

Abstract
Pyruvate kinase M2 (PKM2) is not only a key rate-limiting enzyme that guides glycolysis, but also acts as a non-metabolic protein in regulating gene transcription. In recent years, a series of studies have confirmed that post-translational modification has become an important mechanism for regulating the function of PKM2, which in turn affects tumorigenesis. In this study, we found that K62 residues were deacetylated, which is related to the prognosis of HCC. Further studies indicate that HDAC8 binds and deacetylates the K62 residue of PKM2. Mechanistically, K62 deacetylation facilitate PKM2 transport into the nucleus and bind β-catenin, thereby promoting CCND1 gene transcription and cell cycle progression. In addition, the deacetylation of K62 affects the enzyme activity of PKM2 and the flux of glucose metabolism. Therefore, these results suggest that HDAC8 / PKM2 signaling may become a new target for the treatment of HCC.
Subject terms: Cancer metabolism, Gastrointestinal cancer
Introduction
Liver cancer is predicted to be the sixth most commonly diagnosed cancer and the fourth leading cause of cancer death worldwide in 2018, and its ranks fifth in terms of global cases and second in terms of deaths for males, and hepatocellular carcinoma (HCC) is the most prevalent form one1. New pathophysiology directed therapies are urgently needed. Therefore, with extensive studies, an increasing number of molecular mechanisms of pathogenesis have been discovered2–4.
PKM2 is overexpressed in HCC, and participates in hepatocarcinogenesis and progression through many critical signaling networks, including PRMT6-ERK-PKM2 regulatory axis, circMAT2B/miR-338-3p/PKM2 axis and others5–7. It not only acts as a key enzyme in anaerobic glycolysis in the cytoplasm, but also play a major role in functions as a histone kinase in the nucleus8–10. Further studies have shown these alterations are regulated by various PTMs, including phosphorylation, acetylation, and so on11–15. Aberrant acetylation levels of PKM2 has been linked to the development of several diseases. For example, acetylation of K433 of PKM2 can bind to c-Src-phosphorylated Y333 of β-catenin. Moreover, this interaction is required for both proteins to be recruited to the CCND1 promoter for promoting GBM progression8. The mentioned above site and K305 residues of PKM2 also could be deacetylated by SIRT2 and SIRT 6 respectively, both of kinase and enzyme functions were weakened16,17. Here we try to find new functional site and further characterize PKM2 function in HCC.
Histone deacetylase 8 (HDAC8), one of the crucial HDACs, plays an important role in occurrence and progression of various diseases, including cancer, hereditary disease, and parasitic infections via different signaling pathways18,19. It can drive NAFLD-associated hepatocarcinogenesis through inhibiting p53/p21-mediated apoptosis and cell-cycle arrest and stimulating β-catenin-dependent cell proliferation3. While up to half of HCC patients have activation of Wnt/β-catenin signaling pathway8,20. This signaling pathway plays a significant role in the physiology and pathology of liver and has been a prevalent therapy target21.
Both of PKM2 and HDAC8 proteins are overexpressed in tumor tissues. They can impact cell cycle by influencing β-catenin in hepatocellular carcinoma (HCC). Previous study revealed PKM2 binds with several proteins that regulate glucose and lipid metabolism then promote cell proliferation in HCC22. In this study, we found that HDAC8 directly interacts with PKM2 and deacetylates the conserved K62 residue in the cytoplasm, thereby regulating glucose metabolism and gene transcription. Clinically, the acetylation level of PKM2-K62 residues is significantly correlated with the prognosis of HCC. This study reveals a novel regulatory mechanism of PKM2 protein and suggests its potential significance in HCC treatment.
Materials and methods
Patient tissue specimens
HCC specimens and adjacent tissues obtained from the surgical specimen archives of Renji Hospital, School of Medicine, Shanghai Jiaotong University. This research was approved by the institutional clinical research ethics committee of Renji Hospital, School of Medicine, Shanghai Jiaotong University. None of these patients had received radiotherapy or chemotherapy prior to surgery. And, before surgery, all of them received 18F-FDG PET/CT examination. The diagnosis of all HCC cases was validated by pathological examinations.
Cell lines and reagents
HEK293T, SMMC-7721, HepG2 were obtained from ATCC (Manassas, VA, USA); LO2 was obtained from the Cell Research Institute of the Chinese Academy of Sciences (Shanghai, China); they were cultured in DMEM (GIBCO, Grand Island, NY, USA) supplemented with 10% FBS (GIBCO). The antibodies used were anti-Flag (20543-1-AP, Proteintech), anti-HA (901533, Biolegend), anti-PKM2 (3198S Cell Signaling Technology), anti-HDAC8 (ab187139, Abcam), anti-Cyclin D1 (ab16663, Abcam), β-catenin(8480S, Cell Signaling Technology), Non-phospho (Active) β-Catenin (Ser45) (19807, Cell Signaling Technology), PCI-34051(S2012, Selleck, Houston, USA). TM-2-51(S505528-1G, Sigma-Aldrich, USA).
Co-immunoprecipitation assay
To analyze exogenous protein–protein interaction, 10 μl of anti-HA beads (Sigma-Aldrich, USA) was incubated with the cell lysate that included HA or Flag-tagged PKM2 and HDAC8 protein overnight at 4 °C. For endogenous Co-IP assay, cell lysate was incubated with antibody against PKM2 and 10 μl protein A/G agarose (Pierce, Dallas, USA) overnight at 4 °C. The precipitates were washed four times with lysis buffer, and then, suspended in 5× SDS-PAGE sample loading buffer. After boiling for 5 min, the samples were analyzed by western blotting and detected by the relevant antibodies.
GST pull-down assay
BL21 (DE3, TansGen Biotech, Beijing, China) were used to express GST fusion PKM2 and His fusion HDAC8. The fusion proteins were purified with Glutathione-Sepharose 4B beads (GE Healthcare, Boston, USA) or Ni affinity resins (GE Healthcare) according to the manufacturer’s instructions. The GST fusion proteins were immobilized on glutathione-sepharose beads and then incubated with His-HDAC8 effluent at 4 °C for night. The beads were pelleted, washed by PBST (0.5% Tween-20 in PBS) for 5 times. Samples were analyzed by SAS-PAGE and Coomassie Blue Staining.
Confocal immunofluorescence microscopy and proximity ligation assay (PLA)
SMMC-7721 and LO2 were grown in 24-well plates on glass coverslips. Cell fixation was performed using 4% paraformaldehyde in PBS. After permeabilization with 0.25% Triton X-100, the cells were treated with blocking buffer for 30 min and incubated overnight at 4 °C with the primary antibody, followed by incubation with the secondary antibody at room temperature (RT) for 1 h. The coverslips were mounted onto glass slides and counterstained with DAPI. Confocal laser-scanning microscope (Olympus BX61) was used to observe the image (Sigma-Aldrich, USA).
PLA was performed according to the manufacturer’s protocol (Duolink in situ PLA, Sigma-Aldrich). Before the experiment, the cells were processed as confocal immunofluorescence microscopy, and the end of the experiment, images were obtained as above.
Mass spectrometry for acetylation modification
HepG2 and the corresponding HDAC8 knocked-out cells were overexpressed Flag-tagged PKM2 plasmid. After 48 h, cell lysates were treated as previously mentioned23.
RNA interference and re‐expressed cell lines
Lipofectamine 2000 (Invitrogen, Waltham, MA, USA) was used in transient transfection according to the manufacturer’s protocol. The sequences of siRNA oligos were as follows: PKM2 (sense 5ʹ GCCCGAGGCTTCTTCAAGAAGTT-3ʹ, antisense 5ʹ-CTTCTTGAAGAAGCCTCGGGCTT-3ʹ), HDAC8 (sense 5ʹ-GCAGAUGAGGAUAGUUAAG TT-3ʹ, antisense 5ʹ-CUUAACUAUCCUCAUCUGC TT-3ʹ) and negative control: (sense 5ʹ-UUCUCCGAACGUGUCACGUTT-3ʹ, antisense 5ʹ-ACGUGACACGUUCGGAGAATT-3ʹ).
For PKM2‐re‐expressed cell lines, cells were infected by lentivirus with shPKM2 (sense 5ʹ‐CCGGGCCCGAGGCTTCTTCAAGAAGCTCGAGCTTCTTGAAGAAGCCTCGGGCTTTTTG‐3ʹ, antisense 5ʹ‐AATTCAAAAAGCCCGAGGCTTCTTCAAGAAGCTCGAGCTTCTTGAAGAA GCCTCGGGC‐3ʹ) first. Single colonies were isolated by limited dilution and expansion. Clones were then genotyped by sequencing and validated by immunoblotting. Following by infected by pLenti‐CMV‐Cherry‐3Flag‐PGK‐Puro- PKM2 virus (WT/K62R/K62Q).
RNA extraction and real-time PCR assay
Total RNA was isolated from the cultured cells with TRIzol Kit (Omega, Norcross, GA, USA). Reverse transcription was performed using the cDNA synthesis kit (Takara, Otsu, Japan) following the manufacturer’s instructions. Real-time PCR was carried out by using SYBR green fluorescence (Takara). The StepOnePlus Real-Time PCR System (Thermo Fisher Scientific) was used to carry out the quantitative PCR. The primers for Cyclin D1were as follow: 5ʹ- GCTGCGAAGTGGAAACCATC-3ʹ (sense) and 5ʹ- CCTCCTTCTGCACACATTTGAA - 3ʹ (antisense). PKM2 5ʹ- GGGCCATAATCGTCCTCACC -3ʹ (sense) and 5ʹ-TTGCACAGCACAGGGAAGAT-3ʹ (antisense). HDAC8 5ʹ-TCGCTGGTCCCGGTTTATATC-3ʹ (sense) and 5ʹ-TACTGGCCCGTTTGGGGAT-3ʹ (antisense)
Cell proliferation assay
For colony formation assay, Cells were put into 6-well plates at a density of 1 × 103 per well, stained with Crystal violet after 10 days. For CCK8, according to the instructions of Cell Counting Kit-8 (CCK-8, Bimake, Shanghai, China), 1 × 104 Cells were put in 96-well per well, counting at 450 nm for four consecutive days.
5-Ethynyl-2′-deoxyuridine (EdU) detection kit (RiboBio, Guangzhou, China) experiment was carried according to the manufacturer’s instruction. Cells were putting into 96-well by 1 × 104 per well after transfected by corresponding plasmids 24 h. Another 24 h passed, we fixed the cells by 4% paraformaldehyde in PBS, then other steps according to the manufactures’ instructions. Images were obtained by fluorescence microscopy (Olympus, Tokyo, Japan).
PK activity, lactate, GSH, and NADP(H) assay
Cells were plated in 6-well plate. After different treatment, cell supernatant was collected to measure lactate concentration, while the cell pellets to be lysed and measured PKM2 activity, GSH, and NADP(H). All the experiments were conducted according to (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Extracellular acidification rate (ECAR) assays
The extracellular acidification rate (ECAR) were measured using Seahorse XF Glycolysis Stress Test kit (Agilent Technologies). The Seahorse XF 24 Extracellular Flux Analyzer (Seahorse Bioscience) was used according to a previously described method24.
14CO2 release assay
The incorporation of [1-14C] glucose or [6-14C] glucose into 14CO2 was determined as previously reported25. Briefly, cells were cultured in 10 cm2 dishes, and the cells were exposed to DMEM supplemented with [1-14C] glucose (0.1 µCi/ml) or [6-14C] glucose (0.1 µCi/ml). The dish was placed in a container to collect CO2 produced. Rates of glucose consumption were measured by incubating cells for 120 min at 37 °C. Fresh air was pumped into the container by a ventilator. The [14C] CO2 was driven into a vial and trapped by Hyamine hydroxide. No-cell controls were included to correct for unspecific CO2 trapping.
Mass spectrometry analysis
When Density was 90% per 10 cm dish, Cells were blowed down gently and washed by NS quickly, then transferring to 1.5 ml tube (1/10 was reserved for protein quantification). Samples were extracted by cell metabolite extract (ACN/MeOH/Water: 40/40/20, −20 °C for pre-cooling one night). The cell pellets were put on ice, 300 µl extract buffer was added per tube. The cell precipitation was thoroughly blown and mixed, then it was swirled on the vortex instrument for 10 s and then put into the refrigerator at minus 20 degrees Celsius for 20 min. Finally, supernatant was obtained by centrifugation (12,000 × g, 4 °C for 10 min). In case of precipitation, only 200 µl volume of supernatant was transferred. The samples were tested on LC-MS machine (Agilent Technologies, Calif., USA).
Xenograft tumor studies
4-week male NCG mice were purchased from Renji Hospital Experimental Animal Center (Shanghai, China). All animal experiments were reviewed and approved by Renji Institutional Animal Care and Use Committee (Shanghai, China). Briefly, NCG mice were randomly divided into two groups (n = 4 per group) and inoculated subcutaneously with 1 × 107 HepG2 cells in the right while with 1 × 107 HDAC8-KO cells in the left flank. Similarly, HDAC8-KO/PKM2-WT cells and HDAC8-KO/PKM2-K62R cells were injected subcutaneously into the right and left flank of mice with 1 × 107 cells per injection, respectively. The volumes of tumors were measured as the length × width2 × 0.5. After 5 weeks, PET imaging scans were carried out on a micro-PET/CT scanner (Super Nova®PET/CT, PINGSENG Healthcare Inc., Shanghai, China). 18F-FDG (~200 µl, ~7.4 MBq) was injected into the tail vein of tumor-bearing mice. After 30 min, the animals were anesthetized with 2% isoflurane and immobilized during PET scan acquisition. Maximum standard uptake value (SUVmax) was used to assess the 18F-FDG uptake by tumors. After imaging, mice were sacrificed and tumors were excised.
Statistical analysis
The data analysis was performed by using statistical program SPSS19.0 (IBM, USA) and Graph Pad Prism 7.0 (Graph-Pad Software, Inc., USA). The quantitative data were expressed as mean ± SEM. The variance was similar between the groups that are being statistically compared. No samples or mice were excluded from the analysis as outliers. Student’s t test, Chi-square test and one way ANOVA was used to determine differences between groups upon the data type. Kaplan–Meier method was used to calculate survival curves. p values < 0.05 were regarded as statistically significant.
Results
Acetylation of PKM2 lysine residue 62 impacts prognosis of HCC
PKM2 plays an important role in tumor development and progression through the regulation of both metabolic and nonmetabolic pathways26. To evaluate the expression differences of PKM2 between tumors and normal tissues in HCC, we applied the gene expression profiling interactive analysis (GEPIA) to analyze the data from TCGA and GTEx projects. PKM2 expression was higher in HCC compared with adjacent normal samples (Fig. S1A). We also further used GEPIA to analyze the prognostic value of PKM2 in HCC. Results revealed that high PKM2 expression levels were associated with poorer prognosis of OS in HCC (OS Log-rank p = 0.005, HR = 1.8) (Fig. S1B). In GSE4465, genomic profiling is performed in HepG2 cells after TSA (0.5 µM) treatment. As shown in Fig. S1C, PKM2 levels are upregulated upon HDAC inhibition by TSA treatment. There is clear evidence that the acetylation of PKM2 lysine residues 305 (K305) and 433 (K433) regulates metabolic flux and gene transcription, ultimately affecting the malignant phenotype of the tumor16,17,27–29. There are 24 potential lysine sites in PKM2 that can be modified by acetylation (Fig. S1D, E), but their role in tumors is unclear. Lv, L et al. demonstrate that PKM2 Lysine residue 62 (K62) is acetylated, but not the target of SIRT227. Therefore, there is great interest to test role of K62 acetylation of PKM2 in tumor development and progression. To gain insight of post-translational regulation of PKM2 at K62 residue, we generated an antibody that specifically recognizes K62 acetylation (Fig. S1F, G) to determine its clinical significance in HCC tissues. We evaluated the K62 acetylation level of PKM2 in paired tumors and adjacent normal tissues in 31 HCC patients. The K62 acetylation level of PKM2 in the tumor is lower than that of adjacent tissues, and it is accompanied by rapid cell proliferation (Fig. 1A). Statistical analysis shows that K62 acetylation level of PKM2 is significantly lower than that of adjacent tissues (Fig. 1B). As shown in Table 1, K62 deacetylation level of PKM2 was significantly correlated with tumor size (p = 0.041), sample grade (p = 0.046), glucose uptake (SUVmax, p = 0.036) and hepatocirrhosis (p < 0.019), but not with age, portal vein invasion, or HBV infection (Table 1). More importantly, PKM2 K62 deacetylation level is closely related to the prognosis of HCC patients (5-year survival rate, p < 0.001). The survival time of patients with low K62 acetylation was significantly shorter than that of patients with high K62 acetylation (p < 0.001) (Fig. 1C). Taken together, these data indicate that K62 deacetylation of PKM2 impacts prognosis of HCC, and K62 deacetylation may play an important role in the development and progression of HCC.
Fig. 1. Deacetylation of K62 promotes tumor growth of hepatocellular carcinoma.

A HCC and its adjacent normal tissues were typically histopathologically analyzed using H&E staining and ki67, PKM2-K62-ac antibodies (n = 31). B Comparison of immunohistochemical staining scores of PKM2-K62-ac between cancer and adjacent tissues (**p < 0.01); C Overall survival analysis by Kaplan–Meier (low and high staining groups were categorized according to the immunohistochemistry scores and Scale bar, 100 µm).
Table 1.
Relationship between acetylation of K62and clinicopathological parameters of hepatocellular carcinoma patients.
| PKM2-K62Ac | ||||
|---|---|---|---|---|
| Characteristics | All cases | High | Low | P value |
| Participaints | 31 | 14 | 17 | |
| Age | 0.578 | |||
| < 50 years | 15 | 7 | 8 | |
| ≥ 50 years | 16 | 7 | 9 | |
| Tumor size | 0.041 | |||
| < 5 cm | 16 | 10 | 6 | |
| ≥5 cm | 15 | 4 | 11 | |
| Hepatocirrhosis | 0.019 | |||
| Negative | 7 | 6 | 1 | |
| Positive | 24 | 8 | 16 | |
| Sample grade | 0.046 | |||
| 1 | 5 | 4 | 1 | |
| 2 | 20 | 9 | 10 | |
| 3 | 6 | 1 | 6 | |
| 5-Year survival rate | < 0.001 | |||
| <5 years | 14 | 1 | 13 | |
| ≥5 years | 11 | 10 | 1 | |
| SUVmax | 0.036 | |||
| <5.3 | 18 | 11 | 7 | |
| ≥5.3 | 10 | 2 | 8 | |
| Portal vein invasion | 0.195 | |||
| Negative | 23 | 12 | 11 | |
| Positive | 7 | 2 | 5 | |
| HBV infection | 0.394 | |||
| Negative | 13 | 5 | 8 | |
| Positive | 18 | 9 | 9 | |
HDAC8 regulates K62 acetylation of PKM2
Next, we explored who is responsible for regulating the acetylation level of PKM2-K62 residue. Given that deacetylase or acetylase regulates the level of acetylation after binding to PKM2, we analyzed potential PKM2 binding partners by mass spectrometry and revealed a novel protein interaction between PKM2 and HDAC8 (data not shown). Several studies have shown that HDAC8 deacetylates non-histone proteins, thereby affecting the growth of tumor cells30–32. Therefore, we hypothesize that HDAC8 may be a potential deacetylase of PKM2 protein. Through a series of exogenous or endogenous protein interaction analysis, we found that HDAC8 and PKM2 had protein–protein interaction (Fig. 2A–D). And this interaction localized in both cytoplasm and nuclear of SMMC-7721 and LO2 cells (Fig. 2E). In vitro binding experiments revealed that HDAC8 could directly interact with PKM2 (Fig. 2F). Based on the characteristics of the PKM2 protein domain, we found that the 390–531aa is the key sequence of PKM2 binding to HDAC8 (Fig. 2G). These protein–protein interaction results strongly suggest that HDAC8 may be a regulator of PKM2 protein. Compared with the control group, the overall acetylation level of PKM2 increased after treatment with HDAC8 specific inhibitor PCI-34051or TSA/NAM (Fig. 2H), which is consistent with the previous reports16,27,28.
Fig. 2. HDAC8 deacetylates PKM2 at lysine-62.

HEK293T cells were transfected with Flag- PKM2 / HA-HDAC8 (A) or Flag- HDAC8/HA- PKM2 (B). After 48 h, HA-tagged protein was immunoprecipitated from HEK293T cells lysates. Then, immunoprecipitates and whole-cell extracts were subsequently separated by SDS-PAGE and immunoblotted with Flag and HA antibodies. C HepG2 cells were transfected with HA- PKM2 for 48 h, Extracts were immunoprecipitated with anti-HA (IgG as control). Immunoprecipitates and whole-cell extracts were subsequently analyzed by western blotting with an anti-HDAC8 and HA antibody. D Lysates of HepG2 cells were immunoprecipitated with the antibody against PKM2. Immunoprecipitates and whole-cell extracts were analyzed by western blotting with anti-PKM2 and anti-HDAC8 antibodies. E The Duollink PLA technology showed the interaction(red) between HDAC8 and PKM2 in SMMC-7721 and LO2 cells (Scale bar, 50 µm). F The mixture of recombinant GST- PKM2(GST-vector as a control) and His- HDAC8 were rotated and incubated for 3 h. Bound proteins were eluted, then analyzed with Coomassie Blue staining. G FLAG-HDAC8 was co-expressed in HEK293T cells with HA-PKM2 fragments as indicated. Co-IP followed by western blotting was performed to determine their interaction. H, I After transfected with Flag- PKM2 24 h, PCI-34051 (25 µM) and TSA/NAM (1 µM /10 mM) (H) or different doses of PCI-34051 (I) are added for 24 h. Cell lysates were immunoprecipitated with Flag affinity gel. PKM2 acetylation and protein levels were analyzed by immunoblotting using the indicated antibodies. J–L Interfered by siHDAC8 (J), PCI-34051 (K), and HA-HDAC8 (L) in HepG2 cells or HDAC8-KO HepG2 cells, separately. After immunoprecipitated with Flag affinity gel, acetylation of K62 was determined by western blotting. M LC-MS-MS of PKM2 showing the acetylation of PKM2 K62 in HDAC8-KO HepG2 cells. HepG2 cells and HDAC8-KO HepG2 cells transfected with Flag-PKM2. The immunoprecipitates were sent for LC-MS-MS analysis of the acetylation intensity of PKM2 peptides.
In the meantime, with the increase of HDAC8 inhibitor concentration, the overall level of PKM2 acetylation also moderately upregulated (Fig. 2I). More importantly, knocked down HDAC8 or inhibition its activity significantly increased the level of acetylation at the K62 site of the PKM2 protein, which was more pronounced than the overall level of acetylation (Fig. 2J, K). To further analyze the specificity of HDAC8 on the regulation of PKM2 acetylation, we established HDAC8 knockout HepG2 cells (Fig. S2A). Then, we analyzed the change of K62 acetylation in the absence or presence of HDAC8. As shown in Fig. 2L, the loss of HDAC8 lead to an increase in K62 acetylation levels, which is consistent with the effect of HDAC8 inhibitor. However, in the case of HDAC8 overexpression, the acetylation level of K62 decreased accordingly (Fig. 2L).
Then, we verified the effect of HDAC8 on the acetylation of PKM2 at the K62 residue by mass spectrometry (Fig. 2M and S2B). Therefore, these results indicate that HDAC8 can bind PKM2 and deacetylate its K62 residue.
Deacetylation of K62 reprograms glucose metabolism
PKM2 catalyzes the final and rate-limiting reactions in the glycolysis pathway and plays an important role in glucose metabolism. Through metabolic mass spectrometry, we found that the acetylation status of K62 affects the metabolic phenotype of tumor cells (Fig. 3A). HDAC8 not only regulated the enzyme activity of PKM2 (Fig. 3B), but also affected the protein stability of PKM2 (Fig. 3C). Knockdown of HDAC8 reduced the ubiquitination level of PKM2 (Fig. 3D), resulting in a slight accumulation of PKM2 protein (Fig. 3C). Since HDAC8 was found to be a new upstream regulator of PKM2, then we analyzed the effect of HDAC8 on glucose metabolism of HCC cells. The C1 of glucose mainly flux to PPP and TCA, while the C6 mainly flux to TCA for metabolism. Through [1-14C] glucose or [6–14C] glucose tracer analysis, we found that after HDAC8 knockdown, the glucose metabolism through the PPP pathway decreased, while the metabolism through the TCA pathway increased accordingly (Fig. 3E). HDAC8 can regulate glucose metabolism through enzyme activity dependent or independent manners. As shown in Fig. 3F, the enzyme activity of PKM2 of K62R is significantly lower than that of K62Q. In order to exclude endogenous interference, we expressed PKM2 wild-type, PKM2-K62R or PKM2-K62Q mutant in HDAC8 knockout cells to analyze its impact on cancer cell metabolism (Fig. 3G). The PKM2-K62R mutant was able to partially compromise increase of glycolysis (Fig. 3H) and lactate (Fig. 3I) levels that induced by HDAC8 knockout. It was further supported that by analyzing the levels of NADPH/NADP + and GSH/GSSG (Fig. 3J, K), we found that the PKM2-K62R mutant impaired the ability of cells to respond to oxidative stress after HDAC8 knockout. Therefore, these results indicate that HDAC8 reprograms the glucose metabolism of HCC cells by regulating K62 acetylation of PKM2 protein, so that glucose is mainly metabolized through the PPP pathway to cope with the challenge of oxidative stress (Fig. 3L).
Fig. 3. Deacetylation of K62 reprograms glucose metabolism.

A Mutants at K62 (R/Q) of PKM2 were transferred into HepG2 cells, after 48 h, cells were collected and extracted for LC-MS. B Medium supernatant of HepG2 and HDAC8-KO HepG2 cells in 6-well plate were collected and measured lactate production, protein quantification for calibration. C HepG2 cells expressing si-HDAC8 (si-NC) or HA-HDAC8 (HA-vector) plasmids. After 48 h, cell lysates were analyzed by western blotting with indicated antibodies. D HepG2 cells were transfected with siHDAC8(siNC as a control), after 6 h, followed by co-transfected with Flag-PKM2 and HA-UB plasmid, 42 h after transfection, cells were incubated with MG132 (100 μM) for 6 h. Cell lysates were immunoprecipitated with anti-flag antibody followed by immunoblotting analysis with anti-HA or anti-flag antibody. E After interfering with HDAC8 expression level by siHDAC8 for 48 h, the incorporation of [1-14C] glucose or [6-14C] glucose into 14CO2 was determined. G The construction of cell lines which were stably expressing PKM2 plasmids (WT/K62R/K62Q) in HDAC8-KO HepG2 cell lines. H 15,000 cells per well were planted in Seahorse XF plate, cultured overnight, and then texted ECAR according to protocol. I–K A mass of above-mentioned cells (G) were collected and treatment by the corresponding lysis buffer, and measured lactate (I) NADP+/NADPH (J) and GSH/GSSG (K) according to protocols. l A summary of glucose metabolism pathway regulated by HDAC8 in HCC. (Data are mean ± SEM, n = 3–5, *p < 0.05, **p < 0.01, ***p < 0.001. Student’s t test in (A, B, E), one way ANOVA in (H, I, J, K).
Deacetylation at K62 promotes nuclear translocation of PKM2
Nuclear localized PKM2 plays an important role in regulating the growth of tumor cells through non-metabolic functions. Studies have demonstrated that EGF can stimulate PKM2 to translocate the nucleus, thereby binding and regulating the function of β-catenin8,16. Therefore, we analyzed the effect of EGF on the acetylation level of PKM2. As shown in Fig. S3, we observed that the K62 acetylation level of PKM2 was significantly reduced after EGF treatment. Furthermore, HDAC8 knockdown prevented the accumulation of PKM2 protein in the nucleus (Fig. 4A and S4A). By extracting nuclear proteins, we observed that similar phenomenon that HDAC8 regulated PKM2 nuclear localization (Fig. 4B). In contrast, HDAC8 overexpression was able to promote PKM2 translocation to the nucleus (Fig. 4C, D and S4B). In addition, both confocal fluorescence and nuclear protein analysis showed that the PKM2 mutant (K62R) lacking K62 acetylation had more nuclear enrichment than the acetylated PKM2 mutant (K62Q) (Fig. 4E–G). Therefore, these results indicate that HDAC8 promotes the entry of PKM2 into the nucleus by regulating the deacetylation of K62 residue.
Fig. 4. Deacetylation at K62 promotes nuclear translocation of PKM2.

A–D HepG2 cells were transfected with siHDAC8 (or negative control siRNA) or HA-HDAC8 plasmids (or vector plasmid), after 24 h, cells were placed on glass coverslips, cultured for another 24 h. Anti-PKM2 (red) and anti-k62ac (green) were used for immunofluorescence (scale bar: 50 μm) (A, C). The line profiles of the mean fluorescence intensity of PKM2 and DAPI signals were measured by ImageJ software. The remaining cell pellets were separated by cytosolic and nuclear fractions (B, D) for western blotting. E The cell lines which were stably expressing Flag-mcherry-PKM2 plasmids (WT/K62R/K62Q) in shPKM2-HepG2 cell lines. Immunofluorescence technique was used like above description (F), the remaining cell pellets were separated by cytosolic and nuclear fractions (G) for western blotting (***p < 0.001, ****p < 0.0001, one way ANOVA).
HDAC8 upregulates CCND1 expression and G1-S transition by promoting the binding of PKM2 to β-catenin
PKM2-dependent β-catenin transactivation regulates CCND1 expression and cell cycle in cancer cells8. Therefore, we speculated whether HDAC8 can play a role in regulating the cell cycle through the PKM2/β-catenin complex. As shown in Fig. 5A, we observed that overexpression of HDAC8 promoted the binding of PKM2 to β-catenin. In contrast, HDAC8 inhibitor PCI-34051 inhibited the formation of PKM2/β-catenin complex (Fig. 5B). Compared with wild-type PKM2, the loss of acetylation at the K62 site promoted binding of PKM2 and β-catenin in the nucleus (Fig. 5C). Functionally, HDAC8 knockdown significantly reduced CCND1 expression and lead to G1 phase arrest, which was consistent with the phenomenon caused by PKM2 knockdown (Fig. 5D). In contrast, HDAC8 overexpression promoted CCND1 expression and cell cycle progression (Fig. 5E). However, HDAC8 overexpression and PKM2 knockdown did not result in changes in CCND1 expression and cell cycle (Fig. 5E). In addition, HDAC8 knockdown impaired PKM2-induced CCND1 expression and G1-S transition (Fig. 5F). Therefore, these results indicate that PKM2 as a downstream factor is necessary for HDAC8 to drive CCND1 expression and cell cycle progression.
Fig. 5. HDAC8-dependent deacetylation at K62 promotes the binding with β-catenin and upregulating cyclin D1 expression.

A HepG2 cells were co-transfected with HA-β-catenin with Flag-HADC8 (Flag-vector), after 48 h, cells were collected for immunoprecipitated by HA beads, analyzed by western blotting with PKM2 and HA antibodies. B Co-transfected HA-β-catenin and Flag-PKM2 plasmids for 24 h, then PCI-34051 (25 µM, DMSO as control) added for another 24 h, immunoprecipitated by HA beads, western blotting using Flag and HA antibodies. C Cells were transfected by PKM2 WT or K62R plasmids, separately. After 48 h, the Duollink PLA technology was used to illustrate the interaction (red) of HA-PKM2and β-catenin in HepG2 cells (Scale bar, 50 µm). D HepG2 were transfected with siRNA against HDAC8 or PKM2 (negative control siRNA) for western blotting or qPCR. Another half were analyzed by cytometry. E, F Based on which siRNA were transfected, after 6 h, the cells were overexpressed another protein expression plasmid (controlled by negative siRNA and vector plasmid). Then experiments were carried out as mentioned above. G Cells were transfected PKM2 WT or mutant plasmids, separately. After 48 h, lysates of HepG2 were analyzed by western blotting using the indicated antibodies. H Based on above result. cells were transfected PKM2 wild type or K62 mutant plasmids, separately, then PCI-34051 (25 µM) was added after 24 h and maintained for 24 h. Cell lysates were analyzed by western blotting using the indicated antibodies. (one way ANOVA in (D–F), *p < 0.05, **p < 0.01).
To further elucidate the regulatory effect of K62 acetylation on the cell cycle, we expressed PKM2 wild-type and K62, K305 acetylation-related mutants in HepG2 cells. The result showed that K62 acetylation-deficient mutant (K62R) or K305 acetylation-mimic mutant (K305Q) promoted the expression of CCND1 (Fig. 5G), indicating that these two sites regulate CCND1 gene transcription through different mechanisms. In addition, in the presence of K62 acetylation (K62Q), rather than in the absence of K62 acetylation (K62R), a regulatory effect of HDAC inhibitor on CCDN1 expression was observed (Fig. 5H). In summary, these results indicate that HDAC8 promotes the expression of CCND1 and the transition of the cell cycle from G1 to S phase by deacetylating the K62 site of PKM2 protein.
Deacetylation of PKM2 K62 residue promotes HCC cell growth
Multiple studies have shown that HDAC8 promotes the growth and development of HCC. Consistently, HDAC8 inhibitors PCI-34051 and agonist TM-2-51 inhibited and promoted the growth of liver cancer cells, respectively (Fig. S5A). And, HDAC8 knockout also can significantly inhibit the growth of HCC cells, and the clone formation, and EdU incorporation further demonstrated the regulatory effect of HDAC8 on cell growth (Fig. S5B). Next, we analyzed the relationship between HDAC8-mediated PKM2-K62 deacetylation and HCC cell growth. Compared with the PKM2 wild-type or K62 site acetylation-deficient mutant, the K62 acetylation-deficient mutant significantly promoted the growth, clonal formation and EdU incorporation of HCC cells (Fig. 6A–C). Consistent with in vitro experiments, in vivo experiments have shown that knocking out HDAC8 can significantly inhibit the growth of tumor cells, while PKM2-K62R mutants instead of PKM2 wild-type can restore tumor cell growth defects caused by HDAC8 deficiency partly ((Fig. 6D). In addition, 18F-FDG-PET imaging showed that K62 acetylation-deficient HCC cells displayed glucose avidity phenotype (Fig. 6E). Protein level analysis by immunohistochemistry of confirmed the effect of the K62 acetylation level on promoting proliferation. As shown in Fig. 6F, HDAC8-KO group had the lowest cyclin D1 and GLUT1 protein expression. Just as we expected, HDAC8-KO/PKM2-K62R had a good performance. In conclusion, deacetylation at the K62 site of PKM2 protein plays an important role in regulating the growth of tumor cells (Fig. 7).
Fig. 6. Deacetylation of K62 promotes cells proliferation in vivo and in vitro.

CCK-8 assay (A), colony formation (B), and immunofluorescence analysis with EdU (Scale bar: 20 μm) C were performed to evaluate the effect of HDAC8 knockout and the K62 acetylation deficiency on the proliferation ability of HepG2 cells. Data shown are mean ± SD (n = 3) (*P < 0.05, **P < 0.01, ***P < 0.001). D Image of xenograft tumors resected from tumor-bearing NCG mice (n = 4). E Representative 18F-FDG micro-PET/CT images of two living NCG mice were conducted 5 weeks after subcutaneous inoculation. Images showed the obvious different ability of FDG uptake in different groups of xenografts. F The tumor tissues were examined by hematoxylin and eosin(H/E), anti-ac-lys-62-PKM2 antibodies, HDAC8, Ki-67, Cyclin D1 and Glut1(scale bar, 100 μm). The analysis of the intensity of cyclin D1 expression by image J. (*P < 0.05, one way ANOVA in A, B, D, E, F).
Fig. 7. Schematic deacetylation of PKM2 at K62 in HCC.
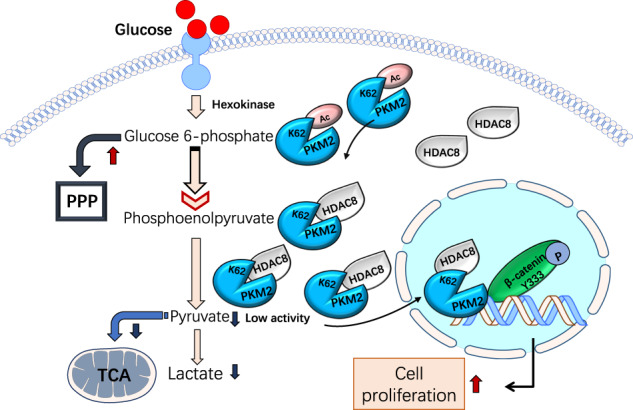
HDAC8 plays a crucial role in deacetylation of PKM2 at K62. Then the metabolic activity and nuclear relocation of PKM2 was narrowed down and increased separately.
Disscusion
PKM2 plays a key role in glucose metabolism and cell proliferation by changing enzyme activity and regulating gene transcription. A large amount of evidence indicates that post-translational modification is an important mechanism for PKM2 function and regulation of tumorigenesis15,33. Here, we found that the deacetylation of K62 is significantly related to the prognosis of liver cancer patients, and also to hepatocirrhosis. In addition, we demonstrated HDAC8 as an upstream deacetylase that regulates PKM2 K62 acetylation levels. Consistently, it has been reported that the HDAC inhibitor suberoylanilide hydroxamic acid can alleviate hepatocirrhosis by inhibiting the TGF-β1 signaling pathway34. Due to HDAC8 activation, K62 acetylation could be masked by the overall acetylation pattern, or could depend on cell type35. In addition, K62 and K305 may play different roles under various nutritional stresses or in different cell types, which also highlights the complex post-translational regulation of PKM2 protein. Especially in the case of HDAC8 activation, deacetylation at position K62 is particularly important for the regulation of PKM2 function. The phenotype of PKM2 K62 deacetylation is consistent with that of HDAC8 overexpression. This indicates that in the case of HDAC8 activation, deacetylation at K62 residue is particularly important for regulating PKM2 function3,36,37.
HDAC8 is localized in the cytoplasm and nucleus. In addition to the deacetylation of histone variants (e.g., H2A/H2B, H3, and H4), many nuclear targets of HDAC8 have been identified (e.g., structural maintenance of chromosome 3 [SMC3])32,37. Similarly, the glycolytic enzyme PGAM1 that catalyzes the conversion of 3-phosphoglycerate to 2-phosphoglycerate in the glycolysis pathway is also a regulatory target of HDAC838. Recent evidence indicate that HDAC8 is phosphorylated by AMPK in lung cancer cells involved in glucose metal metabolism under hypoglycemia induction, and remains in the cytoplasm, resulting in upregulated expression of PGM139. Here, we found that HDAC8 can directly bind and deacetylate to regulate PKM2 protein at K62 residue, which further deepens our understanding of HDAC8 in regulating glucose metabolism and driving tumorigenesis.
Cancer cells display a robust increase in glucose uptake, and there is a higher demand for metabolic intermediates biosynthesized to support rapid cell growth. PKM2 is a critical regulatory factor in directing glucose metabolism and the regulation of pyruvate kinase activity in proliferating cells may be particularly important to coordinate glucose metabolism with the synthesis of deoxynucleotides for DNA replication15. Acutely inhibiting the activity of PKM2 in proliferating cells leads to the temporary accumulation of upstream glycolysis intermediates and reduces oxygen consumption, making glucose metabolites available for anabolic processes40. The regulation of K62 acetylation level by HDAC8 affects the enzyme activity and protein stability of PKM2. As a result, pyruvate can be converted to lactic acid or oxidized to acetyl-CoA into the tricarboxylic acid cycle. The low activity of PKM2 can keep the levels of ATP and GTP low, thereby continuously activating phosphofructokinase-1, so the rate of glycolysis remains high. Therefore, inhibition of PKM2 activity can reprogram glucose metabolism, providing a large number of metabolic intermediates for biosynthesis, such as dihydroxyacetone phosphate for membrane lipid synthesis17,27,41.
In addition, PKM2 plays a carcinogenic role as a nuclear protein kinase8,42,43. Upon EGFR activation, acetylation of PKM2 Lys433 residue promotes its nuclear localization and upregulates the expression of cyclin D18. K62 acetylation has a similar working model. We found that PKM2 K62 residues were deacetylated by HDAC8, causing PKM2 to translocate to nucleus and synergize with β-catenin transcription to upregulate the expression of cyclin D1. Our results confirmed that the regulation of PKM2 entry into the nucleus by HDAC8 is essential for the growth of tumor cells. In summary, we found that PKM2 is deacetylated by HDAC8 at K62 residue, which affects its role in glucose metabolism and gene transcription. The deacetylation status of K62 indicates a poor prognosis in HCC patients. The HDAC8-PKM2 pathway might provide new therapeutic targets for the treatment of HCC.
Supplementary information
Acknowledgements
This study was supported by research grants from National Natural Science Foundation of China (No. 81771858, 81571710, 81874202).
Competing interests
The authors declare no competing interests.
Footnotes
Edited by C. Munoz-Pinedo
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Ruixue Zhang, Mengqin Shen, Chunhua Wu
Contributor Information
Xiaoping Zhao, Email: zxp0856@sina.com.
Jianjun Liu, Email: RJnuclear@126.com.
Supplementary information
Supplementary Information accompanies this paper at (10.1038/s41419-020-03212-3).
References
- 1.Bray F, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Tomas-Loba A, et al. p38gamma is essential for cell cycle progression and liver tumorigenesis. Nature. 2019;568:557–560. doi: 10.1038/s41586-019-1112-8. [DOI] [PubMed] [Google Scholar]
- 3.Tian Y, et al. Histone deacetylase HDAC8 promotes insulin resistance and beta-catenin activation in NAFLD-associated hepatocellular carcinoma. Cancer Res. 2015;75:4803–4816. doi: 10.1158/0008-5472.CAN-14-3786. [DOI] [PubMed] [Google Scholar]
- 4.Zhang M, et al. SOCS5 inhibition induces autophagy to impair metastasis in hepatocellular carcinoma cells via the PI3K/Akt/mTOR pathway. Cell Death Dis. 2019;10:612. doi: 10.1038/s41419-019-1856-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wong TL, et al. CRAF methylation by PRMT6 regulates aerobic glycolysis driven hepatocarcinogenesis via ERK-dependent PKM2 nuclear relocalization and activation. Hepatology. 2019 doi: 10.1002/hep.30923. [DOI] [PubMed] [Google Scholar]
- 6.Li Q, et al. Circular RNA MAT2B promotes glycolysis and malignancy of hepatocellular carcinoma through the miR-338-3p/PKM2 axis under hypoxic stress. Hepatology. 2019 doi: 10.1002/hep.30671. [DOI] [PubMed] [Google Scholar]
- 7.Alquraishi M, et al. Pyruvate kinase M2: a simple molecule with complex functions. Free Radic. Biol. Med. 2019;143:176–192. doi: 10.1016/j.freeradbiomed.2019.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang W, et al. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature. 2011;480:118–122. doi: 10.1038/nature10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang W, et al. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell. 2012;150:685–696. doi: 10.1016/j.cell.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang W, Lu Z. Nuclear PKM2 regulates the Warburg effect. Cell Cycle. 2013;12:3154–3158. doi: 10.4161/cc.26182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu F, et al. PKM2 methylation by CARM1 activates aerobic glycolysis to promote tumorigenesis. Nat. Cell Biol. 2017;19:1358–1370. doi: 10.1038/ncb3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liang J, et al. PKM2 dephosphorylation by Cdc25A promotes the Warburg effect and tumorigenesis. Nat. Commun. 2016;7:12431. doi: 10.1038/ncomms12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang W, et al. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat. Cell Biol. 2012;14:1295–1304. doi: 10.1038/ncb2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu Q, et al. HSP90 promotes cell glycolysis, proliferation and inhibits apoptosis by regulating PKM2 abundance via Thr-328 phosphorylation in hepatocellular carcinoma. Mol. Cancer. 2017;16:178. doi: 10.1186/s12943-017-0748-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Z, et al. PKM2, function and expression and regulation. Cell Biosci. 2019;9:52. doi: 10.1186/s13578-019-0317-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lv L, et al. Mitogenic and oncogenic stimulation of K433 acetylation promotes PKM2 protein kinase activity and nuclear localization. Mol. Cell. 2013;52:340–352. doi: 10.1016/j.molcel.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park SH, et al. SIRT2-mediated deacetylation and tetramerization of pyruvate kinase directs glycolysis and tumor growth. Cancer Res. 2016;76:3802–3812. doi: 10.1158/0008-5472.CAN-15-2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan W, et al. Histone deacetylase inhibitors suppress mutant p53 transcription via histone deacetylase 8. Oncogene. 2013;32:599–609. doi: 10.1038/onc.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chakrabarti A, et al. HDAC8: a multifaceted target for therapeutic interventions. Trends Pharmacol. Sci. 2015;36:481–492. doi: 10.1016/j.tips.2015.04.013. [DOI] [PubMed] [Google Scholar]
- 20.Yuan J, et al. CUL4B activates Wnt/beta-catenin signalling in hepatocellular carcinoma by repressing Wnt antagonists. J. Pathol. 2015;235:784–795. doi: 10.1002/path.4492. [DOI] [PubMed] [Google Scholar]
- 21.Tian, Y., Mok, M. T., Yang, P. & Cheng, A. S. Epigenetic activation of wnt/beta-catenin signaling in NAFLD-associated hepatocarcinogenesis. Cancers.10.3390/cancers8080076 (2016). [DOI] [PMC free article] [PubMed]
- 22.Zhao X, et al. Pyruvate kinase M2 interacts with nuclear sterol regulatory element-binding protein 1a and thereby activates lipogenesis and cell proliferation in hepatocellular carcinoma. J. Biol. Chem. 2018;293:6623–6634. doi: 10.1074/jbc.RA117.000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi L, et al. SIRT5-mediated deacetylation of LDHB promotes autophagy and tumorigenesis in colorectal cancer. Mol. Oncol. 2019;13:358–375. doi: 10.1002/1878-0261.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li L, et al. Transcriptional regulation of the Warburg effect in cancer by SIX1. Cancer cell. 2018;33:368–385. e367. doi: 10.1016/j.ccell.2018.01.010. [DOI] [PubMed] [Google Scholar]
- 25.Tang S, et al. Role of SUMO-specific protease 2 in reprogramming cellular glucose metabolism. PLoS ONE. 2013;8:e63965. doi: 10.1371/journal.pone.0063965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsu MC, Hung WC. Pyruvate kinase M2 fuels multiple aspects of cancer cells: from cellular metabolism, transcriptional regulation to extracellular signaling. Mol. cancer. 2018;17:35. doi: 10.1186/s12943-018-0791-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lv L, et al. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol. cell. 2011;42:719–730. doi: 10.1016/j.molcel.2011.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bhardwaj A, Das S. SIRT6 deacetylates PKM2 to suppress its nuclear localization and oncogenic functions. Proc. Natl Acad. Sci. USA. 2016;113:E538–E547. doi: 10.1073/pnas.1520045113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shanmugasundaram K, et al. NOX4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistance. Nat. Commun. 2017;8:997. doi: 10.1038/s41467-017-01106-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hua WK, et al. HDAC8 regulates long-term hematopoietic stem-cell maintenance under stress by modulating p53 activity. Blood. 2017;130:2619–2630. doi: 10.1182/blood-2017-03-771386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oehme I, et al. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin. Cancer Res. 2009;15:91–99. doi: 10.1158/1078-0432.CCR-08-0684. [DOI] [PubMed] [Google Scholar]
- 32.Wu J, et al. The up-regulation of histone deacetylase 8 promotes proliferation and inhibits apoptosis in hepatocellular carcinoma. Digestive Dis. Sci. 2013;58:3545–3553. doi: 10.1007/s10620-013-2867-7. [DOI] [PubMed] [Google Scholar]
- 33.Prakasam G, Iqbal MA, Bamezai RNK, Mazurek S. Posttranslational modifications of pyruvate kinase m2: tweaks that benefit cancer. Front. Oncol. 2018;8:22. doi: 10.3389/fonc.2018.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, et al. Histone deacetylase inhibitor suberoylanilide hydroxamic acid alleviates liver fibrosis by suppressing the transforming growth factor-beta1 signal pathway. Hepatobiliary Pancreat. Dis. Int. 2018;17:423–429. doi: 10.1016/j.hbpd.2018.09.013. [DOI] [PubMed] [Google Scholar]
- 35.Chakrabarti A, et al. Targeting histone deacetylase 8 as a therapeutic approach to cancer and neurodegenerative diseases. Future Med. Chem. 2016;8:1609–1634. doi: 10.4155/fmc-2016-0117. [DOI] [PubMed] [Google Scholar]
- 36.Ramos TL, et al. HDAC8 overexpression in mesenchymal stromal cells from JAK2+ myeloproliferative neoplasms: a new therapeutic target? Oncotarget. 2017;8:28187–28202. doi: 10.18632/oncotarget.15969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dasgupta T, Antony J, Braithwaite AW, Horsfield JA. HDAC8 inhibition blocks SMC3 deacetylation and delays cell cycle progression without affecting cohesin-dependent transcription in MCF7 cancer cells. J. Biol. Chem. 2016;291:12761–12770. doi: 10.1074/jbc.M115.704627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lopez G, et al. HDAC8, a potential therapeutic target for the treatment of malignant peripheral nerve sheath tumors (MPNST) PLoS ONE. 2015;10:e0133302. doi: 10.1371/journal.pone.0133302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Y, et al. AMPK-dependent phosphorylation of HDAC8 triggers PGM1 expression to promote lung cancer cell survival under glucose starvation. Cancer Lett. 2020;478:82–92. doi: 10.1016/j.canlet.2020.03.007. [DOI] [PubMed] [Google Scholar]
- 40.Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature. 2008;452:181–186. doi: 10.1038/nature06667. [DOI] [PubMed] [Google Scholar]
- 41.Wu S, Le H. Dual roles of PKM2 in cancer metabolism. Acta Biochim. Biophys. Sin. 2013;45:27–35. doi: 10.1093/abbs/gms106. [DOI] [PubMed] [Google Scholar]
- 42.Gao X, Wang H, Yang JJ, Liu X, Liu ZR. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol. Cell. 2012;45:598–609. doi: 10.1016/j.molcel.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Azoitei N, et al. PKM2 promotes tumor angiogenesis by regulating HIF-1alpha through NF-kappaB activation. Mol. Cancer. 2016;15:3. doi: 10.1186/s12943-015-0490-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.