

Abstract
Sexual dimorphism is exhibited remarkably in the female predominance of autoimmune diseases (e.g. systemic lupus erythematosus, female-to-male ratio 9:1). To understand the female bias in autoimmunity, we focused on VGLL3 (Vestigial-like-family-member 3), a female-increased molecule known to promote autoimmunity. We report that VGLL3 mediates cellular stress response by upregulating p53 and IL-17C. Energy stress allows VGLL3 to be induced by IFNα, which ultimately leads to p53-dependent, lupus-associated, inflammatory cell death. Our results suggest that female-biased expression of VGLL3 helps cells adapt to metabolic stress, which intriguingly is known as a significant challenge during the evolution of placental mammals for the need to feed a developing embryo. It uncovers the importance of maintaining metabolic homeostasis in the prevention of autoimmunity.
Keywords: Sexual dimorphism, autoimmunity, immunometabolism
Introduction
Evolution from a common single-celled ancestor into species capable of sexual reproduction is thought to offer genetic diversity and fitness advantages in offspring1. Accordingly, the two sexes, females and males, are defined by differences in the reproductive system2. Beyond sexual organs, females and males also exhibit substantial differences in characteristics such as size and weight, a condition termed sexual dimorphism. Not surprisingly, sexual dimorphism is manifested on the molecular level, now supported by an unprecedented progress in the identification of molecules that are expressed differentially between the two sexes3–5. While the biological significance of sexual dimorphism remains unclear, it is tempting to hypothesize that the sex-biased expression of certain molecules confers evolutionary advantage such that it is selected during evolution.
Paradoxically, however, sexual dimorphism in humans is remarkably manifested in diseases. Many autoimmune diseases feature strikingly increased prevalence in females (e.g. systemic lupus erythematosus [SLE], female-to-male ratio 9:1; systemic sclerosis, female-to-male ratio 11:1; Sjögren’s syndrome, female-to-male ratio 14:1). In contrast, infectious diseases affect more men than women at a ratio of ~2:16–8. Why evolutionary advantage, if any, of sex-biased gene expression associates with obvious disadvantage such as disease is arguably one of the biggest mysteries in biology.
To gain insight into sexually dimorphic diseases, specifically female-biased autoimmunity, our previous study examined gene expression differences between female and male skin9. Given the feasibility of obtaining healthy human skin tissues, skin as a model system offers the unique opportunity to study primary human cells in a sex-stratified manner. In addition, skin is a sensitive indicator of immune function and skin changes are prominently manifested in autoimmune diseases including SLE10,11. Transcriptomic profiling on sex-stratified, normal human skin revealed a female-biased molecular signature associated with susceptibility to autoimmune diseases. Further, a putative transcription factor, VGLL3 (Vestigial Like Family Member 3), was identified as a female-increased molecule upstream of this autoimmunity-associated gene network9. VGLL3 expression level increased in autoimmune diseases including cutaneous lupus, and keratinocyte-specific overexpression of VGLL3 in mice drove lupus-like autoimmunity both in skin and on a systemic level9,12. Collectively, these studies demonstrated the role of VGLL3 in promoting female-biased autoimmunity.
While the above studies uncovered yet another sex-biased molecule, fundamental questions about sexual dimorphism remain unanswered. Why do women express at higher levels of VGLL3, a pro-autoimmune factor? Does female-biased expression of VGLL3 confer any evolutionary advantage? What is the critical trigger that further upregulates VGLL3 in high-risk populations (i.e. women), leading to autoimmune diseases?
Darwin theorized that naturally selected traits favor reproduction13. When placental mammals evolved, the need to feed a developing embryo posed significant challenge to metabolic pathways14. We hypothesize that increased levels of VGLL3 in females help cells adapt to this metabolic challenge. In support of this hypothesis, here we report that VGLL3 is induced by nutritional stress in female but not male human keratinocytes. In female but not male keratinocytes, under growth factor restriction, VGLL3 reprograms the immune profile of keratinocytes, supporting expression of IL-17C to maintain basal defense and limiting expression of IL-1 to prevent systemic inflammation and restrict energy expenditure. In female keratinocytes, energy stress removes c-Fos from VGLL3 chromatin, allowing acetylation of H3K27 and transcriptional induction of VGLL3 upon stimulation by IFNα, a key promoter of autoimmune diseases including SLE15,16. In contrast, in male keratinocytes, VGLL3 promoter is coated with H3K27me3 under both nutrient replete and deficient conditions, which possibly underlies the lack of response of VGLL3 to starvation and IFNα stimulation. Overexpression of VGLL3 in female keratinocytes results in regulated, inflammatory cell death in a manner associated with cutaneous lupus. Collectively, we provide evidence that female-biased expression of VGLL3 helps non-placental tissue adapt to energy stress, which offers an evolutionary rationale for sexual dimorphism in immune regulation. We also identify nutritional deficiency as a trigger that can turn this evolutionary strength into weakness by causing autoimmune pathogenesis. This finding reveals the importance of maintaining metabolic homeostasis in prevention of autoimmunity.
Results
VGLL3 is induced under nutritional stress and reprograms immune profile of keratinocytes
To test the potential role of VGLL3 in cellular stress response, we first examined whether VGLL3 itself is inducible by nutritional deficiency in primary human keratinocytes. Indeed, removal of growth factors from keratinocyte culture media resulted in upregulation of VGLL3 mRNA and protein in female keratinocytes (Figure 1a, g). VGLL3 signal was reduced upon transfection of keratinocytes with siRNA against VGLL3, but not control, scrambled siRNA, demonstrating the specificity of VGLL3 detection (Figure 1a, g). In addition to nutritional stress, we tested for VGLL3 induction under oxidative stress (stimulated by H2O217), ER stress (stimulated by DTT18), and hypoxia (stimulated by CoCl219). We observed VGLL3 upregulation by high levels of DTT but not by other treatment (Supplemental Figure 1a), suggesting that VGLL3 is involved in a specific subset of cellular stress responses.
Figure 1.
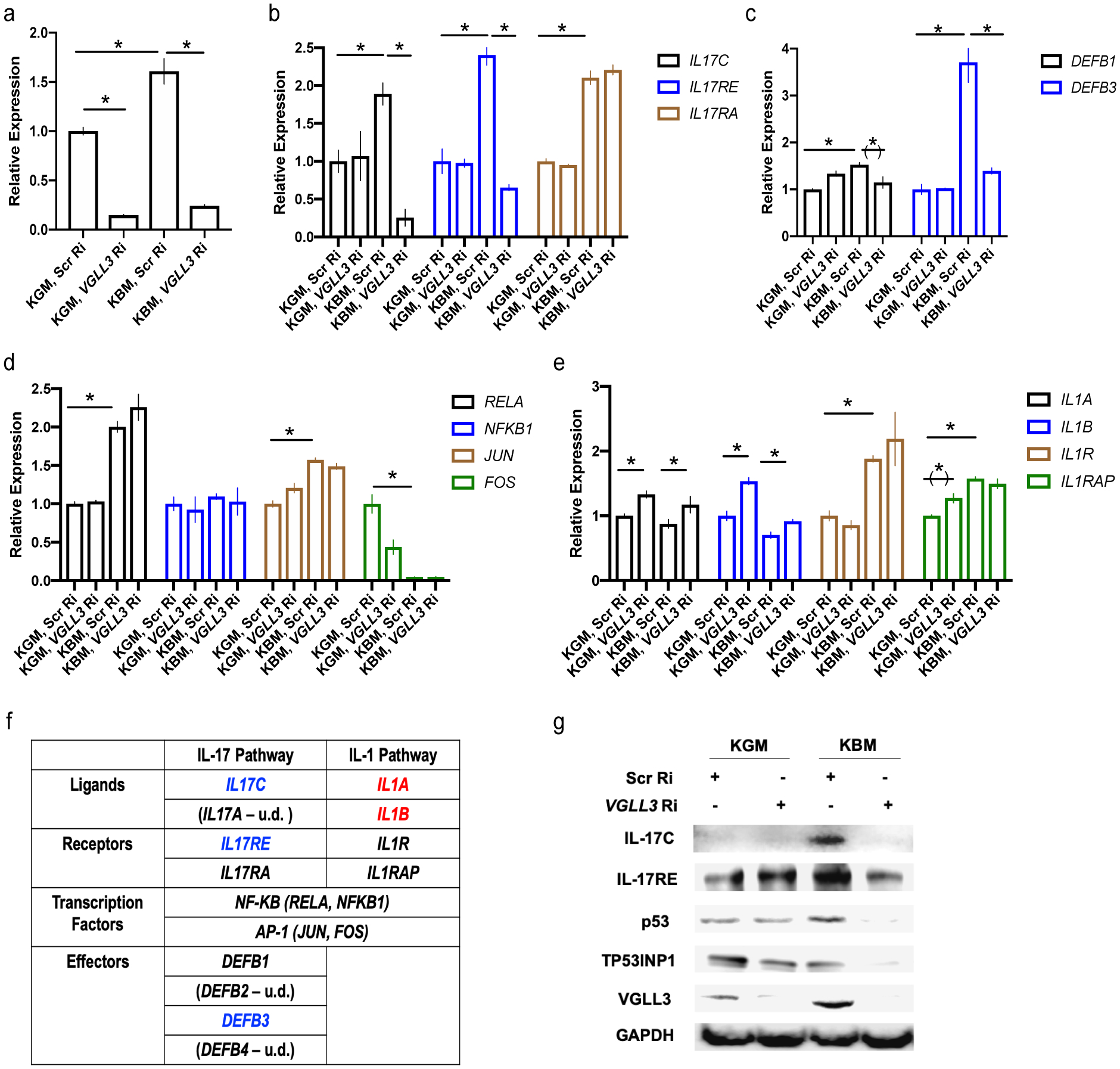
VGLL3 supports keratinocyte adaptation to nutrient deprivation. a-e, qRT-PCR of VGLL3 (a), IL-17 and receptor genes (b), DEFBs (c), NF-κB and AP-1 genes (d), IL-1 and receptor genes (e) under KGM (keratinocyte growth media, complete) or KBM (keratinocyte basal media, removing growth factors) culture conditions in female keratinocytes, with scrambled (Scr Ri) or VGLL3 (VGLL3 Ri) siRNA. n=3, mean ±s.e.m, * P <0 .05, (*) P < 0.1, Student’s t-test. f, summary of the effect of VGLL3 knockdown on IL-17 and IL-1 pathways. Blue, expression downregulated by VGLL3 knockdown. Red, expression upregulated by VGLL3 knockdown. u.d., expression undetermined. g, western blot of indicated proteins under KGM (keratinocyte growth media, complete) or KBM (keratinocyte basal media, removing growth factors) culture conditions, with scrambled (Scr Ri) or VGLL3 (VGLL3 Ri) siRNA in female keratinocytes.
The dynamic regulation of VGLL3 by nutritional stress raises the possibility that VGLL3 functions to help cells adapt to metabolic challenge. Because skin is the first line of defense against infections, we investigated into the potential function of VGLL3 in regulating immune responses in keratinocytes. We focused on two major immune response pathways in keratinocytes, IL-17 and IL-1. The IL-17 family of cytokines plays a critical role in mediating tissue response to injury and infection, with its prototypical member IL-17A being an important driver of autoimmune diseases20–22. IL-17A is mainly produced by Th17 cells. Upon binding to its receptors IL-17RA and IL17-RC in target cells such as keratinocytes, IL-17A activates transcriptional complexes including AP-1 and NF-κB, resulting in strong inflammatory responses20,23,24. In contrast, IL-17C is produced by epithelial cells and functions in a unique, autocrine manner by binding to IL-17RA and IL-17RE. Compared to IL-17A, IL-17C acts on target cells at a much lower potency. Both cytokines activate a similar set of response genes, leading to the model that IL-17C provides local, epithelial defense mechanisms without invoking long-range, pro-inflammatory cascades involving leukocytes, as triggered by IL-17A25,26. In analyzing the response of IL-17 pathway to energy stress in female keratinocytes, we observed that IL-17C and its unique receptor IL17-RE were induced by nutritional stress in a VGLL3-dependent manner (Figure 1b, e). As expected, keratinocytes did not produce IL-17A (data not shown). While IL17-RA, the receptor shared by IL-17A and IL-17C, was upregulated by starvation, the increase was not dependent on VGLL3 (Figure 1b). One major output of IL-17C signaling in epithelial cells is upregulation of defensin, a group of anti-microbial peptides25,26. Consistent with its role in supporting IL-17C-mediated basal defense under energy stress, VGLL3 was required to support the expression DEFB3 under starvation (Figure 1c). Of note, among the four primary β-defensins (DEFB1 to DEFB4), DEFB3 was the major one induced by nutritional stress, with DEFB1 weakly inducible and marginally dependent on VGLL3 and DEFB2 and DEFB4 undetectable under all conditions (Figure 1c; data not shown). Therefore, VGLL3 plays a major role in mediating stress-induced anti-microbial basal defense in keratinocytes. The transcription factor complexes tested, AP-1 and NF-κB, showed limited dependence on VGLL3 (Figure 1d), which is not surprising given that these factors are shared by multiple signaling pathways. Collectively, these lines of evidence demonstrate a specific role of VGLL3 in supporting IL-17C signaling during keratinocyte stress response.
In contrast to a local and restricted defense mechanism mediated by IL-17C, the IL-1 family members are linked to systemic, damaging inflammation, possibly related to their function in acting as damage-associated molecular patterns and increasing nonspecific resistance to infections27–29. We found that in female keratinocytes, VGLL3 inhibition resulted in increase of IL1A and IL1B expression in keratinocytes (Figure 1e), suggesting VGLL3 functions to limit global inflammation.
In contrast, in male keratinocytes, VGLL3 was not induced by nutritional stress (Supplemental Figure 1b). Consistently, male keratinocytes did not show substantial, VGLL3-mediated upregulation of IL-17C, IL17-RE, IL1A, IL1B, and DEFB1 in response to nutritional stress (Supplemental Figure 1b, c). While DEFB3 was induced in male keratinocytes by starvation, the level of induction was less compared to female keratinocytes and the induction was not dependent on VGLL3 (Supplemental Figure 1c, Figure 1c). This suggests that an alternative stress response pathway exists in male keratinocytes in DEFB3 regulation.
In summary, our results demonstrate that under nutritional stress, VGLL3 remodels the immune profile of female but not male keratinocytes to support IL-17C-mediated local defense and limit IL-1-mediated systemic inflammation (Figure 1f). As the latter process is energetically costly30–32, VGLL3 coordinates reduction in energy demand of immune responses during metabolic stress.
VGLL3 supports keratinocyte adaptation to nutritional deprivation via p53
Beyond immune responses, we examined whether VGLL3 played a broad role in helping keratinocytes adapt to energy stress. p53 is known to be a central player that integrates cellular stress response, enabling effective adaptation and survival33–37. In female primary keratinocytes, p53 was upregulated by starvation in a VGLL3-dependent manner (Figure 1g). Consistently, the level of p53-inducible protein TP53INP1 was dependent on VGLL3 under starvation (Figure 1g). While keratinocyte proliferation under standard culture conditions was not dependent on VGLL3, siRNA-mediated knockdown of VGLL3 under prolonged starvation resulted in cell death, which could be partially rescued by Nutlin-3, an MDM2 antagonist activating the p53 pathway (Supplemental Figure 1d, e). In contrast, there was no substantial VGLL3-dependent induction of p53 under nutritional stress (Supplemental Figure 1b). Therefore, VGLL3 supports cellular adaptation to metabolic stress via p53 in female keratinocytes.
Starvation remodels VGLL3 chromatin, enabling VGLL3 induction by IFNα
VGLL3 was initially identified as a factor promoting female-biased autoimmunity9. VGLL3 is upregulated in tissues of multiple autoimmune diseases compared to normal, and its knockdown decreased expression of a network of female-biased, autoimmunity-associated genes9. In addition, keratinocyte-specific overexpression of VGLL3 was sufficient to drive systemic autoimmunity12.
Based on the role of VGLL3 in cellular stress response, it is reasonable to think that higher levels of VGLL3 in females provides competitive advantage in adaptation to the metabolic cost of feeding a developing embryo. Consistently, VGLL3 exhibits increased expression in skin of healthy females compared to males9. However, this level of female-biased expression in the healthy population is clearly insufficient to cause autoimmunity. What, then, is the critical trigger that further elevates VGLL3 level leading to disease onset?
To address this question, we studied the response of female keratinocytes to IFNα, a signature molecule of autoimmune diseases15,16,38, under nutrient-sufficient and deficient conditions. Intriguingly, growth factor deprivation allowed VGLL3 upregulation by IFNα, a response not seen when nutrient was sufficient (Figure 2a, b). The differential response was not due to lack of IFNα signaling with normal growth media, because phospho-STAT3 was induced to a similar level under both conditions (Figure 2b).
Figure 2.
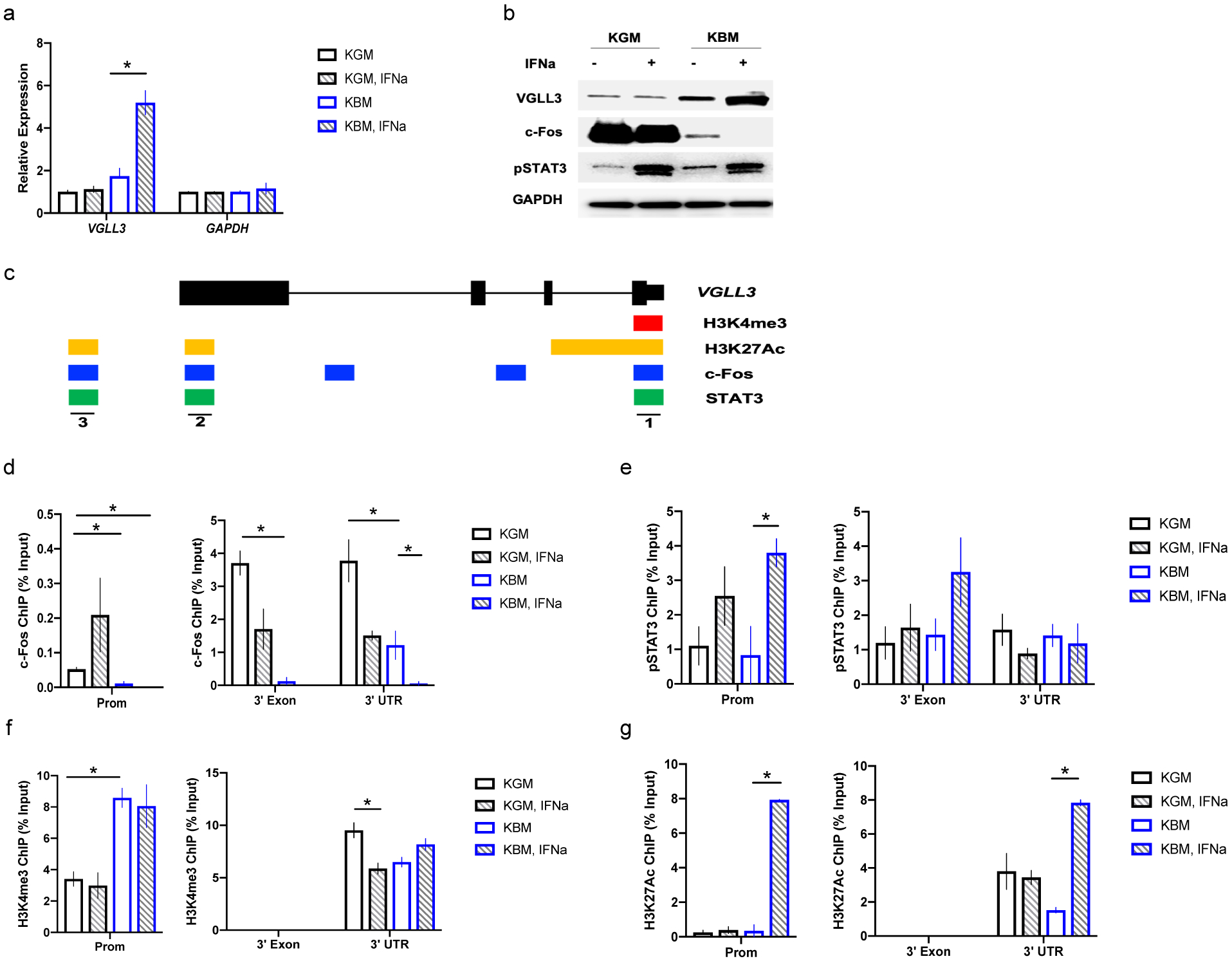
Nutrient stress remodels VGLL3 chromatin, enabling VGLL3 induction by IFNα. a, qRT-PCR of VGLL3 under KGM (keratinocyte growth media, complete) or KBM (keratinocyte basal media, removing growth factors) culture conditions in female keratinocytes, with or without IFNα treatment. b, western blot of indicated proteins under KGM or KBM culture conditions, with or without IFNα treatment in female keratinocytes. c, regulatory sites along VGLL3 gene, summarized from UCSC genome browser. d-g, ChIP-qPCR of c-Fos (d), pSTAT3 (e), H3K4me3 (f), and H3K27ac (g) on the three regulatory sites of VGLL3, under KGM or KBM culture conditions, with or without IFNα treatment in female keratinocytes. n=3, mean ±s.e.m, * P <0 .05, Student’s t-test.
To understand the molecular mechanism underlying the observed selective response to IFNα under energy stress, we analyzed putative transcription factor binding sites along the VGLL3 gene (Figure 2c). We observed three potential regulatory sites at VGLL3 promoter, 3’- exon, and 3’ - UTR, respectively, with activating histone modifications (H3K4me3 and/or H3K27Ac) in keratinocytes and putative STAT-binding sites that lie adjacent to putative c-Fos binding sites (Figure 2c). Fos family members are known to be stimulated by growth factors39. Similarly, we observed significant downregulation of c-Fos in female keratinocytes upon growth factor withdrawal (Figure 1d, 2b). We hypothesized that nutritional deprivation in female keratinocytes results in the removal of c-Fos from VGLL3 chromatin, which subsequently exposes binding sites for STAT3 and/or its associated transcriptional complex, leading to transcriptional induction of VGLL3 upon IFNα stimulation.
We tested this hypothesis by chromatin immunoprecipitation of c-Fos and pSTAT3 at the three regulatory sites of VGLL3 gene under nutrient sufficient- and deficient-conditions, with or without IFNα stimulation in female keratinocytes. As expected, there was loss of c-Fos signal at all three sites under energy stress (Figure 2d), accompanied by a starvation-induced increase of H3K4me3 signal at VGLL3 promoter and loss of H3K27me3 at 3’UTR (Figure 2f, Supplemental Figure 2). The abundance of pSTAT3 under stress was marginally increased (Figure 2e), indicating that transcriptional co-factors other than STAT3 might play a role in IFN-induction. STAT3 is known to complex with CBP/p300, which acetylates histone H3K27 for transcriptional activation40,41. Intriguingly, H3K27 acetylation was only detected with IFNα stimulation under nutrient stress, but not under other conditions (Figure 2g). These observations support a model in which starvation-induced loss of c-Fos results in deposition of the active histone mark, H3K4me3, on VGLL3 promoter. In addition, absence of c-Fos allows binding and functioning of pSTAT3-containing transcriptional complex upon IFNα stimulation. pSTAT3-containing transcriptional complex further acetylates H3K27, which ultimately results in VGLL3 induction.
In contrast, starvation did not sensitize VGLL3 response to IFNα in male keratinocytes (Supplemental Figure 2b). VGLL3 promoter in male keratinocytes was marked with increased H3K27me3 under all conditions tested compared to female cells (Supplemental Figure 2a, c), consistent with previous report of male-biased H3K27me3 on VGLL3 promoter9. This data supports a model in which VGLL3 expression in male cells is inhibited by the repressive histone mark H3K27me3, which needs to be removed before transcriptional activation can occur during both starvation and IFNα stimulation.
We further tested the response of VGLL3 to nutritional stress and IFNα stimulation in human fibroblasts. Similar to keratinocytes, VGLL3 was induced by starvation in female, but not male, fibroblasts (Supplemental Figure 2d, e). However, the effect of nutritional stress on IFNα potentiation is less pronounced compared to keratinocytes. Therefore, VGLL3 senses nutritional stress in fibroblasts similarly to keratinocytes, while its downstream effector pathways exhibit cell-type specificity to some extent.
VGLL3 overexpression causes regulated cell death
Given the two-step upregulation of VGLL3 by nutritional stress and IFNα, we studied the functional consequence of VGLL3 increase in keratinocytes. To this end, we transfected primary keratinocytes with VGLL3 tagged with GFP, whose fluorescence intensity was used to indicate relative level of overexpression between conditions. Female keratinocytes expressing the VGLL3-GFP rounded up and died within 24 hours, while those expressing GFP to the same level and even way above stayed alive and kept proliferating (Figure 3a and data not shown). In addition, we observed chromatin condensation in VGLL3-overexpressing cells (Figure 3b). There was substantial overlap between the VGLL3-GFP and DAPI signals, indicating the association between VGLL3 and condensed chromatin during cell death (Figure 3c). When we attempted to transfect male keratinocytes with VGLL3-GFP, we did not observe significant amount of VGLL3-GFP+ cells (data not shown). We reasoned that male keratinocytes might have a mechanism to downregulate VGLL3, either transcriptionally (e.g. by methylation of H3K27 as shown in supplemental figure 2c) or post translationally (by an unknown mechanism). Nevertheless, we were not able to overexpress VGLL3 in male keratinocytes and therefore focused subsequent studies on female keratinocytes.
Figure 3.
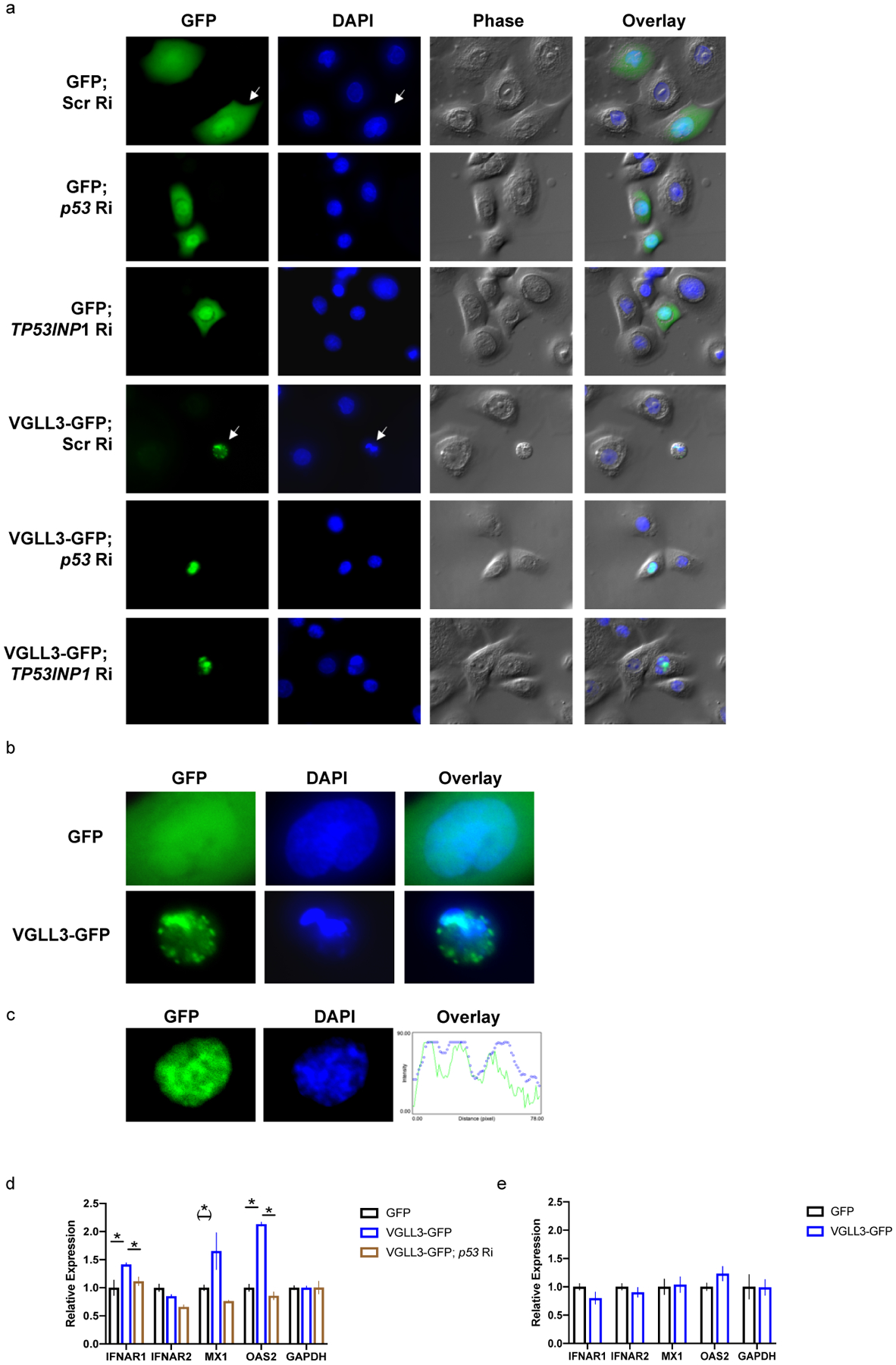
VGLL3 excess causes regulated, inflammatory cell death. a, keratinocyte cell morphology (phase) and DNA status (DAPI) upon transfection with indicated constructs in female keratinocytes. Ri, RNAi. Scr, Scrambled. Zoom-in pictures of arrow-pointed cells are shown in b to demonstrate chromatin condensation. c, overlap between GFP and DAPI signals in female keratinocyte expressing VGLL3-GFP. d, qRT-PCR of indicated genes in THP-1 cells migrated to conditioned media from female keratinocytes overexpressing GFP, VGLL3-GFP, and VGLL3-GFP with siRNA against p53. n=3. e, qRT-PCR of indicated genes in female keratinocytes overexpressing GFP and VGLL3-GFP. n=3. Mean ±s.e.m, * P < 0 .05, (*) P < 0.1, Student’s t-test.
We next asked if VGLL3-induced cell death was programmed and in particular mediated by p53. While p53 supports the adaptation of cells under stress conditions, it also drives the elimination of cells in which stress does not resolve33. Co-transfection of female keratinocytes with VGLL3-GFP and siRNA against p53 or TP53INP1 rescued the death phenotype (Figure 3a), suggesting that increase in VGLL3 levels activated p53-dependent cell death.
Cell death is known to initiate autoimmunity with uncleared cellular debris that contains complex autoantigens and associated inflammatory responses42,43. Specifically, two forms of cell death - apoptosis and pyroptosis - have been observed with human keratinocytes under UVB stimulation, which may explain the photosensitivity seen in subsets of lupus patients44,45. Overexpression of VGLL3 in female keratinocytes did not result in activation of caspase-3, while staurosporine treatment did as positive control (Supplemental Figure 3a–c). In addition, VGLL3 overexpression did not lead to IL-1β cleavage, while UVB treatment did as positive control (Supplemental Figure 3d–f). VGLL3-induced cell death still occurred with addition of Z-VAD-FMK or Z-YVAD-FMK (data not shown), leading us to conclude that VGLL3 does not cause apoptosis or pyroptosis.
VGLL3 overexpression resulted in loss of MitoTracker signal, an indicator of the number of functioning mitochondria (Supplemental Figure 4). This loss was reversed by knockdown of p53 or TP53INP1 (Supplemental Figure 4), consistent with the report that p53 opens mitochondrial permeability transition pore to trigger necrosis46. However, necrostatin-1 only partially inhibited cell death caused by VGLL3 overexpression (data not shown), adding complexity to the form of VGLL3-induced cell death.
VGLL3-induced keratinocyte cell death upregulates IFNα signaling in monocytes
Stressed skin epithelium with VGLL3 upregulation may initiate lupus-associated inflammatory responses by 1) inducing infiltration of monocytes, 2) affecting gene expression profile of infiltrated monocytes, 3) altering the differentiation of monocytes into macrophages and/or myeloid dendritic cells, influencing clearance of dead cells and/or adaptive immunity against self-antigens47,48. To test these possibilities, we first examined the migration of monocyte-like THP-1 cells towards conditioned media from female keratinocytes expressing GFP or VGLL3-GFP. We found that VGLL3 increase in keratinocytes did not have any major effect in stimulating THP-1 migration (Supplemental Figure 5a). Next we analyzed the efficiency of THP-1 differentiation into macrophages or dendritic cells using established markers49,50 with conditioned media from keratinocytes expressing GFP or VGLL3-GFP, and again observed no effect of VGLL3 expression (Supplemental Figure 5b–e and data not shown). Lastly we examined the expression of IFN signaling genes in THP-1 cells that migrated to conditioned media from keratinocytes expressing GFP or VGLL3-GFP. VGLL3 expression in keratinocytes upregulated IFNAR1 and OAS2, a signature, type I interferon-inducible gene upregulated in lupus patients15,16,38,51, in migrated monocytes (Figure 3d). This effect was abolished when p53 siRNA was co-transfected with VGLL3-GFP in keratinocytes (Figure 3d), suggesting that upregulation of IFNα signaling is a result of VGLL3-p53 mediated stress response. In contrast, VGLL3 overexpression was insufficient to upregulate expression of IFNα pathway genes in keratinocytes (Figure 3e), supporting a paracrine but not autocrine effect of keratinocyte stress response.
VGLL3-associated stress signaling is increased in lupus patient skin
The observation that VGLL3-p53 mediated stress response in keratinocytes upregulates IFN response in monocytes raises the possibility that dysregulation of this stress sensing pathway is associated with lupus pathogenesis. To test this possibility, we stained normal skin and lesional skin from discoid lupus erythematosus (DLE) and subacute cutaneous lupus erythematosus (SCLE) patients for VGLL3 and p53. For both DLE and SCLE, there were patients without detectable level of active caspase-3 or cleaved IL-1β (Figure 4a). Consistently, no apoptosis was detected in these patients by TUNEL assay, despite clear signals from positive control samples (Figure 4b). Instead, these patients exhibited increased levels of VGLL3 and p53 (Figure 4a). By contrast, affected skin in vulva lichenoid dermatitis and actinic keratosis did not show increased VGLL3 or p53 levels (Figure 4a). Therefore, dysregulation of the VGLL3-p53 stress sensing pathway is associated with lupus pathogenesis and delineates a distinct subgroup of lupus patients who are negative for apoptosis or pyroptosis.
Figure 4.
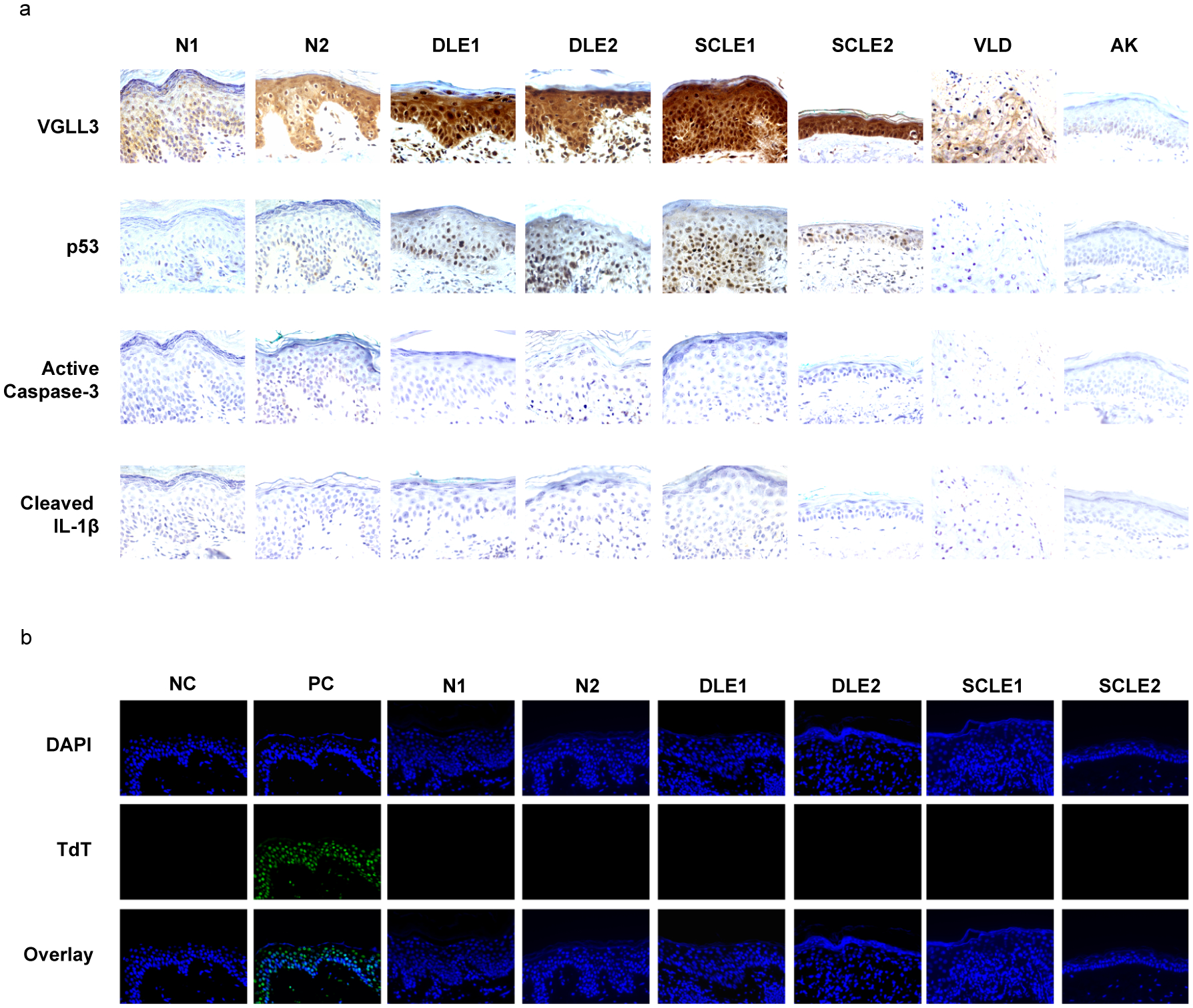
VGLL3-p53 is upregulated in lupus lesional skin. a, immunohistochemistry staining of indicated proteins. b, TUNEL assay of normal and lupus skin, with negative control (NC) and positive control (PC) shown on the left. N1, normal skin, male. N2, normal skin, female. DLE1, discoid lupus erythematosus skin, male. DLE2, discoid lupus erythematosus skin, female. SCLE1, subacute cutaneous lupus erythematosus skin, male. SCLE2, subacute cutaneous lupus erythematosus skin, female. VLD, vulva lichenoid dermatitis, female. AK, actinic keratosis, male.
Discussion
The striking predominance of autoimmune diseases in women (~78% overall and up to ~95% for specific diseases7,8) has long perplexed scientists. Previous studies shed light on the molecular basis of sexual dimorphism in autoimmunity by demonstrating a female-increased, molecular network under the regulation of VGLL39. The role of VGLL3 in autoimmune pathogenesis has been demonstrated on three levels. Firstly, in human keratinocytes, VGLL3 knockdown diminished expression of autoimmune-associated genes that were abnormally upregulated in several female-biased autoimmune diseases, including systemic and cutaneous lupus erythematosus, sclerosis and Sjögren’s syndrome9. Secondly, VGLL3 itself is upregulated in lesional skin of lupus patients9. Thirdly, keratinocyte-specific overexpression of VGLL3 in mice was sufficient to recapitulate lupus-associated cutaneous inflammation and drive systemic autoimmunity12. Why, then, would females express a pathogenic gene at a high level?
To answer this question, we need to first better understand the function of VGLL3. VGLL3 (Vestigial like family member 3) was identified in humans based on its homology with the Drosophila gene vg (vestigial). In Drosophila, vg encodes a cofactor of Scalloped, homolog of the transcription factor TEF-152. Besides the conjecture that VGLL3 may function similar to vg, we lack understanding on the exact function of VGLL3 in humans.
Addressing this knowledge gap, in this study we report a previously unknown role of VGLL3 in sensing nutritional stress. We provide evidence that under nutritional deficiency, VGLL3 is upregulated in primary human female, but not male, keratinocytes and functions to upregulate IL-17C and restrict IL-1 signaling. A major difference between these two pro-inflammatory pathways is that IL-17C has low potency and provides local, basal defense25,26. In contrast, IL-1 drives systemic, damaging inflammation, which is an energetically costly process27,28. Therefore, VGLL3 remodels the immune profile of human keratinocytes so that skin, as the front line of defense, can maintain basic antimicrobial activities while restricting energy expenditure. In this way VGLL3 helps cells and on a larger scale, the organism, adapt to metabolic stress.
Our finding offers an evolutionary explanation to female-biased expression of VGLL3. When placental mammals evolved, the need to feed a developing embryo posed significant challenge to metabolism and forced metabolic pathways to adapt14. We now show that increased levels of VGLL3 provides a competitive advantage for females by helping non-placental, maternal tissues adapt to competing demands for energy. Meanwhile, it is worth noting that according to the hygiene hypothesis, decrease in the infectious burden associates with the rise of autoimmune diseases53, which suggests that lifestyle changes may lead to increase in autoimmune disease incidence. To the best of our knowledge, there has been no evidence that suggests autoimmune diseases can, in turn, affect the evolution of humans.
Consistent with a protective role of VGLL3 in females, its level in healthy women is not sufficient to cause disease. In other words, there is a critical trigger that further elevates VGLL3, leading to autoimmune onset. Intriguingly, we have found that nutritional stress remodels the chromatin landscape on the VGLL3 gene in female keratinocytes, allowing it to be further induced by IFNα, a signature cytokine in autoimmune diseases including SLE15,16,38. Human keratinocytes have low tolerance for VGLL3 increase and once threshold is reached, undergo inflammatory, regulated cell death that has been strongly implicated in autoimmune pathogenesis54,55. The same pathway (VGLL3-p53) operates in both adaptation, below stress threshold, and cell death, when stress exceeds the critical level. Consistently, both VGLL3 and p53 are upregulated in lesional skin of lupus patients. Importantly, majority of recent studies demonstrate that SLE worsens during pregnancy56, when maternal and fetal tissues compete for energy. Together with clinical observation of malnutrition in SLE patients57–59, these lines of evidence support the model that energy stress is an important trigger of autoimmune pathogenesis in high-risk populations, and pinpoint VGLL3 as the key molecule at the intersection of metabolic and immune regulation.
With nutritional deficiency as a newly identified autoimmune trigger, it is reasonable to think that nutritional monitoring strategies can be developed to prevent and/or treat autoimmune diseases. For lack of effect preventative and treatment strategies, autoimmune diseases as a category now affects more than seven percent of the general population, is the second highest cause of chronic illness, and the top cause of morbidity in women in the US60–63. Our findings may ultimately lead to novel clinical approaches that address this significant public health issue.
In addition to autoimmunity, our finding on the regulation of p53 by VGLL3 has implications for other diseases including cancer. Malignancies affect more males than females across a range of cancer types including skin cancers for unknown reasons64, and the female-biased regulation of the tumor suppressor p53 may shed light on its molecular basis. Recently, VGLL3 expression has been associated with a tumor suppressor phenotype65, strengthening its link to sex bias in cancer biology.
In summary, in this study we have demonstrated the role of the female-biased, pro-autoimmune factor VGLL3 in stress sensing and delineated how dysregulation of VGLL3-mediated stress sensing could lead to autoimmune pathogenesis. These findings provide an evolutionary rationale for female-biased autoimmunity, highlight the intricate interaction between metabolic and immune regulation, and offer novel strategies for autoimmune disease prevention or treatment.
Materials and methods
Cell culture, siRNA and overexpression
Normal primary human keratinocytes were purchased from Lonza and grown in Lonza KGM-gold keratinocyte growth medium according to manufacturer instructions. For KBM treatment, growth factors were removed from KGM-gold keratinocyte growth medium. Cells were used at passage 1 or 2. THP-1 cells were purchased from ATCC and cultured in RPMI-1640 medium, 10% fetal bovine serum, and 0.05 mM 2-mercaptoethanol (ATCC) according to ATCC recommendations. siRNA and overexpression vectors were introduced by electroporation using Lonza 4D-nucleofector following manufacturer’s recommendations.
Gene and protein expression analysis
Cells were collected in Trizol (Thermo Fisher Scientific) and RNA was isolated using the Purelink RNA Mini kit (Thermo Fisher Scientific) following recommended manual. Reverse transcription was performed with Superscript IV first-strand synthesis system (Thermo Fisher Scientific) and qPCR was performed on Quantstudio-7 with Power SYBR green master mix (Thermo Fisher Scientific) following manufacturer’s protocol. For protein level analysis, cells were lysed in 2x Laemmli buffer and resolved by SDS-PAGE gel (Bio-rad) following manufacturer’s instructions. Proteins were transferred to PVDF membrane, blocked with blocking buffer (Cell Signaling Technologies), incubated with primary antibodies overnight, washed in TBST, incubated with HRP-conjugated secondary antibodies, and imaged on Odyssey Imaging System.
Immunofluorescence
Cells were fixed with 4% formaldehyde in PBS for 5 minutes, blocked with 1% BSA for 20 minutes, permeabilized with 0.1% Triton X-100, and incubated with primary antibodies overnight at 4 degrees. Cells were washed with PBS and 0.1% Triton X-100, incubated with fluorophore-conjugated secondary antibodies in dark for 1 hour at room temperature, washed with PBS, stained for DNA and imaged.
Immunohistochemistry
Formalin-fixed, paraffin-embedded specimens on slides were heated for 30 minutes at 55 degrees, rehydrated and epitope-retrieved with Tris-EDTA, pH9. Slides were blocked, incubated with primary antibody overnight at 4 degrees, washed, incubated with secondary antibody, developed with DAB (3, 3’ diaminobenzidine) and counterstained using hematoxylin.
TUNEL assay
TUNEL staining was performed using the TACS TdT-Fluor in situ apoptosis detection kit (R&D systems).
Flow cytometry
Cells were collected, blocked with human TruStain FcX (BioLegend), incubated with fluorescent conjugated antibody for 20 minutes on ice in dark, washed with FACS buffer, stained with DAPI and analyzed on Attune Flow Cytometer.
Supplementary Material
Supplemental Figure 1. Role of VGLL3 in keratinocyte stress response. a, qRT-PCR of VGLL3 under indicated treatment conditions in female keratinocytes. n=3, mean ±s.e.m, * P <0 .05, (*) Student’s t-test. b, western blot of indicated proteins under KGM (keratinocyte growth media, complete) or KBM (keratinocyte basal media, removing growth factors) culture conditions, with scrambled (Scr Ri) or VGLL3 (VGLL3 Ri) siRNA in male keratinocytes. c, qRT-PCR of indicated genes under KGM (keratinocyte growth media, complete) or KBM (keratinocyte basal media, removing growth factors) culture conditions, with scrambled (Scr Ri) or VGLL3 (VGLL3 Ri) siRNA in male keratinocytes. n=3, mean ±s.e.m, * P <0 .05, Student’s t-test. d, keratinocyte cell number under KGM (keratinocyte growth media, complete) culture condition with. e, keratinocyte cell number under KBM (keratinocyte basal media, removing growth factors) culture condition with scrambled (Scr Ri) or VGLL3 (VGLL3 Ri) siRNA, with or without Nutlin treatment conditions, with scrambled (Scr Ri) or VGLL3 (VGLL3 Ri) siRNA.
Supplemental Figure 2. Nutrient stress modifies VGLL3 chromatin. a, ChIP-qPCR of H3K27me3 on the three regulatory sites of VGLL3, under KGM (keratinocyte growth media, complete) or KBM (keratinocyte basal media, removing growth factors) culture conditions, with or without IFNα treatment in female keratinocytes. n=3, mean ±s.e.m. b, western blot of indicated proteins under KGM (keratinocyte growth media, complete) or KBM (keratinocyte basal media, removing growth factors) culture conditions, with or without IFNα treatment in male keratinocytes. c, ChIP-qPCR of H3K27me3 on VGLL3 promoter, under KGM (keratinocyte growth media, complete) or KBM (keratinocyte basal media, removing growth factors), with or without IFNα treatment in male keratinocytes. d, western blot of indicated proteins under KGM (keratinocyte growth media, complete) or KBM (keratinocyte basal media, removing growth factors) culture conditions, with or without IFNα treatment in female fibroblasts. e, western blot of indicated proteins under KGM (keratinocyte growth media, complete) or KBM (keratinocyte basal media, removing growth factors) culture conditions, with or without IFNα treatment in male fibroblasts.
Supplemental Figure 3. VGLL3 does not induce apoptosis or pyroptosis. a, immunofluorescence images of keratinocytes overexpressing GFP or VGLL3-GFP (red, active caspase-3; green, GFP; blue, DNA). b, immunofluorescence images of keratinocytes with or without staurosporine treatment (red, active caspase-3; blue, DNA). c, quantification of percentage of cells with active caspase-3 staining under indicated conditions. mean ±s.e.m, * P <0 .05, Student’s t-test. d, immunofluorescence images of keratinocytes overexpressing GFP or VGLL3-GFP (red, cleaved IL-1β; green, GFP; blue, DNA). b, immunofluorescence images of keratinocytes with or without UVB stimulation (red, cleaved IL-1β; blue, DNA). f, quantification of percentage of cells with cleaved IL-1β staining under indicated conditions. mean ± s.e.m, * P <0 .05, Student’s t-test.
Supplemental Figure 4. VGLL3 overexpression caused loss of mitochondria function. Immunofluorescence images of keratinocytes under indicated conditions (red, MitoTracker; green, GFP; blue, DNA). Arrows point to a representative VGLL3-overexpressing cell with loss of MitoTracker signal. Ri, RNAi. Scr, scrambled.
Supplemental Figure 5. VGLL3 upregulation in keratinocytes does not affect THP-1 chemotaxis or differentiation. a, number of monocytes migrated towards conditioned media from keratinocytes overexpressing GFP or VGLL3-GFP. n=5. b, percentage of CD14+ cells upon macrophage differentiation of THP-1 cells in the presence of conditioned media from keratinocytes overexpressing GFP or VGLL3-GFP. c, percentage of CD14+ and CD86+ cells upon dendritic cell differentiation of conditioned media from keratinocytes overexpressing GFP or VGLL3-GFP. n=3. d,e, Flow cytometry of indicated markers upon THP-1 differentiation into macrophages (d) or dendritic cells (e) after gating for singlets and viability.
Acknowledgments
We thank the UW-Madison Skin Disease Resource Center for providing de-identified normal and lupus skin tissues. We thank the UWCCC flow cytometry core for technical support. Y.L. is funded by the NIH grant K01 AR073340.
Footnotes
Conflict of interests
The authors declare no financial conflict of interests.
References
- 1.Crow JF Advantages of sexual reproduction. Dev Genet 15, 205–213, doi: 10.1002/dvg.1020150303 (1994). [DOI] [PubMed] [Google Scholar]
- 2.Klein SL & Flanagan KL Sex differences in immune responses. Nat Rev Immunol 16, 626–638, doi: 10.1038/nri.2016.90 (2016). [DOI] [PubMed] [Google Scholar]
- 3.Li B et al. A Comprehensive Mouse Transcriptomic BodyMap across 17 Tissues by RNA-seq. Sci Rep 7, 4200, doi: 10.1038/s41598-017-04520-z (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trabzuni D et al. Widespread sex differences in gene expression and splicing in the adult human brain. Nat Commun 4, 2771, doi: 10.1038/ncomms3771 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gershoni M & Pietrokovski S The landscape of sex-differential transcriptome and its consequent selection in human adults. BMC Biol 15, 7, doi: 10.1186/s12915-017-0352-z (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghosh S & Klein RS Sex Drives Dimorphic Immune Responses to Viral Infections. J Immunol 198, 1782–1790, doi: 10.4049/jimmunol.1601166 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whitacre CC Sex differences in autoimmune disease. Nat Immunol 2, 777–780, doi: 10.1038/ni0901-777 (2001). [DOI] [PubMed] [Google Scholar]
- 8.Whitacre CC, Reingold SC & O’Looney PA A gender gap in autoimmunity. Science 283, 1277–1278, doi: 10.1126/science.283.5406.1277 (1999). [DOI] [PubMed] [Google Scholar]
- 9.Liang Y et al. A gene network regulated by the transcription factor VGLL3 as a promoter of sex-biased autoimmune diseases. Nat Immunol 18, 152–160, doi: 10.1038/ni.3643 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cervera R et al. Systemic lupus erythematosus: clinical and immunologic patterns of disease expression in a cohort of 1,000 patients. The European Working Party on Systemic Lupus Erythematosus. Medicine (Baltimore) 72, 113–124 (1993). [PubMed] [Google Scholar]
- 11.Nestle FO, Di Meglio P, Qin JZ & Nickoloff BJ Skin immune sentinels in health and disease. Nat Rev Immunol 9, 679–691, doi: 10.1038/nri2622 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Billi AC et al. The female-biased factor VGLL3 drives cutaneous and systemic autoimmunity. JCI Insight 4, doi: 10.1172/jci.insight.127291 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Darwin C The descent of man, and selection in relation to sex. (1871).
- 14.Della Torre S, Benedusi V, Fontana R & Maggi A Energy metabolism and fertility: a balance preserved for female health. Nat Rev Endocrinol 10, 13–23, doi: 10.1038/nrendo.2013.203 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Banchereau J & Pascual V Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity 25, 383–392, doi: 10.1016/j.immuni.2006.08.010 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Crow MK, Olferiev M & Kirou KA Type I Interferons in Autoimmune Disease. Annu Rev Pathol 14, 369–393, doi: 10.1146/annurev-pathol-020117-043952 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Sies H Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol 11, 613–619, doi: 10.1016/j.redox.2016.12.035 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oslowski CM & Urano F Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol 490, 71–92, doi: 10.1016/B978-0-12-385114-7.00004-0 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cervellati F et al. Hypoxia induces cell damage via oxidative stress in retinal epithelial cells. Free Radic Res 48, 303–312, doi: 10.3109/10715762.2013.867484 (2014). [DOI] [PubMed] [Google Scholar]
- 20.McGeachy MJ, Cua DJ & Gaffen SL The IL-17 Family of Cytokines in Health and Disease. Immunity 50, 892–906, doi: 10.1016/j.immuni.2019.03.021 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McInnes IB et al. Efficacy and safety of secukinumab, a fully human anti-interleukin-17A monoclonal antibody, in patients with moderate-to-severe psoriatic arthritis: a 24-week, randomised, double-blind, placebo-controlled, phase II proof-of-concept trial. Ann Rheum Dis 73, 349–356, doi: 10.1136/annrheumdis-2012-202646 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Trial watch: targeting IL-17A shows broad promise in autoimmune diseases. Nat Rev Drug Discov 9, 908, doi: 10.1038/nrd3327 (2010). [DOI] [PubMed] [Google Scholar]
- 23.Wu NL, Huang DY, Tsou HN, Lin YC & Lin WW Syk mediates IL-17-induced CCL20 expression by targeting Act1-dependent K63-linked ubiquitination of TRAF6. J Invest Dermatol 135, 490–498, doi: 10.1038/jid.2014.383 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Ha HL et al. IL-17 drives psoriatic inflammation via distinct, target cell-specific mechanisms. Proc Natl Acad Sci U S A 111, E3422–3431, doi: 10.1073/pnas.1400513111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ramirez-Carrozzi V et al. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat Immunol 12, 1159–1166, doi: 10.1038/ni.2156 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Pappu R, Rutz S & Ouyang W Regulation of epithelial immunity by IL-17 family cytokines. Trends Immunol 33, 343–349, doi: 10.1016/j.it.2012.02.008 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Dinarello CA Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol Rev 281, 8–27, doi: 10.1111/imr.12621 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dinarello CA Blocking IL-1 in systemic inflammation. J Exp Med 201, 1355–1359, doi: 10.1084/jem.20050640 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rivers-Auty J, Daniels MJD, Colliver I, Robertson DL & Brough D Redefining the ancestral origins of the interleukin-1 superfamily. Nat Commun 9, 1156, doi: 10.1038/s41467-018-03362-1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sadd BM & Schmid-Hempel P Principles of ecological immunology. Evol Appl 2, 113–121, doi: 10.1111/j.1752-4571.2008.00057.x (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Odegaard JI & Chawla A Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science 339, 172–177, doi: 10.1126/science.1230721 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ganeshan K et al. Energetic Trade-Offs and Hypometabolic States Promote Disease Tolerance. Cell 177, 399–413 e312, doi: 10.1016/j.cell.2019.01.050 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kruiswijk F, Labuschagne CF & Vousden KH p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol 16, 393–405, doi: 10.1038/nrm4007 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Kastenhuber ER & Lowe SW Putting p53 in Context. Cell 170, 1062–1078, doi: 10.1016/j.cell.2017.08.028 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jones RG et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell 18, 283–293, doi: 10.1016/j.molcel.2005.03.027 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Tajan M et al. A Role for p53 in the Adaptation to Glutamine Starvation through the Expression of SLC1A3. Cell Metab 28, 721–736 e726, doi: 10.1016/j.cmet.2018.07.005 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reid MA et al. The B55alpha subunit of PP2A drives a p53-dependent metabolic adaptation to glutamine deprivation. Mol Cell 50, 200–211, doi: 10.1016/j.molcel.2013.02.008 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Banchereau R et al. Personalized Immunomonitoring Uncovers Molecular Networks that Stratify Lupus Patients. Cell 165, 1548–1550, doi: 10.1016/j.cell.2016.05.057 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Brown JR et al. Fos family members induce cell cycle entry by activating cyclin D1. Mol Cell Biol 18, 5609–5619, doi: 10.1128/mcb.18.9.5609 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gray MJ et al. HIF-1alpha, STAT3, CBP/p300 and Ref-1/APE are components of a transcriptional complex that regulates Src-dependent hypoxia-induced expression of VEGF in pancreatic and prostate carcinomas. Oncogene 24, 3110–3120, doi: 10.1038/sj.onc.1208513 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Tie F et al. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development 136, 3131–3141, doi: 10.1242/dev.037127 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Munoz LE, Lauber K, Schiller M, Manfredi AA & Herrmann M The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol 6, 280–289, doi: 10.1038/nrrheum.2010.46 (2010). [DOI] [PubMed] [Google Scholar]
- 43.Rock KL & Kono H The inflammatory response to cell death. Annu Rev Pathol 3, 99–126, doi: 10.1146/annurev.pathmechdis.3.121806.151456 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Raj D, Brash DE & Grossman D Keratinocyte apoptosis in epidermal development and disease. J Invest Dermatol 126, 243–257, doi: 10.1038/sj.jid.5700008 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sand J et al. Expression of inflammasome proteins and inflammasome activation occurs in human, but not in murine keratinocytes. Cell Death Dis 9, 24, doi: 10.1038/s41419-017-0009-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vaseva AV et al. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 149, 1536–1548, doi: 10.1016/j.cell.2012.05.014 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heath WR & Carbone FR The skin-resident and migratory immune system in steady state and memory: innate lymphocytes, dendritic cells and T cells. Nat Immunol 14, 978–985, doi: 10.1038/ni.2680 (2013). [DOI] [PubMed] [Google Scholar]
- 48.Rosenblum MD, Remedios KA & Abbas AK Mechanisms of human autoimmunity. J Clin Invest 125, 2228–2233, doi: 10.1172/JCI78088 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Park EK et al. Optimized THP-1 differentiation is required for the detection of responses to weak stimuli. Inflamm Res 56, 45–50, doi: 10.1007/s00011-007-6115-5 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Berges C et al. A cell line model for the differentiation of human dendritic cells. Biochem Biophys Res Commun 333, 896–907, doi: 10.1016/j.bbrc.2005.05.171 (2005). [DOI] [PubMed] [Google Scholar]
- 51.Tang J et al. Increased expression of the type I interferon-inducible gene, lymphocyte antigen 6 complex locus E, in peripheral blood cells is predictive of lupus activity in a large cohort of Chinese lupus patients. Lupus 17, 805–813, doi: 10.1177/0961203308089694 (2008). [DOI] [PubMed] [Google Scholar]
- 52.Maeda T, Chapman DL & Stewart AF Mammalian vestigial-like 2, a cofactor of TEF-1 and MEF2 transcription factors that promotes skeletal muscle differentiation. J Biol Chem 277, 48889–48898, doi: 10.1074/jbc.M206858200 (2002). [DOI] [PubMed] [Google Scholar]
- 53.Strachan DP Hay fever, hygiene, and household size. BMJ 299, 1259–1260, doi: 10.1136/bmj.299.6710.1259 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nagata S & Tanaka M Programmed cell death and the immune system. Nat Rev Immunol 17, 333–340, doi: 10.1038/nri.2016.153 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Yatim N, Cullen S & Albert ML Dying cells actively regulate adaptive immune responses. Nat Rev Immunol 17, 262–275, doi: 10.1038/nri.2017.9 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Clowse ME Lupus activity in pregnancy. Rheum Dis Clin North Am 33, 237–252, v, doi: 10.1016/j.rdc.2007.01.002 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Correa-Rodriguez M et al. The Prognostic Nutritional Index and Nutritional Risk Index Are Associated with Disease Activity in Patients with Systemic Lupus Erythematosus. Nutrients 11, doi: 10.3390/nu11030638 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Idborg H et al. TNF-alpha and plasma albumin as biomarkers of disease activity in systemic lupus erythematosus. Lupus Sci Med 5, e000260, doi: 10.1136/lupus-2018-000260 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cohen MJ et al. Severe malnutrition due to systemic lupus erythematosus associated protein losing enteropathy. Nutrition 28, 220–223, doi: 10.1016/j.nut.2011.07.017 (2012). [DOI] [PubMed] [Google Scholar]
- 60.NIH. Biennial Report of the Director, National Institutes of Health Fiscal Years 2006 & 2007, Summary of Activities by Disease Category, Autoimmune Diseases. (2007).
- 61.Tobias L A Briefing Report on Autoimmune Diseases and AARDA: Past, present, and future. (2010).
- 62.Faustman D Institute of Medicine Report, Women’s health research: progress, pitfalls, and promise’. (2010).
- 63.AARDA. The Cost Burden of Autoimmune Disease: The Latest Front in the War on Healthcare Spending. (2011).
- 64.Dorak MT & Karpuzoglu E Gender differences in cancer susceptibility: an inadequately addressed issue. Front Genet 3, 268, doi: 10.3389/fgene.2012.00268 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gambaro K et al. VGLL3 expression is associated with a tumor suppressor phenotype in epithelial ovarian cancer. Mol Oncol 7, 513–530, doi: 10.1016/j.molonc.2012.12.006 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Role of VGLL3 in keratinocyte stress response. a, qRT-PCR of VGLL3 under indicated treatment conditions in female keratinocytes. n=3, mean ±s.e.m, * P <0 .05, (*) Student’s t-test. b, western blot of indicated proteins under KGM (keratinocyte growth media, complete) or KBM (keratinocyte basal media, removing growth factors) culture conditions, with scrambled (Scr Ri) or VGLL3 (VGLL3 Ri) siRNA in male keratinocytes. c, qRT-PCR of indicated genes under KGM (keratinocyte growth media, complete) or KBM (keratinocyte basal media, removing growth factors) culture conditions, with scrambled (Scr Ri) or VGLL3 (VGLL3 Ri) siRNA in male keratinocytes. n=3, mean ±s.e.m, * P <0 .05, Student’s t-test. d, keratinocyte cell number under KGM (keratinocyte growth media, complete) culture condition with. e, keratinocyte cell number under KBM (keratinocyte basal media, removing growth factors) culture condition with scrambled (Scr Ri) or VGLL3 (VGLL3 Ri) siRNA, with or without Nutlin treatment conditions, with scrambled (Scr Ri) or VGLL3 (VGLL3 Ri) siRNA.
Supplemental Figure 2. Nutrient stress modifies VGLL3 chromatin. a, ChIP-qPCR of H3K27me3 on the three regulatory sites of VGLL3, under KGM (keratinocyte growth media, complete) or KBM (keratinocyte basal media, removing growth factors) culture conditions, with or without IFNα treatment in female keratinocytes. n=3, mean ±s.e.m. b, western blot of indicated proteins under KGM (keratinocyte growth media, complete) or KBM (keratinocyte basal media, removing growth factors) culture conditions, with or without IFNα treatment in male keratinocytes. c, ChIP-qPCR of H3K27me3 on VGLL3 promoter, under KGM (keratinocyte growth media, complete) or KBM (keratinocyte basal media, removing growth factors), with or without IFNα treatment in male keratinocytes. d, western blot of indicated proteins under KGM (keratinocyte growth media, complete) or KBM (keratinocyte basal media, removing growth factors) culture conditions, with or without IFNα treatment in female fibroblasts. e, western blot of indicated proteins under KGM (keratinocyte growth media, complete) or KBM (keratinocyte basal media, removing growth factors) culture conditions, with or without IFNα treatment in male fibroblasts.
Supplemental Figure 3. VGLL3 does not induce apoptosis or pyroptosis. a, immunofluorescence images of keratinocytes overexpressing GFP or VGLL3-GFP (red, active caspase-3; green, GFP; blue, DNA). b, immunofluorescence images of keratinocytes with or without staurosporine treatment (red, active caspase-3; blue, DNA). c, quantification of percentage of cells with active caspase-3 staining under indicated conditions. mean ±s.e.m, * P <0 .05, Student’s t-test. d, immunofluorescence images of keratinocytes overexpressing GFP or VGLL3-GFP (red, cleaved IL-1β; green, GFP; blue, DNA). b, immunofluorescence images of keratinocytes with or without UVB stimulation (red, cleaved IL-1β; blue, DNA). f, quantification of percentage of cells with cleaved IL-1β staining under indicated conditions. mean ± s.e.m, * P <0 .05, Student’s t-test.
Supplemental Figure 4. VGLL3 overexpression caused loss of mitochondria function. Immunofluorescence images of keratinocytes under indicated conditions (red, MitoTracker; green, GFP; blue, DNA). Arrows point to a representative VGLL3-overexpressing cell with loss of MitoTracker signal. Ri, RNAi. Scr, scrambled.
Supplemental Figure 5. VGLL3 upregulation in keratinocytes does not affect THP-1 chemotaxis or differentiation. a, number of monocytes migrated towards conditioned media from keratinocytes overexpressing GFP or VGLL3-GFP. n=5. b, percentage of CD14+ cells upon macrophage differentiation of THP-1 cells in the presence of conditioned media from keratinocytes overexpressing GFP or VGLL3-GFP. c, percentage of CD14+ and CD86+ cells upon dendritic cell differentiation of conditioned media from keratinocytes overexpressing GFP or VGLL3-GFP. n=3. d,e, Flow cytometry of indicated markers upon THP-1 differentiation into macrophages (d) or dendritic cells (e) after gating for singlets and viability.