Availability Statement
Data can be obtained by request to the corresponding author.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barcomb K, Goodell DJ, Arnold DB, Bayer KU, 2015. Live imaging of endogenous Ca2+/calmodulin-dependent protein kinase II in neurons reveals that ischemia-related aggregation does not require kinase activity. J. Neurochem. 135, 666–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H, 2001. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature. 411, 801–5. [DOI] [PubMed] [Google Scholar]
- Borza I, Kolok S, Galgoczy K, Gere A, Horvath C, Farkas S, Greiner I, Domany G, 2007. Kynurenic acid amides as novel NR2B selective NMDA receptor antagonists. Bioorg Med Chem Lett. 17, 406–9. [DOI] [PubMed] [Google Scholar]
- Bourinet E, Altier C, Hildebrand ME, Trang T, Salter MW, Zamponi GW, 2014. Calcium-permeable ion channels in pain signaling. Physiol Rev. 94, 81–140. [DOI] [PubMed] [Google Scholar]
- Boyce S, Wyatt A, Webb JK, O’Donnell R, Mason G, Rigby M, Sirinathsinghji D, Hill RG, Rupniak NM, 1999. Selective NMDA NR2B antagonists induce antinociception without motor dysfunction: correlation with restricted localisation of NR2B subunit in dorsal horn. Neuropharmacology. 38, 611–23. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Malmberg AB, Yaksh TL, 1997. Efficacy of spinal NMDA receptor antagonism in formalin hyperalgesia and nerve injury evoked allodynia in the rat. J Pharmacol Exp Ther. 280, 829–38. [PubMed] [Google Scholar]
- Chen Y, Luo F, Yang C, Kirkmire CM, Wang ZJ, 2009. Acute inhibition of Ca2+/calmodulin-dependent protein kinase II reverses experimental neuropathic pain in mice. J Pharmacol Exp Ther. 330, 650–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SS, Seo YJ, Shim EJ, Kwon MS, Lee JY, Ham YO, Suh HW, 2006. Involvement of phosphorylated Ca2+/calmodulin-dependent protein kinase II and phosphorylated extracellular signal-regulated protein in the mouse formalin pain model. Brain Res. 1108, 28–38. [DOI] [PubMed] [Google Scholar]
- D’Mello R, Marchand F, Pezet S, McMahon SB, Dickenson AH, 2011. Perturbing PSD-95 interactions with NR2B-subtype receptors attenuates spinal nociceptive plasticity and neuropathic pain. Mol Ther. 19, 1780–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Wang H, Ogawa A, Yamanaka H, Obata K, Tokunaga A, Noguchi K, 2005. Ca2+/calmodulin-dependent protein kinase II in the spinal cord contributes to neuropathic pain in a rat model of mononeuropathy. Eur J Neurosci. 21, 246774. [DOI] [PubMed] [Google Scholar]
- Fang L, Wu J, Lin Q, Willis WD, 2002. Calcium-calmodulin-dependent protein kinase II contributes to spinal cord central sensitization. J Neurosci. 22, 4196–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedder KN, Sabo SL, 2015. On the role of glutamate in presynaptic development: Possible contributions of presynaptic NMDA receptors. Biomolecules. 5, 3448–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher K, Coderre TJ, Hagen NA, 2000. Targeting the N-methyl-D-aspartate receptor for chronic pain management. Preclinical animal studies, recent clinical experience and future research directions. J Pain Sympton Manag. 20, 358–73. [DOI] [PubMed] [Google Scholar]
- Galan A, Laird JM, Cervero F, 2004. In vivo recruitment by painful stimuli of AMPA receptor subunits to the plasma membrane of spinal cord neurons. Pain. 112, 315–23. [DOI] [PubMed] [Google Scholar]
- Gardoni F, Caputi A, Cimino M, Pastorino L, Cattabeni F, Di Luca M, 1998. Calcium/calmodulin-dependent protein kinase II is associated with NR2A/B subunits of NMDA receptor in postsynaptic densities. J Neurochem. 71, 1733–41. [DOI] [PubMed] [Google Scholar]
- Gardoni F, Schrama LH, Kamal A, Gispen WH, Cattabeni F, Di Luca M, 2001. Hippocampal synaptic plasticity involves competition between Ca2+/calmodulin-dependent protein kinase II and postsynaptic density 95 for binding to the NR2A subunit of the NMDA receptor. J Neurosci. 21, 1501–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazewski S, Giese KP, Silva A, Fox K, 2000. The role of α-CaMKII autophosphorylation in neocortical experience-dependent plasticity. Nat Neurosci. 3, 911–918. [DOI] [PubMed] [Google Scholar]
- Haley JE, Dickenson AH, 2016. Evidence for spinal N-methyl-d-aspartate receptor involvement in prolonged chemical nociception in the rat. Brain Res. 1645, 58–60. [DOI] [PubMed] [Google Scholar]
- Halt AR, Dallapiazza RF, Zhou Y, Stein IS, Qian H, Juntti S, Wojcik S, Brose N, Silva AJ, Hell JW, 2012. CaMKII binding to GluN2B is critical during memory consolidation. EMBO J. 31, 1203–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton AM, Oh WC, Vega-Ramirez H, Stein IS, Hell JW, Patrick GN, Zito K, 2012. Activity-dependent growth of new dendritic spines is regulated by the proteasome. Neuron. 74, 1023–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt DJ, 2000. The use of NMDA-receptor antagonists in the treatment of chronic pain. Clin J Pain. 16, S73–S79. [DOI] [PubMed] [Google Scholar]
- Hildebrand ME, Pitcher GM, Harding EK, Li H, Beggs S, Salter MW, 2014. GluN2B and GluN2D NMDARs dominate synaptic responses in the adult spinal cord. Sci Rep. 4, 4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand ME, Xu J, Dedek A, Li Y, Sengar AS, Beggs S, Lombroso PJ, Salter MW, 2016. Potentiation of Synaptic GluN2B NMDAR Currents by Fyn Kinase Is Gated through BDNF-Mediated Disinhibition in Spinal Pain Processing. Cell Rep. 17, 2753–2765. [DOI] [PubMed] [Google Scholar]
- Hurley RW, Hammond DL, 2000. The analgesic effects of supraspinal mu and delta opioid receptor agonists are potentiated during persistent inflammation. J Neurosci. 20, 1249–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hylden JL, Wilcox GL, 1980. Intrathecal morphine in mice: a new technique. Eur J Pharmacol. 67, 313–6. [DOI] [PubMed] [Google Scholar]
- Incontro S, Díaz-Alonso J, Iafrati J, Vieira M, Asensio CS, Sohal VS, Roche KW, Bender KJ, Nicoll RA, 2018. The CaMKII/NMDA receptor complex controls hippocampal synaptic transmission by kinase-dependent and independent mechanisms. Nat Comm. 9, 2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Kohno T, Moore KA, Woolf CJ, 2003. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci. 26, 696–705. [DOI] [PubMed] [Google Scholar]
- Jiang H, Jiang W, Zou J, Wang B, Yu M, Pan Y, Lin Y, Mao Y, Wang Y, 2015. The GluN2B subunit of N-methyl-D-asparate receptor regulates the radial migration of cortical neurons in vivo. Brain Res. 1610, 20–32. [DOI] [PubMed] [Google Scholar]
- Jung J, Shin JS, Lee SY, Hwang SW, Koo J, Cho H, Oh U, 2004. Phosphorylation of vanilloid receptor 1 by Ca2+/calmodulin-dependent kinase II regulates its vanilloid binding. J Biol Chem. 279, 7048–54. [DOI] [PubMed] [Google Scholar]
- Kreutzwiser D, Tawfic QA, 2019. Expanding role of NMDA receptor antagonists in the management of pain. CNS Drugs. 33, 347–374. [DOI] [PubMed] [Google Scholar]
- Le Bars D, Gozariu M, Cadden SW, 2001. Animal models of nociception. Pharmacol Rev. 53, 597–652. [PubMed] [Google Scholar]
- Leonard AS, Lim IA, Hemsworth DE, Horne MC, Hell JW, 1999. Calcium/calmodulin-dependent protein kinase II is associated with the N-methyl-D-aspartate receptor. Proc Natl Acad Sci U S A. 96, 3239–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Chen B, Ge Q, Wang ZW, 2007. Presynaptic Ca2+/calmodulin-dependent protein kinase II modulates neurotransmitter release by activating BK channels at Caenorhabditis elegans neuromuscular junction. J Neurosci. 27, 10404–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Liang Y, Hou B, Liu M, Yang X, Liu C, Zhang J, Zhang W, Ma Z, Gu X, 2014. The inhibitor of calcium/calmodulin-dependent protein kinase II KN93 attenuates bone cancer pain via inhibition of KIF17/NR2B trafficking in mice. Pharmacol Biochem Behav. 124, 19–26. [DOI] [PubMed] [Google Scholar]
- Luo F, Yang C, Chen Y, Shukla P, Tang L, Wang LX, Wang ZJ, 2008. Reversal of chronic inflammatory pain by acute inhibition of Ca2+calmodulin-dependent protein kinase II. J Pharmacol Exp Ther. 325, 267–275. [DOI] [PubMed] [Google Scholar]
- MacDermott AB, 2014. Synaptic GluN2A and GluN2B containing NMDA receptors within the superficial dorsal horn activated following primary afferent stimulation. J Neurosci. 34, 10808–10820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura S, Kunori S, Mabuchi T, Katano T, Nakazawa T, Abe T, Watanabe M, Yamamoto T, Okuda-Ashitaka E, Ito S, 2010. Impairment of CaMKII activation and attenuation of neuropathic pain in mice lacking NR2B phosphorylated at Tyr1472. Eur J Neurosci. 32, 798–810. [DOI] [PubMed] [Google Scholar]
- McKenna JE, Melzack R, 2001. Blocking NMDA receptors in the hippocampal dentate gyrus with AP5 produces analgesia in the formalin pain test. Exp Neurol. 172, 92–9. [DOI] [PubMed] [Google Scholar]
- McKinney RA, Lüthi A, Bandtlow CE, Gähwiler BH, Thompson SM, 1999. Selective glutamate receptor antagonists can induce or prevent axonal sprouting in rat hippocampal slice cultures. Proc Nat Acad Sci USA. 96, 11631–11636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRoberts JA, Ennes HS, Marvizon JC, Fanselow MS, Mayer EA, Vissel B, 2011. Selective knockdown of NMDA receptors in primary afferent neurons decreases pain during phase 2 of the formalin test. Neuroscience. 172, 474–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momiyama A, 2000. Distinct synaptic and extrasynaptic NMDA receptors identified in dorsal horn neurones of the adult rat spinal cord. The Journal of Physiology. 523, 621–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa T, Komai S, Watabe AM, Kiyama Y, Fukaya M, Arima-Yoshida F, Horai R, Sudo K, Ebine K, Delawary M, Goto J, Umemori H, Tezuka T, Iwakura Y, Watanabe M, Yamamoto T, Manabe T, 2006. NR2B tyrosine phosphorylation modulates fear learning as well as amygdaloid synaptic plasticity. EMBO J. 25, 2867–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarian A, Gu G, Gracias NG, Wilkinson K, Hua XY, Vasko MR, Yaksh TL, 2008. Spinal N-methyl-D-aspartate receptors and nociception-evoked release of primary afferent substance P. Neuroscience. 152, 119–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninan I, Arancio O, 2004. Presynaptic CaMKII is necessary for synaptic plasticity in cultured hippocampal neurons. Neuron. 42, 129–41. [DOI] [PubMed] [Google Scholar]
- Parsons CG, Quack G, Bresink I, Baran L, Przegalinski E, Kostowski W, Krzascik P, Hartmann S, Danysz W, 1995. Comparison of the potency, kinetics and voltage-dependency of a series of uncompetitive NMDA receptor antagonists in vitro with anticonvulsive and motor impairment activity in vivo. Neuropharmacology. 34, 1239–58. [DOI] [PubMed] [Google Scholar]
- Pellicena P, Schulman H, 2014. CaMKII inhibitors: from research tools to therapeutic agents. Front Pharmacol. 5, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrenko AB, Yamakura T, Baba H, Shimoji K, 2003. The role of N-methyl-D-aspartate (NMDA) receptors in pain: A review. Anesth Analg. 97, 1108–16. [DOI] [PubMed] [Google Scholar]
- Petrenko AB, Yamakura T, Askalany AR, Kohno T, Sakimura K, Baba H, 2006. Effects of ketamine on acute somatic nociception in wild-type and N-methyl-D-aspartate (NMDA) receptor epsilon1 subunit knockout mice. Neuropharmacology. 50, 741–7. [DOI] [PubMed] [Google Scholar]
- Price GW, Ahier RG, Middlemiss DN, Singh L, Tricklebank MD, Wong EH, 1988. In vivo labelling of the NMDA receptor channel complex by [3H]MK-801. Eur J Pharmacol. 158, 279–82. [DOI] [PubMed] [Google Scholar]
- Price TJ, Jeske NA, Flores CM, Hargreaves KM, 2005. Pharmacological interactions between calcium/calmodulin-dependent kinase II alpha and TRPV1 receptors in rat trigeminal sensory neurons. Neurosci Lett. 389, 94–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurada T, Wako K, Sugiyama A, Sakurada C, Tan-No K, Kisara K, 1998. Involvement of spinal NMDA receptors in capsaicin-induced nociception. Pharmacol Biochem Behav. 59, 339–45. [DOI] [PubMed] [Google Scholar]
- Schroeder C, Schroeder K, 2019. NMDA Receptor Antagonists In: Pain. Abd-Elsayed A, ed. Springer International Publishing, Cham, pp. 285–287. [Google Scholar]
- She K, Rose JK, Craig AM, 2012. Differential stimulus-dependent synaptic recruitment of CaMKIIα by intracellular determinants of GluN2B. Mol Cell Neurosci. 51, 68–78. [DOI] [PubMed] [Google Scholar]
- Shirahama M, Ushio S, Egashira N, Yamamoto S, Sada H, Masuguchi K, Kawashiri T, Oishi R, 2012. Inhibition of Ca2+/calmodulin-dependent protein kinase II reverses oxaliplatin-induced mechanical allodynia in rats. Mol Pain. 8, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soliman AC, Yu JS, Coderre TJ, 2005. mGlu and NMDA receptor contributions to capsaicin-induced thermal and mechanical hypersensitivity. Neuropharmacology. 48, 325–32. [DOI] [PubMed] [Google Scholar]
- Stein IS, Donaldson MS, Hell JW, 2014. CaMKII binding to GluN2B is important for massed spatial learning in the Morris water maze. F1000Res. 3, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack S, Colbran RJ, 1998. Autophosphorylation-dependent targeting of calcium/ calmodulin-dependent protein kinase II by the NR2B subunit of the N-methyl-D-aspartate receptor. J Biol Chem. 273, 20689–92. [DOI] [PubMed] [Google Scholar]
- Taniguchi K, Shinjo K, Mizutani M, Shimada K, Ishikawa T, Menniti FS, Nagahisa A, 1997. Antinociceptive activity of CP-101,606, an NMDA receptor NR2B subunit antagonist. Br J Pharmacol. 122, 809–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tullis JE, Rumian NL, Brown CN, Bayer KU, 2020. The CaMKII K42M and K42R mutations are equivalent in suppressing kinase activity and targeting. PLoS One. 15, e0236478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vest RS, O’Leary H, Coultrap SJ, Kindy MS, Bayer KU, 2010. Effective post-insult neuroprotection by a novel Ca2+/ Calmodulin-dependent protein kinase II (CaMKII) inhibitor. J Biol Chem. 285, 20675–20682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong MH, Samal AB, Lee M, Vlach J, Novikov N, Niedziela-Majka A, Feng JY, Koltun DO, Brendza KM, Kwon HJ, Schultz BE, Sakowicz R, Saad JS, Papalia GA, 2019. The KN-93 molecule inhibits calcium/calmodulin-dependent protein kinase II (CaMKII) activity by binding to Ca2+/CaM. J Mol Biol. 431, 1440–1459. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, 2011. Central sensitization: implications for the diagnosis and treatment of pain. Pain. 152, S2–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitz KP, Giese KP, Silva AJ, Basbaum AI, 2004. The contribution of autophosphorylated alpha-calcium-calmodulin kinase II to injury-induced persistent pain. Neuroscience. 128, 889–98. [DOI] [PubMed] [Google Scholar]
- Zhang RX, Yan XB, Gu YH, Huang D, Gan L, Han R, Huang LH, 2013. Gene silencing of NR2B-containing NMDA receptor by intrathecal injection of short hairpin RNA reduces formalin-induced nociception in C57BL/6 mouse. Int J Neurosci. 123, 650–6. [DOI] [PubMed] [Google Scholar]
- Zhou YQ, Liu DQ, Chen SP, Sun J, Zhou XR, Luo F, Tian YK, Ye DW, 2017. Cellular and molecular mechanisms of calcium/calmodulin-dependent protein kinase II in chronic pain. J Pharmacol Exp Ther. 363, 176–183. [DOI] [PubMed] [Google Scholar]
- Zhu YB, Jia GL, Wang JW, Ye XY, Lu JH, Chen JL, Zhang MB, Xie CS, Shen YJ, Tao YX, Li J, Cao H, 2020. Activation of CaMKII and GluR1 by the PSD-95-GluN2B coupling-dependent phosphorylation of GluN2B in the spinal cord in a rat model of Type-2 diabetic neuropathic pain. J Neuropathol Exp Neurol. 79, 800–808. [DOI] [PubMed] [Google Scholar]