

Abstract
The CRISPR/Cas9 system allows scarless, marker-free genome editing. Current CRISPR/Cas9 systems for the fission yeast Schizosaccharomyces pombe rely on tedious and time-consuming cloning procedures to introduce a specific sgRNA target sequence into a Cas9-expressing plasmid. In addition, Cas9 endonuclease has been reported to be toxic to fission yeast when constitutively overexpressed from the strong adh1 promoter. To overcome these problems we have developed an improved system, SpEDIT, that uses a synthesised Cas9 sequence codon-optimised for S. pombe expressed from the medium strength adh15 promoter. The SpEDIT system exhibits a flexible modular design where the sgRNA is fused to the 3’ end of the self-cleaving hepatitis delta virus (HDV) ribozyme, allowing expression of the sgRNA cassette to be driven by RNA polymerase III from a tRNA gene sequence. Lastly, the inclusion of sites for the BsaI type IIS restriction enzyme flanking a GFP placeholder enables one-step Golden Gate mediated replacement of GFP with synthesized sgRNAs for expression. The SpEDIT system allowed a 100% mutagenesis efficiency to be achieved when generating targeted point mutants in the ade6 + or ura4 + genes by transformation of cells from asynchronous cultures. SpEDIT also permitted insertion, tagging and deletion events to be obtained with minimal effort. Simultaneous editing of two independent non-homologous loci was also readily achieved. Importantly the SpEDIT system displayed reduced toxicity compared to currently available S. pombe editing systems. Thus, SpEDIT provides an effective and user-friendly CRISPR/Cas9 procedure that significantly improves the genome editing toolbox for fission yeast.
Keywords: CRISPR/Cas9, gene editing, gene tagging, S. pombe, fission yeast
Introduction
The fission yeast Schizosaccharomyces pombe is a powerful model organism widely used in cellular and molecular biology ( Fantes & Hoffman, 2016). Traditionally, gene manipulation in S. pombe is achieved by transforming a DNA construct that includes the desired change alongside a selectable marker. The DNA construct integrates in the genome via flanking regions that target the genomic locus of interest. DNA constructs vary depending on the application but commonly consist of a sole PCR product that comprises an insertion, deletion or tagging cassette amplified from an existing plasmid ( Bähler et al., 1998). Albeit convenient, this approach results in a selectable marker integrated at the target locus, consequently disrupting the local genomic context and limiting the availability of markers for subsequent manipulations.
The prokaryotic CRISPR/Cas9 system enables flexible and scarless genome editing without the necessity of selectable markers escorting the introduced DNA change and disturbing the local genomic environment ( Hsu et al., 2014; Jinek et al., 2012). Adapted from a genome defence mechanism against invading DNA, the engineered minimal CRISPR/Cas9 system consists of a Cas9 endonuclease and a single-guide RNA (sgRNA) chimera that contains both the trans-activating CRISPR RNA and the targeting CRISPR RNA ( Jinek et al., 2012). The sgRNA sequence targets the system to a defined genomic location where the Cas9 endonuclease binds to a protospacer adjacent motif (PAM). The Cas9 enzyme then creates a double-strand break (DSB) three base pairs upstream of the PAM site in the protospacer sequence ( Jinek et al., 2012). Following DSB generation, repair is executed either through error-prone non-homologous end-joining (NHEJ), where strand end resection and subsequent repair frequently induce indels, or through high-fidelity homology-directed repair (HDR). HDR involves recombination via sequence homology and therefore can be exploited to generate precise mutations by providing a DNA editing template that contains the required DNA change and engages in homologous recombination (HR) with the cleaved region ( Hsu et al., 2014).
Implementation of the CRISPR/Cas9 system in S. pombe has been previously described in several reports ( Fernandez & Berro, 2016; Hayashi & Tanaka, 2019; Jacobs et al., 2014; Rodríguez-López et al., 2016; Zhao & Boeke, 2018; Zhang et al., 2018). A useful web-tool, CRISPR4P, was also developed to support the design of sgRNAs and oligonucleotides required to perform CRISPR/Cas9-mediated gene deletions ( Rodríguez-López et al., 2016).
To date, all CRISPR/Cas9 systems for S. pombe utilise the promoter/leader sequence of K RNA ( rrk1) and a hammerhead ribozyme (HHR) for sgRNA cassette expression ( Jacobs et al., 2014). A humanised Cas9 endonuclease expressed from the strong adh1 promoter was used in the original S. pombe system ( Jacobs et al., 2014). The resulting high levels of the Cas9 endonuclease were found to be detrimental for S. pombe growth. This Cas9 toxicity was partially bypassed by co-expression of the sgRNA and the Cas9 enzyme from a single plasmid ( Jacobs et al., 2014).
Cloning of a sgRNA target into the single sgRNA/Cas9 plasmid was originally executed via CspCI digestion ( Jacobs et al., 2014). This procedure proved to be extremely inefficient due to the large plasmid size and the inconsistency in available commercial CspCI preparations ( Rodríguez-López et al., 2016). A subsequent study attempted to overcome this problem by implementing a PCR-based method in which a sgRNA target is introduced into a single sgRNA/Cas9 plasmid by using overlapping PCR primers that contain the sgRNA sequence (pMZ379 plasmid, Rodríguez-López et al., 2016). Although an improvement over the initial system, this PCR-based method generated only a low frequency of bacterial colonies that contain correct and intact constructs. As a consequence, the required screening makes the entire process inefficient and time consuming.
To circumvent issues pertaining to Cas9 toxicity and inefficient sgRNA cloning procedures, here we report the development of SpEDIT, an improved CRISPR/Cas9 system for the efficient manipulation of the fission yeast genome. SpEDIT employs a highly effective one-step Golden Gate cloning strategy for the insertion of sgRNAs, that in combination with the use of a GFP placeholder allows visual screening for identification of positive clones. The Cas9 endonuclease gene implemented in this system is codon-optimised for expression in S. pombe and driven by the medium strength promoter adh15, resulting in reduced toxicity associated with Cas9 levels. SpEDIT can generate targeted ade6 and ura4 point mutants in asynchronous cells with 100% mutagenesis efficiency. Moreover, SpEDIT allows simultaneous editing at two non-homologous genes at distinct locations in the S. pombe genome, as well as seamless insertion, deletion and tagging at S. pombe loci. SpEDIT provides an efficient and simple CRISPR/Cas9 method to easily manipulate the genome of the fission yeast.
Results
The SpEDIT system
The SpEDIT system has been developed to address the two main complications associated with existing CRISPR/Cas9 methods for S. pombe: toxicity associated with Cas9 overexpression, and laborious cloning procedures required to insert a specific sgRNA target sequence into a Cas9-containing plasmid. An overview of the SpEDIT system is provided ( Figure 1) along with a full protocol (see Methods).
Figure 1. SpEDIT provides a fast and effective CRISPR/Cas9 method to manipulate the genome of Schizosaccharomyces pombe.

Diagram illustrating the required steps for S. pombe strain construction using SpEDIT. For a full protocol see methods. sgRNA, single guide RNA. HR template, homologous recombination donor template.
High levels of human codon-optimised Cas9 endonuclease constitutively expressed from the exceptionally strong adh1 promoter (400 RNA molecules/cell, PomBase, Lock et al., 2019) lead to reduced cell growth in S. pombe ( Jacobs et al., 2014). A recent report attempted to solve this toxicity problem by expressing the human codon-optimised Cas9 under control of the repressible nmt41 promoter ( Hayashi & Tanaka, 2019). Although this approach does generate mutations, it requires the non-ideal use of minimal media and relies on auxotrophic ( ura4 - or leu1 -) strains to allow plasmid selection. Moreover, the mutagenesis efficiency obtained was dependent on the selectable marker employed.
To overcome the toxicity related to high levels of humanised Cas9, we synthesized a Cas9 gene codon-optimised for expression in S. pombe (SpCas9) that is transcribed from the medium strength adh15 promoter ( Yamagishi et al., 2008). This adh15-SpCas9 gene is carried on a new plasmid, pLSB, that contains a choice of dominant selectable markers. Versions bearing natMX6, kanMX6 or hphMX6 markers are available, thereby allowing the SpEDIT system to be employed on fission yeast strains that harbour various manipulations where other selectable markers are already present ( Figure 2A).
Figure 2. The SpEDIT pLSB plasmid allows one-step insertion of sgRNAs via Golden Gate cloning.

A. Map of pLSB plasmid .Full sequence is available scanning the QR code in Figure 1 or at allshirelab.com/spedit. Versions with natMX6 (cloNAT), kanMX6 (G418) or hphMX6 (hygromycin) S. pombe resistance markers are available. A Cas9 codon optimised for S. pombe (SpCas9) is expressed from the adh15 promoter ( Padh15). B. Diagram of sgRNA cassette and cloning procedure. sgRNA cassette expression is driven by a tRNA Ser Pol III promoter (purple block arrow). A self-cleaving hepatitis delta virus (HDV) ribozyme is located at the 5’ end of the sgRNA cassette ( Ryan et al., 2014) (red block arrow). A superfolder green fluorescent protein (sfGFP) is used as placeholder (green block arrow). BsaI sites flanking sfGFP allow one-step insertion of a sgRNA target (light blue block arrow) into the sgRNA scaffold (grey block arrow) via Golden Gate cloning. The Pol III terminator sequence from S. cerevisiae SUP4 (tRNA Tyr) is present at the 3’ end of the sgRNA cassette (black block). C. The sfGFP placeholder allows cultures carrying empty (green) pLSB plasmids to be distinguished from sgRNA-loaded (non-green) pLSB plasmids. sgRNA, single guide RNA.
Eukaryotic CRISPR/Cas9 systems usually rely on snRNA or snoRNA RNA polymerase III (RNAPIII) promoters for transcription of the sgRNA cassette ( Cong et al., 2013). However, characterised S. pombe RNAPIII genes contain promoter elements within the transcribed region, preventing their use for generating accurately positioned 5’ ends, and RNA polymerase II (RNAPII) promoters generally do not generate transcripts with precise 5’ and 3’ ends. Consequently, all sgRNA expression systems for S. pombe so far utilise the rrk1 promoter plus its downstream 5’ untranslated region which generates a RNAPII transcript with a cleavable leader RNA ( Jacobs et al., 2014). Insertion of sgRNA sequences targeting genomic regions of interest into the rrk1 sgRNA expression cassette in current CRISPR/Cas9 systems for S. pombe relies on slow and arduous cloning procedures involving either traditional restriction digestion ( Hayashi & Tanaka, 2019; Jacobs et al., 2014) or PCR over a long template ( Rodríguez-López et al., 2016). An alternative method that uses in vivo gap repair to assemble a gapped Cas9-encoding plasmid and a PCR-amplified sgRNA fragment into a single circular plasmid has been reported ( Zhang et al., 2018). Although this method provided an advance, this system still utilises humanised Cas9 expressed from the very strong adh1 promoter, which consequently reduces cell growth due to Cas9-associated toxicity.
It has previously been shown that the upstream tRNA Ser gene of an S. pombe tRNA Ser-tRNA Met gene pair drives RNAPIII expression of the downstream tRNA Met gene ( Hottinger-Werlen et al., 1985). We therefore use this tDNA Ser to drive sgRNA expression in S. pombe. The resulting SpEDIT system employs a modular design where sgRNAs are expressed from this tDNA Ser sequence and fused to the hepatitis delta virus ribozyme (HDV), as previously described for Saccharomyces cerevisiae ( Ryan et al., 2014). The tDNA Ser acts as a RNAPIII promoter and the self-cleaving ribozyme protects and defines the 5’ end of the resulting sgRNA ( Figure 2A and B). The presence of the HDV ribozyme was shown to result in a six-fold increase in sgRNA abundance and this correlated with high targeting efficiency in S. cerevisiae ( Ryan et al., 2014). To facilitate cloning of sgRNA target sequences into this tDNA/HDV expression cassette, we placed sites for the BsaI type IIS restriction enzyme on each side of a GFP placeholder, thereby allowing one-step insertion of sgRNAs via Golden Gate cloning. Importantly, this strategy also permits visual screening to identify colonies that have lost the green GFP fluorescence signal indicating that the GFP has been successfully replaced with an incoming sgRNA ( Figure 2B and C).
SpEDIT can generate targeted ade6 and ura4 point mutants in asynchronous cells with 100% mutagenesis efficiency
To assess the performance of the SpEDIT system in comparison to the existing pMZ379 system ( Rodríguez-López et al., 2016), we targeted the ade6 + and ura4 + genes and provided HR templates that disable the PAM (NGG) sequence downstream of the sgRNA target to generate premature STOP codon mutations ( Figure 3A). ade6 + and ura4 + mutations can be easily scored due to their characteristic phenotypes: ade6 mutants, pink colonies develop on low (1/10) adenine-containing plates; ura4 mutants, cannot grow in the absence of supplementing uracil (uracil auxotrophy) but can grow in the presence of counterselective 5-fluoroorotic acid (FOA resistant) ( Figure 3B).
Figure 3. SpEDIT can generate targeted ade6 and ura4 point mutants in asynchronous cells with 100% mutagenesis efficiency.

A. Schematic of experiment to generate targeted ade6 and ura4 point mutants. A sgRNA-loaded pLSB or pMZ379 plasmid was co-transformed with an HR template that creates a premature STOP codon by disabling the PAM (NGG) sequence. sgRNA and HR template sequences for ade6 and ura4 are shown. Full HR template sequences can be found in Table 1. B. After transformation, cloNAT-resistant colonies were picked and re-streaked to non-selective YES plates. Cells were then replica-plated to indicated media to assess their phenotype. Representative plates from two independent experiments are shown. Quantification is shown in C– D. C– D. Percentage of cloNAT-resistant transformants displaying a mutant phenotype (pink cells, ade6; uracil auxotrophy and FOA resistance, ura4) after asynchronous ( C) or G1-synchronized ( D) wild-type cells were transformed with a sgRNA-loaded SpEDIT/pLSB (developed here, Figure 2) or pMZ379 ( Rodríguez-López et al., 2016) plasmid targeting ade6 + or ura4 + (or no sgRNA plasmid as control). An HR template targeting the same or a different gene was co-transformed as indicated. n = number of cloNAT-resistant colonies assayed. Note that when an HR template targeting a different gene or no HR template was co-transformed into asynchronous cells, the number of cloNAT-resistant colonies obtained was drastically reduced. Experiment was repeated twice with similar results. E. For each condition in C– D, five colonies displaying the mutant phenotype (or 5 cloNAT-resistant colonies for no sgRNA plasmids) were taken and the gene targeted by the sgRNA was sequenced to confirm changes in its DNA sequence. Both ade6 and ura4 were sequenced when no sgRNA was used. Edited clones harbour the change contained in the corresponding HR template. Other mutations at PAM disrupt the PAM (NGG) sequence and the corresponding gene coding sequence. For asynchronous cells transformed with pLSB- ura4 (no HR template) and pMZ379- ade6 (no HR template) only two and one colonies were respectively obtained and analysed. * No colonies were obtained for these conditions. sgRNA, single guide RNA. PAM, proto-spacer adjacent motif. HR, homologous recombination donor template. N/S, non-selective medium. FOA, 5-fluoroorotic acid.
We scored cloNAT-resistant colonies after electroporation of asynchronous cultures with either SpEDIT/pLSB or pMZ379 plasmids expressing sgRNA designed to mediate cleavage within the ade6 + or ura4 + genes in the presence or absence of an HR template homologous to ade6 + or ura4 +, respectively. The results revealed that both pLSB and pMZ379 plasmids can generate targeted mutations in ade6 + and ura4 + with 100% efficiency when a matching HR template was co-transformed ( Figure 3C). However, when an HR template targeting a different gene or no HR template was provided, the number of cloNAT-resistant colonies obtained was dramatically reduced. This decrease in transformant number in the absence of an HR template is consistent with futile cleavage-repair cycles where the persistence of a double strand break prevents cell division and thus colony formation ( Figure 3C).
Sequence analysis of the resulting ade6 and ura4 mutants showed that when a matching HR template was co-transformed, all clones analysed harboured the mutation provided by the HR template ( Figure 3E, left). However, a variety of mutations that disable the PAM sequence and ultimately disrupt the coding sequence of each gene were detected in mutants generated when a non-homologous HR template (targeting a different gene to the sgRNA) or no HR template was provided ( Figure 3E, left).
A previous study suggested that G1 synchronization of S. pombe cultures by nitrogen starvation prior to CRISPR/Cas9-mediated genome editing enhances transformation and deletion efficiencies ( Rodríguez-López et al., 2016). The rationale for this was that in G1 only one copy of a target locus would need to be modified as opposed to the two copies that are present in G2 cells. The remodelled transcriptional programme of G1 cells was also expected to render genomic regions more open ( Mata et al., 2002), and thus increase accessibility to the editing machinery.
We therefore compared the performance of the SpEDIT system (pLSB plasmid) versus the existing pMZ379 system (pMZ379 plasmid) at generating ade6 and ura4 mutations using G1-synchronized S. pombe cells as previously described ( Rodríguez-López et al., 2016). This comparison revealed that the pLSB plasmid is more efficient than the pMZ379 plasmid at generating mutations in G1-synchronized cells ( Figure 3D). However, even when a matching HR template was co-transformed, the mutagenesis efficiencies obtained with G1 cells were lower than that of asynchronous cells (pLSB→ ade6, 92% G1 versus 100% Asynchronous; pLSB→ ura4, 85% G1 versus 100% Asynchronous) ( Figure 3D).
Moreover, sequence analysis revealed that even when a matching HR template was co-transformed into G1 cells, not all mutant clones harboured the anticipated mutation that was presented by the HR template ( Figure 3E, right). This lack of accuracy is likely due to the suppression of the HR pathway that is known to occur in G1 cells ( Orthwein et al., 2015; Trickey et al., 2008).
Taken together, our results show that the SpEDIT system can generate targeted mutations at ade6 + and ura4 + with 100% mutagenesis efficiency using asynchronous cell cultures. Notably, our experiments show that G1 synchronization of S. pombe cells prior transformation has a detrimental effect on mutagenesis efficiency regardless of the CRISPR/Cas9 system used.
SpEDIT shows reduced toxicity compared with the current pMZ379 CRISPR/Cas9 system in asynchronous cells
To determine if the SpEDIT system leads to reduced toxicity compared to the current pMZ379 system, we measured colony area on selection plates after transforming asynchronous or G1-synchronized cultures with pLSB or pMZ379 plasmids targeting ade6 + or ura4 + (or empty plasmid controls) in the presence of a matching HR template. The colony area (equivalent to colony size) was found to be greater when asynchronous cells were transformed with pLSB as opposed to pMZ379 ( Figure 4A). The resulting difference in colony size was independent of the presence of a sgRNA, indicating that excessive levels of Cas9 alone, and not Cas9 targeting to a genomic locus, are sufficient to cause the observed toxicity ( Figure 4A). Consistent with this, the toxicity of adh1-Cas9 has also been shown to be independent of Cas9 catalytic activity ( Ciccaglione, 2015).
Figure 4. SpEDIT shows reduced toxicity compared with the current pMZ379 S. pombe CRISPR/Cas9 system in asynchronous cells.
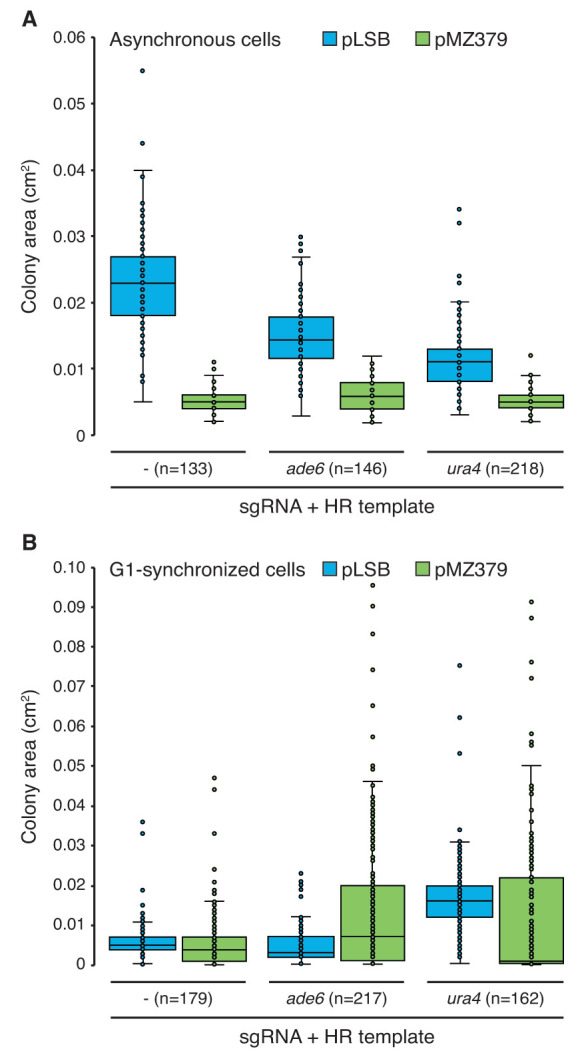
A– B. Colony area measurements of asynchronous ( A) or G1-synchronized ( B) wild-type cells transformed with pLSB or pMZ379 plasmids and indicated HR templates growing on selective cloNAT-containing plates (same experiment as Figure 3). Colony area was quantified (cm 2) using ImageJ. sgRNA, single guide RNA. HR, homologous recombination donor template.
In contrast, colony area measurements of pLSB and pMZ379 transformants obtained from G1-synchronized cells revealed no major difference in resulting colony size ( Figure 4B). This indicates that the toxicity related to high levels of catalytically active Cas9 is more prominent when transforming asynchronous cells. The lack of apparent toxicity in G1 cells is likely due to the known upregulation of non-homologous end joining-mediated repair and the suppression of homologous recombination repair that is known to occur at this cell cycle stage ( Orthwein et al., 2015).
SpEDIT allows simultaneous editing at two non-homologous genes at distinct locations in the S. pombe genome
The availability of pLSB versions bearing different dominant selectable markers presented the opportunity to test if the simultaneous editing of two different non-homologous loci by co-transformation with and selection of two distinct pLSB plasmids expressing different sgRNAs was possible.
To evaluate the possibility of simultaneous editing, we targeted two non-homologous genes, clr5 + and meu27 +, located on different chromosomes (I and III, respectively) in wild-type cells using pLSB-cloNAT and pLSB-hygromycin plasmids that express sgRNAs designed to target clr5 + and meu27 +, respectively ( Figure 5A). We co-transformed two HR templates that generate point mutations in clr5 + ( clr5-Q264STOP) and meu27 + ( meu27-S100Y), and concomitantly disabled both corresponding PAM sequences ( Figure 5A and B). These mutations ( clr5-Q264STOP and meu27-S100Y) had previously been identified in a proportion of heterochromatin-dependent epimutants resistant to caffeine ( Torres-Garcia et al., 2020).
Figure 5. SpEDIT allows simultaneous editing at two non-homologous genes at distinct locations in the S. pombe genome.

A. Schematic of experiment to simultaneously generate targeted point mutations in clr5 + and meu27 +. Two sgRNA-loaded pLSB plasmids (with different selection markers) were co-transformed with two HR templates that create the desired point mutations and disable the corresponding PAM (NGG) sequence. Transformed cells were then selected on selective plates containing both cloNAT and hygromycin. B. sgRNA and HR template sequences for clr5 (left) and meu27 (right) are shown, along with Sanger sequencing chromatograms for a successfully edited clone. Full HR template sequences can be found in Table 1. C. Percentage of cloNAT- and hygromycin-resistant transformants harbouring the targeted mutations in clr5 and meu27 as revealed by Sanger sequencing. sgRNA, single guide RNA. HR, homologous recombination donor template.
Sequencing of clr5 and meu27 in ten resulting cloNAT and hygromycin doubly resistant co-transformants revealed that two harboured both of the expected DNA changes. Five clones carried mutations in only one of the two targeted genes or bore mutations that uniquely affected the PAM sequence, and three clones displayed neither of the anticipated changes ( Figure 5C). Importantly, whole-genome sequencing of one of the isolates that contained both expected gene editing events revealed no additional genetic changes (SNPs or indels) in coding regions of the genome ( Torres-Garcia et al., 2020).
These results demonstrate that our improved system can be utilised to perform simultaneous gene editing at two distant, non-homologous S. pombe loci, albeit with reduced efficiency relative to the frequency of editing of a single locus.
Seamless insertion, deletion and tagging at S. pombe loci using SpEDIT
To assess the capabilities of SpEDIT in additional gene editing tasks, we utilised it to perform insertion, deletion and tagging at single S. pombe loci. Specifically, using SpEDIT, we inserted tetO binding sites downstream of the cup1 + (SPBC17G9.13c) gene ( Figure 6A). tetO binding sites allow tethering of proteins such as TetR-Clr4* and heterochromatin formation in the vicinity of the tethering site ( Audergon et al., 2015; Ragunathan et al., 2015). A fusion-PCR construct containing 4xtetO sites with 120 bp homology arms flanking the desired insertion site was used as the HR template (see Table 1 for sgRNA and HR template sequences). Correct insertion of the cup1:4xtetO HR template resulted in ablation of the PAM sequence. Furthermore, we used SpEDIT to seamlessly fuse GFP in frame with the 3’ end of the of cup1 + gene to produce Cup1-GFP ( Figure 6B). To generate the cup1-gfp HR template, the GFP open reading frame was amplified with oligonucleotides that had long extensions homologous to sequence immediately up-stream and down-stream of the normal cup1 + STOP codon. This HR template also carried a DNA change (in the 5’ long oligo) designed to disable the PAM sequence without altering the Cup1 protein sequence. Resulting strains were confirmed to carry the planned cup1:4xtetO or Cup1-GFP insertions in the absence of any associated selectable marker and have been utilised to show that heterochromatin-mediated silencing of cup1 + is sufficient to drive caffeine resistance in wild-type cells and that Cup1-GFP localises to mitochondria ( Torres-Garcia et al., 2020).
Figure 6. SpEDIT allows seamless insertion, deletion and tagging at S. pombe loci.

For sgRNA and HR template sequences see Table 1. A. 4xtetO binding sites were inserted downstream of cup1 +. Sanger sequencing chromatograms covering the insert junctions are shown for a successfully edited clone. PCR primers (half arrows) flanking the insert were used to amplify products from wild-type (wt) and edited strains. B. Cup1 was C-terminally tagged with a green fluorescent protein (GFP). Sanger sequencing chromatogram covering the gene-tag junction is shown for a successfully edited clone. Western blot using anti-GFP antibody was performed on wild-type (wt) and edited strains. C. The coding sequence of epe1 + was deleted. Sanger sequencing chromatogram covering the deletion junction is shown for a successfully edited clone. PCR primers (half arrows) flanking the deletion (and within the epe1 + coding sequence as control) were used to amplify products from wild-type (wt) and edited strains. D. Epe1 was N-terminally tagged with three FLAG epitopes. Sanger sequencing chromatogram covering the gene-tag junction is shown for a successfully edited clone. Western blot using anti-FLAG antibody was performed on wild-type (wt) and edited strains.
Table 1. sgRNA and HR templates used in this study.
All sgRNA sequences used were obtained using CRISPR4P ( Rodríguez-López et al., 2016).
| Name | Sequence |
|---|---|
| ade6 - sgRNA | TTGATAGCAACAGTGGCGAC |
| ura4 - sgRNA | CCTTGTATAATACCCTCGCC |
| meu27 - sgRNA | TATTAGCCTTTGAAGGATTT |
| clr5 - sgRNA | AGCTTGTGGCTGACCGTTAA |
| cup1:4xtetO - sgRNA | ATTTCTTTTGCTTTACGGTC |
| cup1-GFP - sgRNA | GCTCAGGCTAAACGTCGGAA |
| epe1 - sgRNA | GGACTTTTAAGATGGATTCC |
| ade6 - HR template | GCAATGACACCTCTTCCAGTAATCGGCGTTCCTGTAAAAGGAAGCACTCTTGACGGAGTTGACTCTCTTTAGTCTATTGTTCA
GATGCCTCGAGGTGTCTAGGTCGCCACTGTTGCTATCAATAATAGCCAAAATGCCGGTATTTTAGCCTGTCGTATACTTGCTACA TTTCAACCCTCC |
| ura4 - HR template | TTGGAAGACATTTCAGCCAAAAGCAAGAGACCACGTCCCAAAGGTAAACCAACTTCTTTGAGGCCTTGTATAATACCCTCGCCC
TACACTGTATGGCAATTTGTGATATGAGCCCAAGACTAAATTTTGTACACACCAGATGCATATTGTAGCTTGACGGTATTTCCAAT GTCTGCGAAT |
| meu27 - HR template | GCCAAAATCAATAGAGAACAATTATACTTTAAAAAAAAAAAATGAAGAAGGCTTCTTAAGTCAACAGGAAAATAAGTATTCAAATC
AAAATCCTTCAAAGGCTAATAACCTTTTCGACAAGCTCGGCGTAAGCAAGCCAATAACATTCATAGCTTGTATACTGCAATTGGGC GGCACCTC |
| clr5 - HR template | CTTATTTGCAGCAGCCTTTCCAAATACCCTCTCAACGTTTCTCTCGACAGCAACAATCTCATCCATTCCCTGCTGCTCAACATGCA
GTTAACGGTTAGCCACAAGCTTTGTATCCTTTCATCTACCAATCTAGAAATGTCCCAATGGGCTCCACCATGTTTGCTTCTTCAAA CCAATCTG |
|
cup1:4xtetO - HR
template |
TTGAATTAATTCATAGAGTATGATAAAAATTGATAGTAAATTCATTGGTATACTAAAGTGATGTAGAAAATTAAGAAATCACATAGAC
TACTTGAGTTACGATGATTTATTAGCATGCATcaaggcctactagtgcatgcatcgatagatcctctatcactgatagggagatctccctatcagtgata gagaggatctccctatcagtgatagagaggatctccctatcagtgatagagaggatctccctatcagtgatagagaggatcctctctatcactgataggg agatcttcaacttggtggtgaggtaaacgaaatccagTAAATATTTATGAAAAAAAAAATAAATGATTCATAACAAGCAGATGAAAATGATGA CGAATTAGGACTCTTCAAAATAAATGAAGATTATACATTACAAA |
| cup1-GFP - HR
template |
ATGACGAATTAGGACTCTTCAAAATAAATGAAGATTATACATTACAAACTTTGGTCTGACTTTTTAAAGCACACGATTTGctatttgta
tagttcatccatgccatgtgtaatcccagcagctgttacaaactcaagaaggaccatgtggtctctcttttcgttgggatctttcgaaagggcagattgt gtggacaggtaatggttgtctggtaaaaggacagggccatcgccaattggagtattttgttgataatggtctgctagttgaacgcttccatcttcaatgt tgtgtctaattttgaagttaactttgattccattcttttgtttgtctgccatgatgtatacattgtgtgagttatagttgtattccaatttgtgtccaag aatgtttccatcttctttaaaatcaataccttttaactcgattctattaacaagggtatcaccttcaaacttgacttcagcacgtgtcttgtagttcccg tcatctttgaaaaatatagttctttcctgtacataaccttcgggcatggcactcttgaaaaagtcatgccgtttcatatgatctgggtatcttgaaaagc attgaacaccataagtgaaagtagtgacaagtgttggccatggaacaggtagttttccagtagtgcaaataaatttaagggtaagttttccgtatgttgc atcaccttcaccctctccactgacagaaaatttgtgcccattaacatcaccatctaattcaacaagaattgggacaactccagtgaaaagttcttctcct ttactCCTTTTTTTGACTGACTTTTTAAGAACCTTTCCGACGTTTAGCCTGAGCTGAATGTCTGACAAAGAGTCCCACGATACAA |
| epe1Δ - HR template | GTGAACTACTCAAGAATCATAAGCACGTGGGGATAAATATTCAATGGTAGCCGAAGGAAATAAAAAGTGCCGAGGTACTTCTTA
AAAGTCCCAAAAATTA |
|
3xFLAG-epe1 - HR
template |
GAATATCAATGTCTTGATTTATAATGTCATCGTATTCAAGCCAGGAATCGCTGCCTCCTCCCTTGTCATCGTCATCCTTGTAGTCG
ATGTCATGATCTTTATAATCACCGTCATGGTCTTTGTAGTCCATCTTAAAAGTCCCAAAAATTAATTGCTTACTAGCAAAAAGGTAA CTATAAA |
Epe1 is a putative histone demethylase that acts to prevent heterochromatin island formation ( Sorida et al., 2019; Wang et al., 2015; Zofall et al., 2012). Using SpEDIT, we deleted the epe1 + coding sequence ( Figure 6C). The epe1Δ HR template employed contained 80 bp arms homologous to sequences immediately flanking the epe1 + coding sequence as previously used for the deletion of other sequences ( Rodríguez-López et al., 2016). Correct deletion of the epe1 + coding sequence results in loss of the sgRNA target and PAM sequences. In addition, we seamlessly inserted a sequence to encode a 3xFLAG epitope tag between the epe1 + gene promoter and the 5’ end of the epe1 + coding sequence to allow the production of N-terminally 3xFLAG-tagged Epe1 without any associated selectable marker ( Figure 6D). To accomplish this, an in-frame epe1-3xflag-epe1 HR template containing 50 bp arms homologous to the sequence immediately flanking the epe1 + start codon was used. Correct insertion of the epe1-3xflag-epe1 sequence resulted in loss of both the sgRNA target and the PAM sequence. The resulting epe1Δ and 3xFLAG-Epe1 strains were recently utilised to study the role of Epe1 in ectopic heterochromatin island formation following caffeine exposure ( Torres-Garcia et al., 2020). Notably, whole-genome sequencing of the cup1:4xtetO and epe1Δ strains revealed no additional genetic changes (SNPs or indels) in coding regions of the genome ( Torres-Garcia et al., 2020).
In the four distinct genome editing scenarios described above, a maximum of eight primary transformants needed to be screened to obtain at least one that exhibited the desired sequence change. We therefore conclude that SpEDIT markedly speeds up the process of generating accurate insertion, deletion and tagging events at a variety of S. pombe loci.
Discussion
Here we report the development of SpEDIT, an optimized CRISPR/Cas9 editing system and method for the fission yeast, S. pombe ( Figure 1 and Figure 2). SpEDIT makes use of Cas9 codon-optimised for expression in S. pombe that, coupled with the incorporation of a tDNA Ser/HDV ribozyme sgRNA expression cassette ( Ryan et al., 2014), achieves 100% efficiency in generating mutations at targeted ade6 + or ura4 + genes in asynchronous cells ( Figure 3). A high mutagenesis efficiency was also obtained with the pre-existing pMZ379 system in asynchronous cells ( Jacobs et al., 2014; Rodríguez-López et al., 2016). However, SpEDIT displayed reduced toxicity by removing the detrimental physiological effects associated with high humanised Cas9 endonuclease expression and consequently speeds up the genome editing process ( Figure 4). In addition, our analysis indicates that the use of G1-synchronized cell cultures for CRISPR/Cas9-mediated genome manipulation reduces the efficiency of targeted mutagenesis, with both the SpEDIT and pMZ379 systems, relative to asynchronous cultures ( Figure 3). G1 synchronization therefore represents an unnecessary time-consuming step in the genome editing process.
SpEDIT can be used to introduce simultaneous mutations at two non-homologous genes at distinct locations in the S. pombe genome ( Figure 5), and allows flexible engineering of seamless insertion, deletion and tagging events at S. pombe loci in the absence of linked selectable markers and without observed off-target sequence changes ( Figure 6). It is worth noting that many traditional S. pombe transformation protocols involve the use of carrier DNA. We advise against the use of carrier DNA as it has been shown to insert at many locations in resulting transformants, causing unplanned off-target mutations ( Longmuir et al., 2019).
Besides achieving high mutagenesis efficiency, the greatest advance of SpEDIT is a very simple cloning protocol allowing sgRNA target sequences to be inserted with minimal effort into the Cas9-bearing pLSB plasmid through a one-step Golden Gate reaction. Candidate sgRNA-bearing clones are easily visualized by loss of the GFP placeholder, negating the need for repetitive screening of numerous E. coli colonies by laborious plasmid purification and inspection.
Recently it was reported that homology arms of as short as 25 bp flanking each side of a cleavage site can be used to successfully introduce point mutations and epitope tags at S. pombe loci ( Hayashi & Tanaka, 2019). Further analyses will be required to determine whether HR templates with such short homology arms are as efficient as longer arms when combined with SpEDIT. In addition, the tDNA/HDV ribozyme sgRNA expression cassette that was originally developed for S. cerevisiae has been used to express up to three tandem HDV-sgRNAs from a single tDNA RNAPIII promoter with 80% mutagenesis efficiency ( Ryan et al., 2014). This suggests that a similar approach could be used with SpEDIT to simultaneously express multiple different sgRNAs that target a single locus or many distinct loci.
In summary, the combination of the CRISPR4P algorithm ( Rodríguez-López et al., 2016), that conveniently aids the identification of suitable sgRNAs across the S. pombe genome, with SpEDIT, which provides a straightforward and user-friendly experimental method, markedly enhances the capabilities of CRISPR/Cas9-mediated genome editing in S. pombe. We anticipate SpEDIT will permit the broad application of genome editing procedures to fission yeast in order to explore diverse biological questions in this model fungal system.
Methods
pLSB construction
For pLSB-NAT construction, the Cas9 gene and the strong adh1 promoter present in pMZ379 (plasmid generated by Mikel Zaratiegui and provided by Jürg Bähler ( Rodríguez-López et al., 2016)), were replaced by a Cas9 gene codon-optimised for S. pombe (custom synthesised, Gen9) and the medium strength adh15 promoter (from pRAD15, gift from Yoshi Watanabe), respectively, via Gibson Assembly (New England Biolabs, Cat# E2611). Next, the rrk1/HHR sgRNA cassette present in pMZ379 was replaced by the tDNA/HDV sgRNA cassette (custom synthesised, Gen9) via Gibson Assembly (New England Biolabs, Cat# E2611). BsaI sites flanking the GFP placeholder in the tDNA/HDV sgRNA cassette were then introduced using Q5 Site-Directed Mutagenesis Kit (New England Biolabs, Cat# E0554).
To construct pLSB versions with kanMX6 (pLSB-Kan, G418 resistance) or hphMX6 (pLSB-Hyg, hygromycin resistance) selectable markers, the natMX6 gene from pLSB-NAT was replaced by the kanMX6 or hphMX6 genes from pFA6a-kanMX6 ( Bähler et al., 1998) or pFA6a-hphMX6 ( Hentges et al., 2005) plasmids, respectively, by Gibson Assembly (New England Biolabs, Cat# E2611).
sgRNA and HR template sequences can be found in Table 1. All sgRNA sequences used were obtained using CRISPR4P ( Rodríguez-López et al., 2016). Oligonucleotide sequences are listed in Table 2.
Table 2. Oligonucleotides used in this study.
| Name | Sequence |
|---|---|
| Making ade6 – sgRNA - F | CtagaGGTCTCgGACTTTGATAGCAACAGTGGCGACGTTTcGAGACCcttCC |
| Making ade6 – sgRNA - R | GGaagGGTCTCgAAACGTCGCCACTGTTGCTATCAAAGTCcGAGACCtctaG |
| Making ura4 – sgRNA - F | CtagaGGTCTCgGACTCCTTGTATAATACCCTCGCCGTTTcGAGACCcttCC |
| Making ura4 – sgRNA - R | GGaagGGTCTCgAAACGGCGAGGGTATTATACAAGGAGTCcGAGACCtctaG |
| Making meu27 - sgRNA - F | CtagaGGTCTCgGACTTATTAGCCTTTGAAGGATTTGTTTcGAGACCcttCC |
| Making meu27 - sgRNA - R | GGaagGGTCTCgAAACAAATCCTTCAAAGGCTAATAAGTCcGAGACCtctaG |
| Making clr5 – sgRNA - F | CtagaGGTCTCgGACTAGCTTGTGGCTGACCGTTAAGTTTcGAGACCcttCC |
| Making clr5 – sgRNA - R | GGaagGGTCTCgAAACTTAACGGTCAGCCACAAGCTAGTCcGAGACCtctaG |
| Making cup1:4xtetO – sgRNA - F | CtagaGGTCTCgGACTATTTCTTTTGCTTTACGGTCGTTTcGAGACCcttCC |
| Making cup1:4xtetO – sgRNA - R | GGaagGGTCTCgAAACGACCGTAAAGCAAAAGAAATAGTCcGAGACCtctaG |
| Making cup1-GFP – sgRNA - F | CTAGAGGTCTCGGACTGCTCAGGCTAAACGTCGGAAGTTTCGAGACCCTTCC |
| Making cup1-GFP – sgRNA - R | GGAAGGGTCTCGAAACTTCCGACGTTTAGCCTGAGCAGTCCGAGACCTCTAG |
| Making epe1 – sgRNA - F | CtagaGGTCTCgGACTGGACTTTTAAGATGGATTCCGTTTcGAGACCcttCC |
| Making epe1 – sgRNA - R | GGaagGGTCTCgAAACGGAATCCATCTTAAAAGTCCAGTCcGAGACCtctaG |
| Making ade6 - HR template - F | GCAATGACACCTCTTCCAGTAATCGGCG TTCCTGTAAAAGGAAGCACTCTTGACGG AGTTGACTCTCTTTA
GTCTATTGTTCAGA TGCCTCGAGGTGTCT |
| Making ade6 - HR template - R | GGAGGGTTGAAATGTAGCAAGTATACGA CAGGCTAAAATACCGGCATTTTGGCTATT TATTGATAGCAACA
GTGGCGACCTAGACA CCTCGAGGCATCTGA |
| Making ura4 - HR template - F | TTGGAAGACATTTCAGCCAAAAGCAAGA GACCACGTCCCAAAGGTAAACCAACTTC TTTGAGGCCTTGTAT
AATACCCTCGCCCT ACACTGTATGGCAAT |
| Making ura4 - HR template - R | ATTCGCAGACATTGGAAATACCGTCAAG CTACAATATGCATCTGGTGTGTACAAAAT TTAGTCTTGGGCTCA
TATCACAAATTGCC ATACAGTGTAGGGC |
| Making meu27 - HR template - F | GCCAAAATCAATAGAGAACAATTATACTTTAAAAAAAAAAAATGAAGAAGGCTTCTTAAGTCAACAGGAAAAT
AAGTATTCAAATCAAAATCCTTCAAAG |
| Making meu27 - HR template - R | GAGGTGCCGCCCAATTGCAGTATACAAGCTATGAATGTTATTGGCTTGCTTACGCCGAGCTTGTCGAAAAGG
TTATTAGCCTTTGAAGGATTTTGATTTG |
| Making clr5 - HR template - F | CTTATTTGCAGCAGCCTTTCCAAATACCCTCTCAACGTTTCTCTCGACAGCAACAATCTCATCCATTCCCTGCT
GCTCAACATGCAGTTAACGGTTAGCC |
| Making clr5 - HR template - R | CAGATTGGTTTGAAGAAGCAAACATGGTGGAGCCCATTGGGACATTTCTAGATTGGTAGATGAAAGGATAC
AAAGCTTGTGGCTAACCGTTAACTGCATG |
| Making
cup1:4xtetO - HR template
- 1 - F |
TTGAATTAATTCATAGAGTATGATAAAAATTGATAGTAAATTCATTGG |
| Making
cup1:4xtetO - HR template
- 1 - R |
cactagtaggccttgATGCATGCTAATAAATCATCGTAACTCAAGTAG |
| Making
cup1:4xtetO - HR template
- 2 - F |
TTTATTAGCATGCATcaaggcctactagtgcatgca |
| Making
cup1:4xtetO - HR template
- 2 - R |
TTTTTTTTTTCATAAATATTTActggatttcgtttacctcaccacc |
| Making
cup1:4xtetO - HR template
- 3 - F |
tggtgaggtaaacgaaatccagTAAATATTTATGAAAAAAAAAATAAATGATTCATAACAAGCAGATGAAAA |
| Making
cup1:4xtetO - HR template
- 3 - R |
TTTGTAATGTATAATCTTCATTTATTTTGAAGAGTCCTAATTCGT |
| Making cup1-GFP - HR template - F | ATGACGAATTAGGACTCTTCAAAATAAATGAAGATTATACATTACAAACTTTGGTCTGACTTTTTAAAGCACAC
GATTTGCTATTTGTATAGTTCATCCA |
| Making cup1-GFP - HR template - R | TTGTATCGTGGGACTCTTTGTCAGACATTCAGCTCAGGCTAAACGTCGGAAAAGTTCTTAAAAAGTCAGTCA
AAAAAAGGAGTAAAGGAGAAGAACTTTT |
| Making epe1Δ - HR template - F | GTGAACTACTCAAGAATCATAAGCACGTGGGGATAAATATTCAATGGTAGCCGAAGGAAATAAAAAGTGCCG
AGGTACTTCTTAAAAGTCCCAAAAATTA |
| Making epe1Δ - HR template - R | CCATAGAATCTCCTTAGTTTGCATCGCAATTTTATAGTTACCTTTTTGCTAGTAAGCAATTAATTTTTGGGACTT
TTAAGAAGTACCTCGGCACTTTTTA |
| Making 3xFLAG-epe1 - HR template - F | TTTATAGTTACCTTTTTGCTAGTAAGCAATTAATTTTTGGGACTTTTAAGATGGACTACAAAGACCATGACGGT
GATTATAAAGATCATGACATCGACTA |
| Making 3xFLAG-epe1 - HR template - R | GAATATCAATGTCTTGATTTATAATGTCATCGTATTCAAGCCAGGAATCGCTGCCTCCTCCCTTGTCATCGTCAT
CCTTGTAGTCGATGTCATGATCTTT |
| Checking mutations ade6 - F | TTGTTTCAGCTCACCGCACA |
| Checking mutations ade6 - R | AAAGCAAGCAAAATCATTTAACAGT |
| Checking mutations ura4 - F | GCTCCATAGACTCCACGACC |
| Checking mutations ura4 - R | TTGTCAGTCGCGGTCGATTT |
| Checking mutations meu27 - F | AAATTTGCGCTCCTCTCTGC |
| Checking mutations meu27 - R | GTTTGGTATTTACGAGCTGCCA |
| Checking mutations clr5 - F | CACACAATGCGCACTCTTCT |
| Checking mutations clr5 - R | ACAGCAGTTGGTCCGTTAGA |
| Checking cup1:4xtetO – F (A1) | GGTTAGGCAGAAGACTTGAGCA |
| Checking cup1:4xtetO – R (A2) | ATCATCACTTGCATTCACTTCTCT |
| Checking cup1-GFP - F | GGCGAAGCTTTTAAGTCTGAAGG |
| Checking cup1-GFP - R | GCTGTCCCACTCTTACCACA |
| Checking epe1Δ – F (C1) | CAAATCTAACGAGTTTGCCTGC |
| Checking epe1Δ – R (C3) | GCAAACAACGAGTCAAAGTGGA |
| Checking epe1 cds – F (C2) | GGGCGAGCGGACAATCATAA |
| Checking 3xFLAG-epe1 - F | AGTGAGGCTGTGCAAAGGAA |
| Checking 3xFLAG-epe1 - R | TCTAACGAGTTTGCCTGCTT |
| M13F | GTAAAACGACGGCCAGT |
Yeast strains and manipulations
Standard methods were used for fission yeast growth, genetics and manipulation ( Moreno et al., 1991). S. pombe strains generated in this study are described in Table 3. 972 h - wild-type cells were used for all experiments.
Table 3. Schizosaccharomyces pombe strains used in this study.
| Strain number | Name | Description |
|---|---|---|
| 143 | wt | h- ED972 wild-type |
| B4752 | Clr5-Q264STOP Meu27-S100Y | h- clr5-Q264STOP meu27-S100Y |
| B3808 | cup1:4xtetO | h- 4xtetO 3' of cup1 leu1-32 (cup1=SPBC17G9.13c) |
| B4567 | Cup1-GFP | h- cup1-GFP (cup1=SPBC17G9.13c) |
| B4621 | epe1Δ | h- epe1Δ |
| B4958 | 3xFLAG-Epe1 | h- 3xFLAG-epe1 |
Competent cryopreserved G1-synchronized S. pombe cells were prepared as described in Rodríguez-López et al. (2016). Wild-type cells were grown to a concentration of 1×10 7 cells per mL in EMM media and harvested at 3500 x g for 2 min. Cells were then resuspended in EMM-N and incubated for 2 hrs at 25°C until cells were of a small round morphology. Following three washes with ice-cold H 2O, cells were resuspended in 2 mL of ice-cold 30% glycerol, 0.1 M lithium acetate (pH 4.9) and 50 μL aliquots were made and stored at -80°C until further usage.
Competent cryopreserved G1-synchronized S. pombe cells were transformed using the lithium acetate/PEG method ( Sabatinos & Forsburg, 2010), as described in Rodríguez-López et al., 2016. Aliquots of cryopreserved, G1-synchronised cells were thawed at 40°C for 2 min. 200 ng of empty or sgRNA-containing pLSB or pMZ379 plasmid DNA, 250–1000 ng of HR template DNA (when applicable) and 145 μL of 50% PEG 4000 were added to the cells and mixed thoroughly. Note that herring sperm DNA was omitted due to concerns regarding the erroneous integration of these fragments at the targeted loci as described in Longmuir et al. (2019). Cells were then resuspended in 1 mL of EMM-N and incubated for 16 hrs at room temperature. Following incubation, cells were resuspended in 500 μL H 2O and plated onto YES plus cloNAT media. Plates were incubated at 32°C for 3–5 days.
Asynchronous cultures of S. pombe cells were transformed by electroporation. 50 mL cultures were grown to log phase (5×10 6 to 1×10 7 cells per mL) in YES media and harvested at 3500 x g for 2 min. Cells were resuspended in 5 mL of pre-transformation buffer (25 mM DTT, 0.6 M sorbitol, 20 mM HEPES, pH 7.6) and incubated at 32°C for 10 min. Cells were then washed three times in 10 mL of 1.2 M ice-cold sorbitol and resuspended in 500 μL of 1.2 M ice-cold sorbitol. 200 μL cells were added to 200 ng of empty or sgRNA-loaded pLSB or pMZ379 plasmid DNA and 250–1000 ng of HR template DNA (when applicable) in an ice-cold electroporation cuvette. Cells were pulsed using a Gene Pulser II electroporation system (Bio-Rad) with S. pombe settings: 2.25 kV, 200 Ω and 25 μF. Immediately following pulse, cells were rapidly mixed with 500 μL of 1.2 M ice-cold sorbitol. Cells were grown overnight in 10 mL of non-selective YES medium before plating on selection (YES plus cloNAT, YES plus G418 or YES plus hygromycin depending on pLSB version used). Plates were incubated at 32°C for 3-5 days. cloNAT, nourseothricin.
To assess colony area of pLSB- or pMZ379-harbouring cloNAT-resistant colonies, plates were scanned after four days of incubation. Images were then analysed using ImageJ (v1.51) (analyse particles) with default settings.
Assessing mutations at the ade6 and ura4 loci
Colonies harbouring mutations at the target genes ade6 + or ura4 + were identified through a replica-plating assay. cloNAT-resistant colonies were individually picked from YES plus cloNAT plates, re-streaked onto YES plates without selection and incubated at 32°C for two days. Isolates were then replica-plated onto the following plates: YES, YES 1/10 adenine (to examine ade6 + mutations), PMG minus uracil and PMG plus 5-fluoroorotic acid (to examine ura4 + mutations). Plates were incubated at 32°C for 2–4 days and then visually examined.
Colony PCR and Sanger sequencing
A small amount of fission yeast cells (~1 × 10 4) from individual single colonies was incubated in 10 μL of SPZ buffer (1.2 M sorbitol, 100 mM sodium phosphate, 2.5 mg/mL Zymolyase 100T (AMS Biotechnology)) at 37°C for approximately 1 hr. 40 μL H 2O was added to the cells and 5 μL of the extract was used as a PCR template. PCRs were performed using 0.5U Taq Polymerase (Roche, Cat# 11147633103), 1 μM oligonucleotides, and the following conditions on a standard thermocycler: 94°C for 4 min; 29 cycles of 94°C for 30 sec, 55°C for 30 sec and 72°C for 1 min; and a final step of 72°C for 5 min. PCR products were analysed on a 1% agarose gel, purified using Monarch PCR Cleanup Kit (New England Biolabs, Cat# T1030) and sequenced using BigDye Terminator Cycle sequencing kit (Thermo Scientific, Cat# 4337458).
Protein extraction and western analysis
Protein samples were prepared as previously described ( Torres-Garcia et al., 2020). Western blotting detection was performed using mouse monoclonal anti-FLAG-HRP (Sigma, Cat# A8591, RRID:AB_439702), rabbit polyclonal anti-GFP (Invitrogen, Cat# A11122, RRID:AB_2307355) and goat polyclonal anti-rabbit-HRP (Sigma, Cat# A6154, RRID:AB_258284).
SpEDIT protocol
A convenient protocol card of this procedure can be found by visiting allshirelab.com/spedit or by scanning the QR code in Figure 1.
Before you begin
● Download required DNA sequences at allshirelab.com/spedit , on Zenodo ( Torres-Garcia, 2020) or by scanning the QR code in Figure 1
Required reagents
● pLSB vector (75 ng/μL) – Available on request
● NEB Golden Gate Assembly Kit ( BsaI-HF v2) – NEB #E1601S
● sgRNA fragment for Golden Gate assembly (1 ng/μL) – See below for design and preparation
● HR template (250-1000 ng) – See below for design and preparation
sgRNA design
● Find a suitable sgRNA targeting the gene of interest using CRISPR4P ( Rodríguez-López et al., 2016)
● Copy the 20 bp sgRNA sequence in place of ‘your 20 bp targeting RNA’ in ‘GG_sgRNA_template.dna’ file
● Order 52-nt forward and reverse sgRNA oligonucleotides as indicated in the ‘GG_sgRNA_template.dna’ file
sgRNA fragment preparation
● Anneal sgRNA oligonucleotides
-
○
Mix 5 μL of 100 μM forward and reverse sgRNA oligonucleotides
-
○
Heat to 95°C for 3 min and cool down to room temperature slowly (e.g. -1°C/30 sec)
-
○
Add 1 μL of annealed mix to 1.5 mL of H 2O. This dilution corresponds to approximately 1 ng/μL annealed sgRNA fragment.
Golden Gate reaction
● Mix the following components in a PCR tube:
-
○
pLSB plasmid (75 ng/μL) – 0.5 μL
-
○
Annealed sgRNA fragment (1 ng/μL) – 0.5 μL
-
○
T4 DNA Ligase Buffer (10x) – 1 μL
-
○
NEB Golden Gate Assembly Mix ( BsaI-HF v2) – 0.5 μL
-
○
H 2O – 7.5 μL
● Incubate at 37°C for 1 hr
● Incubate at 60°C for 5 min
● Transform into Escherichia coli by heat shock:
-
○
1 μL of Golden Gate reaction to 25 μL of DH5-alpha or 10-Beta E. coli cells
-
○
Place the mixture on ice for 30 min
-
○
Heat shock at exactly 42°C for exactly 30 sec
-
○
Place on ice for 5 min
-
○
Add 475 μL SOC media and recover cells at 37°C for 1 hr
-
○
Plate 200 μL / 100 μL / 50 μL / rest on LB plus ampicillin
● Select four E. coli colonies and set up liquid cultures in LB plus ampicillin
● Isolate plasmid (miniprep)
-
○
IMPORTANT: do not miniprep culture if green – these are GFP-containing clones where the Golden Gate reaction did not occur
-
○
One miniprep (approximately 200 ng/μL) should be sufficient for many S. pombe transformations (200 ng/transformation)
Plasmid check
● Digest plasmid using NcoI
-
○
This digest allows sgRNA-containing plasmids to be distinguished from those containing GFP
● Sequence sgRNA-containing plasmids using M13F oligonucleotide (see Table 2) to confirm sgRNA insertion
HR template design and preparation
● The HR template should contain:
-
○
Your desired mutation: point mutation, insertion, deletion or tag
-
○
At least 80 bp homology on each side relative to the cleavage site (3 bp upstream of PAM sequence (NGG))
-
○
A mutation that disrupts the PAM sequence to avoid repeated DSBs
-
▪
If the total size of the HR template is equal or smaller than 180 bp, we recommend generating HR templates by PCR using oligonucleotides with overlapping at their 3’ end as described in Rodríguez-López et al., 2016
-
▪
If the total size of the HR template is larger than 180 bp, we recommend using a fusion PCR construct containing homology arms to the target site (see HR template for cup1:4xtetO in Table 1)
-
▪
S. pombe transformation
● Transform S. pombe cells using your preferred method with sgRNA-loaded pLSB plasmid (200 ng) and HR template (500-1000 ng)
● Grow non-selectively o/n on YES plates or liquid
● Plate on YES plus cloNAT (or YES plus G418 or YES plus hygromycin depending on pLSB version used)
● Re-streak transformant colonies to non-selective media to allow loss of plasmid
● Amplify region of interest by colony PCR and sequence amplicon to confirm mutation
Data availability
Underlying data
Whole-genome sequencing data for relevant strains are available on GEO, Accession number GSE138436: https://identifiers.org/geo:GSE138436 ( Torres-Garcia et al., 2020).
Zenodo: SpEDIT: A fast and efficient CRISPR/Cas9 method for fission yeast.
https://doi.org/10.5281/zenodo.4247568 ( Torres-Garcia, 2020)
This project contains the following underlying data:
-
-
Fig_3C.csv (Raw mutagenesis efficiency data underlying Figure 3C)
-
-
Fig_3D.csv (Raw mutagenesis efficiency data underlying Figure 3D)
-
-
Fig_4A.csv (Raw colony area measurements underlying Figure 4A)
-
-
Fig_4B.csv (Raw colony area measurements underlying Figure 4B)
-
-
F6_uncropped.pdf (Original, uncropped PCR gel and western blot images from Figure 6)
Extended data
Zenodo: SpEDIT: A fast and efficient CRISPR/Cas9 method for fission yeast.
https://doi.org/10.5281/zenodo.4247568 ( Torres-Garcia, 2020)
This project contains the following extended data:
-
-
GG_sgRNA_template.dna (sgRNA sequence)
-
-
pLSB_Allshire.dna (pLSB plasmid sequence)
Data are available under the terms of the Creative Commons Zero "No rights reserved" data waiver (CC0 1.0 Public domain dedication).
Acknowledgements
We thank Gabor Varga for technical assistance. We thank members of the Allshire lab for valuable discussions.
Funding Statement
S. T-G. is supported by the Darwin Trust of Edinburgh. S. T-G., L. D. P., L. E., and B. G. were supported by Erasmus+ traineeships. I. Y. is supported by an EMBO Long Term Fellowship (EMBO ALTF 130-2018). R. C. A. is a Wellcome Principal Research Fellow (095021, 200885); the Wellcome Centre for Cell Biology is supported by core funding from Wellcome (203149).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
[version 1; peer review: 3 approved]
References
- Audergon PNCB, Catania S, Kagansky A, et al. : Epigenetics. Restricted epigenetic inheritance of H3K9 methylation. Science. 2015;348(6230):132–135. 10.1126/science.1260638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bähler J, Wu JQ, Longtine MS, et al. : Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 1998;14(10):943–951. [DOI] [PubMed] [Google Scholar]
- Ciccaglione KM: Adaptation of CRISPR/Cas9 to improve the experimental utility of the model system Schizosaccharomyces pombe. Master’s thesis Rutgers University.2015. 10.7282/T3N58P92 [DOI] [Google Scholar]
- Cong L, Ran FA, Cox D, et al. : Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. 10.1126/science.1231143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantes PA, Hoffman CS: A Brief History of Schizosaccharomyces pombe Research: A Perspective Over the Past 70 Years. Genetics. 2016;203(2):621–629. 10.1534/genetics.116.189407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez R, Berro J: Use of a fluoride channel as a new selection marker for fission yeast plasmids and application to fast genome editing with CRISPR/Cas9. Yeast. 2016;33(10):549–557. 10.1002/yea.3178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi A, Tanaka K: Short-Homology-Mediated CRISPR/Cas9-Based Method for Genome Editing in Fission Yeast. G3 (Bethesda). 2019;9(4):1153–1163. 10.1534/g3.118.200976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentges P, Van Driessche B, Tafforeau L, et al. : Three novel antibiotic marker cassettes for gene disruption and marker switching in Schizosaccharomyces pombe. Yeast. 2005;22(13):1013–1019. 10.1002/yea.1291 [DOI] [PubMed] [Google Scholar]
- Hottinger-Werlen A, Schaack J, Lapointe J, et al. : Dimeric tRNA gene arrangement in Schizosaccharomyces pombe allows increased expression of the downstream gene. Nucleic Acids Res. 1985;13(24):8739–8747. 10.1093/nar/13.24.8739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, Zhang F: Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157(6):1262–1278. 10.1016/j.cell.2014.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs JZ, Ciccaglione KM, Tournier V, et al. : Implementation of the CRISPR-Cas9 system in fission yeast. Nat Commun. 2014;5:5344. 10.1038/ncomms6344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, et al. : A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–821. 10.1126/science.1225829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock A, Rutherford K, Harris MA, et al. : PomBase 2018: user-driven reimplementation of the fission yeast database provides rapid and intuitive access to diverse, interconnected information. Nucleic Acids Res. 2019;47(D1):D821–D827. 10.1093/nar/gky961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longmuir S, Akhtar N, MacNeill SA: Unexpected insertion of carrier DNA sequences into the fission yeast genome during CRISPR-Cas9 mediated gene deletion. BMC Res Notes. 2019;12(1):191. 10.1186/s13104-019-4228-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata J, Lyne, R, Burns G, et al. : The transcriptional program of meiosis and sporulation in fission yeast. Nat Genet. 2002;32(1):143–147. 10.1038/ng951 [DOI] [PubMed] [Google Scholar]
- Moreno S, Klar A, Nurse P, et al. : Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 1991;194:795–823. 10.1016/0076-6879(91)94059-l [DOI] [PubMed] [Google Scholar]
- Orthwein A, Noordermeer SM, Wilson MD, et al. : A mechanism for the suppression of homologous recombination in G1 cells. Nature. 2015;528(7582):422–426. 10.1038/nature16142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragunathan K, Jih G, Moazed D, et al. : Epigenetics. Epigenetic inheritance uncoupled from sequence-specific recruitment. Science. 2015;348(6230):1258699. 10.1126/science.1258699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-López M, Cotobal C, Fernández-Sánchez O, et al. : A CRISPR/Cas9-based method and primer design tool for seamless genome editing in fission yeast [version 3; peer review: 2 approved]. Wellcome Open Res. 2016;1:19. 10.12688/wellcomeopenres.10038.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan OW, Skerker JM, Maurer MJ, et al. : Selection of chromosomal DNA libraries using a multiplex CRISPR system. eLife. 2014;3:e03703. 10.7554/eLife.03703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatinos SA, Forsburg SL: Molecular genetics of Schizosaccharomyces pombe. Methods Enzymol. 2010;470:759–795. 10.1016/S0076-6879(10)70032-X [DOI] [PubMed] [Google Scholar]
- Sorida M, Hirauchi T, Ishizaki H, et al. : Regulation of ectopic heterochromatin-mediated epigenetic diversification by the JmjC family protein Epe1. PLoS Genet. 2019;15(6):e1008129. 10.1371/journal.pgen.1008129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Garcia S, Yaseen I, Shukla M, et al. : Epigenetic gene silencing by heterochromatin primes fungal resistance. Nature. 2020;585(7825):453–458. 10.1038/s41586-020-2706-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Garcia S: SpEDIT: A fast and efficient CRISPR/Cas9 method for fission yeast [Data set]. Zenodo. 2020. 10.5281/zenodo.4247568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trickey M, Grimaldi M, Yamano H, et al. : The anaphase-promoting complex/cyclosome controls repair and recombination by ubiquitylating Rhp54 in fission yeast. Mol Cell Biol. 2008;28(12):3905–3916. 10.1128/MCB.02116-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Reddy BD, Jia S, et al. : Rapid epigenetic adaptation to uncontrolled heterochromatin spreading. eLife. 2015;4:e06179. 10.7554/eLife.06179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi Y, Sakuno T, Shimura M, et al. : Heterochromatin links to centromeric protection by recruiting shugoshin. Nature. 2008;455(7210):251–255. 10.1038/nature07217 [DOI] [PubMed] [Google Scholar]
- Zhang XR, He JB, Wang YZ, et al. : A Cloning-Free Method for CRISPR/Cas9-Mediated Genome Editing in Fission Yeast. G3 (Bethesda). 2018;8(6):2067–2077. 10.1534/g3.118.200164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Boeke JD: Construction of Designer Selectable Marker Deletions with a CRISPR-Cas9 Toolbox in Schizosaccharomyces pombe and New Design of Common Entry Vectors. G3 (Bethesda). 2018;8(3):789–796. 10.1534/g3.117.300363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zofall M, Yamanaka S, Reyes-Turcu FE, et al. : RNA elimination machinery targeting meiotic mRNAs promotes facultative heterochromatin formation. Science. 2012;335(6064):96–100. 10.1126/science.1211651 [DOI] [PMC free article] [PubMed] [Google Scholar]