
Figure 1: Molecular Mechanisms of the UPR.
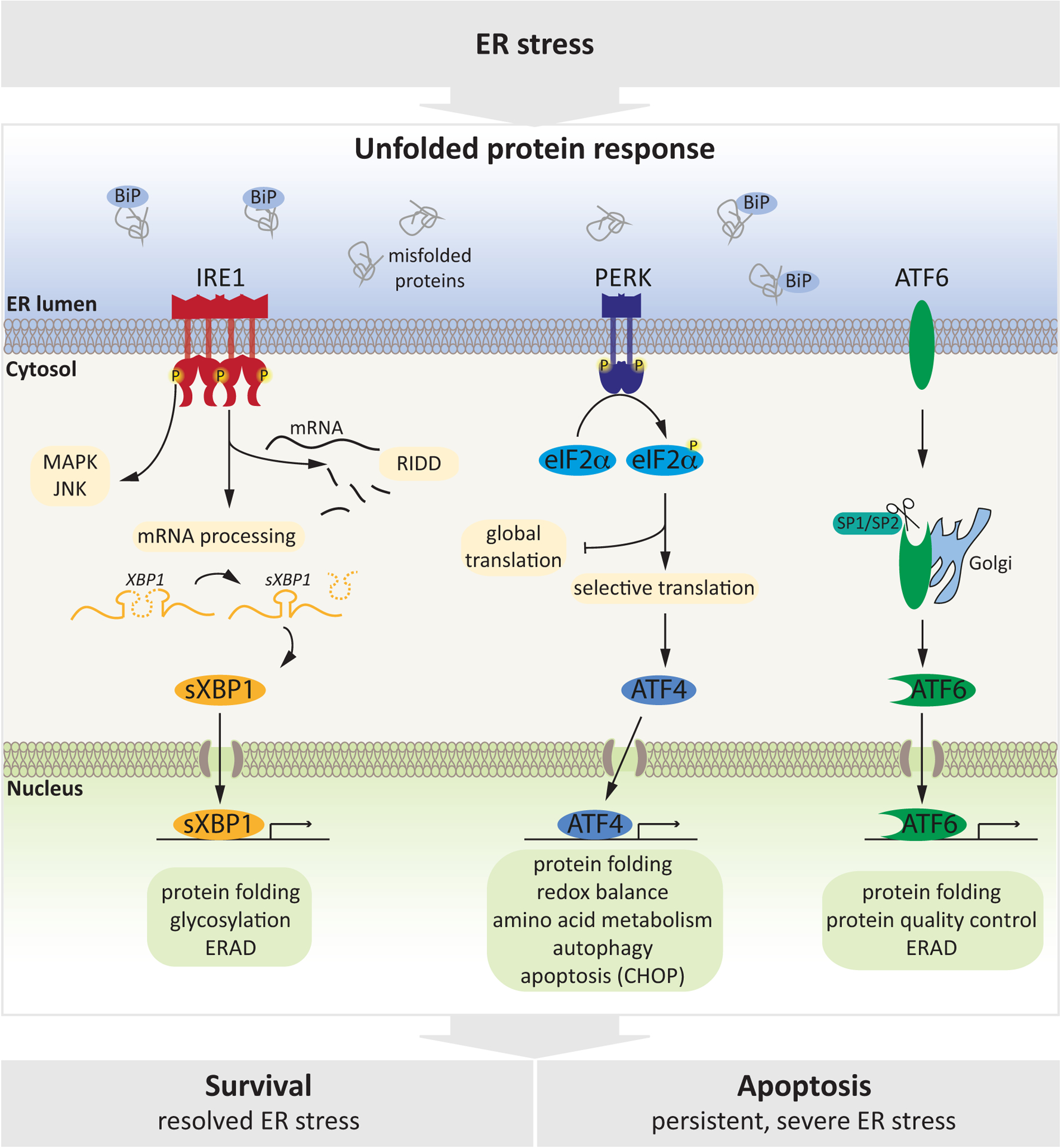
ER stress, triggered by accumulation of un- or mis-folded proteins in the ER lumen is sensed by three transmembrane proteins (IRE1, PERK, ATF6) that serve as ER stress sensors and cooperatively coordinate the UPR. IRE1α and PERK are type I ER transmembrane proteins and are activated by oligo/dimerization and autophosphorylation. IRE1α has a cytoplasmic kinase and endoribonuclease domain and once activated, it catalyzes excision of a 26-nucleotide intron from the mRNA encoding X-box binding protein-1 (XBP-1) which is translated to form the transcription factor spliced XBP1 (sXBP1), and also degrades a subset of ER-associated RNAs through a process called IRE1 dependent decay (RIDD). Through its kinase domain, IRE1 activates further stress-related pathways to modulate response to stress. PERK is composed of an ER luminal stress sensor and a cytosolic protein kinase domain. Activated PERK attenuates global protein synthesis by phosphorylating serine 51 of the initiation factor eukaryotic translation initiator factor 2α (eIF2α), globally decreasing initiation of mRNA translation while enabling selective translation of stress-related mRNAs including activating transcription factor 4 (ATF4). Finally, ATF6 is a transmembrane protein whose cytosolic domain has transcriptional activity. Following ER stress, ATF6 is transported to the Golgi apparatus where it is processed by site 1 protease (S1P) and S2P, releasing a transcriptionally active ATF6 protein which then enters the nucleus. All three arms of the UPR mediate a transcriptional response (by activating sXBP1, ATF4 or ATF6) to increase ER capacity to resolve stress, by upregulating target genes implicated in protein folding, quality control and ERAD. Upon severe or prolonged stress, the UPR orchestrates a pro-apoptotic response that eliminates damaged cells.