

Abstract
Complex coacervation refers to the formation of distinct liquid phases that arise when polyelectrolytes are mixed under appropriate polymer and salt concentrations. Molecular-level studies of coacervation have been limited. In this work, a coarse-grained model of the polymers and the corresponding counterions is proposed and used to simulate coacervation as a function of polymer length and overall salt concentration. Several sampling methods are used to determine the phase behavior of the underlying polymers. In particular, the results of simulations in different ensembles are shown to be consistent and to reproduce a number of phenomena observed in experiments, including the disruption of complexation by increasing ionic strength or by decreasing molecular weight. The coacervate concentrations determined from phase behavior calculations are then used to examine the rheology of the corresponding materials. By relying on long dynamic simulations, we are able to generate the dynamic response of the material in the form of dynamic moduli as a function of frequency, which are also found to compare favorably with experimental measurements.
Graphical Abstract
INTRODUCTION
Charged polymers in solution can adopt a wide variety of conformations and morphologies. Examples include films,1 micelles,2,3 hydrogels,4,5 and coacervates.6–8 Such morphologies depend on molecular mass, polymer charge density, ratio of cationic and anionic groups, and the overall concentration of the polymer and added salts. They also depend on pH, which can be tuned to create reversible materials.9 Charged biocompatible polymers, in particular, are of interest for biomedical applications.10,11 For example, they can be used as wet adhesives12,13 that aid tissue healing and bone growth processes. They can also be used as dental implants or for drug delivery.14,15 Bunderberg de Jong and Kruyt16 originally introduced the term “coacervation” to describe the assembly of polyelectrolytes in dilute solution. They considered the formation of an aqueous phase rich in both polymers and salt, in equilibrium with an aqueous, polymer-free aqueous salt phase. There is now a growing interest in quantifying the thermodynamics and dynamics of coacervates in a more systematic manner.8,17–26 Spruijt et al.,17 in particular, examined a dilute mixture of poly(acrylic acid) (PAA) and poly(N,N-dimethylaminoethyl methacrylate) (PDMAEMA). That study is relevant from a modeling perspective because it considered polymer chains of opposite charge, but comparable molecular mass, and because the molecules were relatively small, i.e., in the range of 1.5–36 kg/mol for PAA. Furthermore, the pH was adjusted to remain in a range in which the polymers were fully ionized. A later study by Priftis et al.20 considered a ternary coacervate mixture of PAA, PDMAEMA, and poly-(ethylenimine) (PEI) or poly(allylamine) (PAH). These authors performed an extensive analysis of stoichiometry, salt concentration, and pH. That work is also highly relevant because it included a rheological characterization of the coacervate phase.
At the simplest level of theory, coacervation can be described in terms of the Voorn–Overbeek (VO) model,27 which provided the first theoretical description of coacervation. A number of features, however, are absent from this model, and recent theoretical and computational efforts have sought to develop more complete representations of polyelectrolytes in solution.
The “random phase approximation” (RPA) was first applied to polyelectrolyte solutions by Borue and Erukhimovich,28 who showed its validity for intermediate charge concentrations. RPA was applied to coacervate by Castelnovo and Joanny29 by implementing attractive electrostatic interaction. Later it was further developed by Kudlay and Olvera de la Cruz.30 Other theoretical work examples include the “restricted primitive model” (RPM),31 more recent studies based on liquid-state theory and approximate closures,32–35 and theoretical work aimed at incorporating the effects of charge connectivity and polymer architecture into the VO theory.36 Work by Wang37 included effects of chain connectivity as well as dielectric and charge correlations on the polyelectrolyte self-energy. Fluctuations beyond RPA have been captured by field-theoretic simulations (FTS) by Fredrickson and co-workers.38–42 Applicability of FTS to coacervates was first shown in the works of Popov and Lee.38,39 Recent works on FTS study phase behavior and interfacial tension of complexating homopolymers as well as copolyelectrolytes.40–42 Finally, a recent study of coacervation with Monte Carlo simulations was able to reproduce the phase diagram qualitatively, but the authors did not perform dynamical simulations to characterize the rheology of their systems.43 An extensive discussion of theoretical approaches to polyelectrolytes and specifically to polyelectrolyte complexation has been presented in a recent review by Muthukumar.44
Molecular simulations of coacervation have been limited. To the best of our knowledge, only one previous study45 considered coacervation involving relatively small chain molecules (≤24 beads) and this study of phase stability of coascervates involved Monte Carlo simulation rather than molecular dynamics. We also note the interesting work of Ou and Muthukumar46 used Langevin dynamics simulations to address polyelectrolyte complexation. That study highlighted the importance of entropic effects on complexation, but it was limited to only two chains. In this work we perform molecular simulations of a many-chain system and rely on simulations in various ensembles to determine the phase boundaries for coacervation. An important feature of our work is that having determined the composition of the coacervate phase, we proceed to predict its rheological properties. Specifically, by relying on long molecular dynamics simulations, we also calculate the linear response—or dynamic modulus—of the coacervate phase. The coarse-grained model considered here was parametrized to reproduce the experimental data of Spruijt et al.17,47 In general, its predictions are found to be in agreement with experimental observations, serving to underscore the validity of the approximations implicit in the model and providing a platform that might be of use for future studies of complex coacervation.
METHODS AND MODEL
The model considered here represents an extension of the so-called “theoretically informed coarse-grained model” (TICG)48 that has been used in the past to describe neutral polymer melts consisting of homopolymers, copolymers, composites, and their blends.49–51 The model uses a bead–;spring representation of polymer chains and single beads to represent ions and solvent. All of the polymer beads are charged. They are connected by stiff harmonic bonds, and their number per chain corresponds to the number of monomers. At short distances, the TICG model relies on soft-core interaction potentials. A Hamiltonian with a soft-core repulsion is written as,
| (1) |
where ri denotes the positions of particles and w(r) is a spherical step function:
Beads representing polymers, ions, and solvent have the same size (rcut) and the same interaction strength. Charged species—polymers and ions—also interact via the Coulomb potential. Its strength is expressed in terms of the Bjerrum length for water at room temperature). However, instead of a conventional Coulomb functional form, we introduce a modified soft Coulomb given by,
| (2) |
where r0 is a characteristic distance where repulsive forces become constant. The repulsion strength and rcut parameters are set by comparison to the combination of traditional hardcore LJ and Coulomb potentials, as shown in Figure 1.
Figure 1.

Modification of Coulomb force and comparison to inclusion of pairwise forces. “Soft Coulomb & Repulsion” are set to capture the interplay of long-range Coulomb and short-range LJ interactions “Coulomb+LJ”. The attractive Coulomb force is shown.
For oppositely charged particles, the combination of LJ and Coulomb potentials considered here has a minimum at a distance r0, where the force is equal to 0. Note that forces labeled as “Coulomb+LJ” and “Soft Coulomb & Repulsion” have the same root at a distance r0. An overall comparison of the potentials is given in Figure 1. The strength for the LJ potential is set to , and the cutoff radius is set to 2.5σ. As one can appreciate in the figure, the long-range nature of the electrostatic interactions is unchanged, and it is only the close-range packing behavior that is affected by the revised potential. We anticipate that such short-range behavior plays a minor role in coacervation, which occurs primarily in dilute or semidilute solutions. Electrostatic interactions between charged species are implemented in the context of a particle–particle particle–mesh (PPPM) Ewald summation method.52–54
We use a Langevin thermostat under constant volume conditions to evolve the system through modified velocity-Verlet algorithm55 (see the Supporting Information for details).
RESULTS
We seek to validate the model by comparing its results to the experimental data Spruijt et al.;17 as mentioned above, they considered mixtures of PAA and PDMAEMA with degrees of polymerization (N) of 20, 50, and 140. Following experiments, our system consists of a complex coacervate formed by the stoichiometric blend of oppositely charged polyelectrolytes with the same number of segments. In what follows, we refer to these mixtures as N20, N50, and N140. To set a length scale for our model, we rely on data from atomistic molecular dynamics simulations, which place the end-to-end distance of a 140 monomer PAA chain at 10.2 nm.56 We use that length as the simulation unit for our model. A monomer concentration of 0.11 mol L−1 corresponds to 71 polymer beads per unit of volume (10.23 nm3).
We follow two different strategies to determine the phase behavior of our model system. We do so because simulations of the coexistence of long, charged polymers have not been attempted before, and it is therefore important to cross-validate the corresponding results. In the first of these, we rely on an iterative process to equate the pressure and chemical potentials of salt ions and solvent in the two phases. The concentrations of polymer beads, salt ions, and solvent beads are the key variables in the coacervate phase (, , and ); we assume that the supernatant contains no polymer , and we fix the salt concentration . For all salt concentrations, the supernatant’s total bead density is held constant by altering the solvent content. We use large simulation boxes, and the results presented here represent an average over uncorrelated configurations separated by 105 timesteps. The chemical potential of the system is determined through a Widom insertion technique.57 Ideal gas contributions to the chemical potentials and the pressure are included after the fact according to and , where i is salt or solvent, n is the concentration, and PMD is a negative one-third of the trace of the stress tensor. Typical configurations of simulation boxes are presented in Figure S1. Figure S2 illustrates a typical workflow for the iterative calculation.
In the second strategy, we rely on simulations in the Gibbs ensemble. In that case, simulation boxes communicate directly via Monte Carlo exchange moves with traditional Metropolis acceptance criterion. The goal of the exchanges is to equilibrate a species’ chemical potential between the boxes. Exchanges are allowed for all species except the polymers; all polymers are restricted to remain in the “coacervate” box. In our Gibbs ensemble calculations, we fix the polymer concentration at the values provided by the previous iterative Widom-insertion-based calculations. For ions, exchanges are performed in pairs to ensure electroneutrality. Additionally, in between Monte Carlo steps, the boxes relax via molecular dynamics at constant pressure condition. As shown in the Supporting Information (Figure S3), both strategies adopted here lead to the same results, serving to establish the validity of the underlying methods and codes.
Spruijt et al.17 reported that the concentration of polymers in supernatant is on the order of 0.01–0.1 mol L−1. Simulating 100 chains of 50 monomers at 0.01 mol L−1 with our model requires a simulation box with V = 142 × 10.23 nm3 containing 213000 beads total. Our calculations are focused on the region far away from the critical point, and we therefore assume that the polymer concentration in the supernatant is negligible—a feature that is consistent with experiments and that simplifies our calculations considerably. Note that such an assumption was also justified by a recent work of Delaney and Fredrickson,42 where very low polymer concentration in the supernatant was observed with field-theoretic simulations.
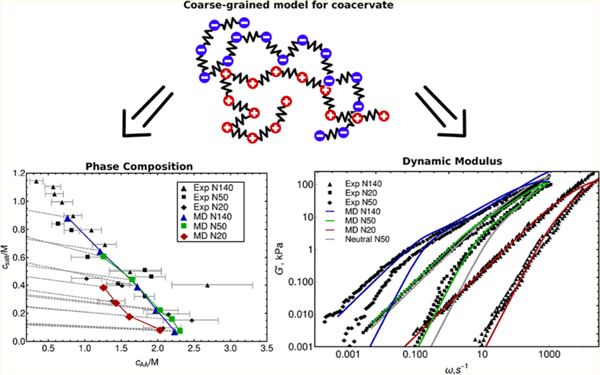
The results of our simulations are presented in Figure 2. We compare the phase diagram extracted from simulations to experimental data. Gray dashed lines represent tie-lines for the salt concentration in the coacervate and the corresponding supernatant phase. In general, one can appreciate that salt concentration in the coacervate phase is lower than in the supernatant. The simulated phase diagram appears to reproduce the experimentally observed effect of molecular weight on coacervation. Shorter chains (N20) show a significantly lower concentration of polymer than longer chains and exhibit a much stronger sensitivity to the overall salt content. The N20 coacervate phase formation is disrupted by high salt content; coacervation is no longer possible above 0.5 M salt. Overall, the effect of polymer molecular mass on phase behavior is more pronounced when comparing the short- and intermediate-length polyelectrolytes N20 and N50; the difference in coacervate composition in going from N50 to N140 is less evident. It seems a stronger correlation between like-charge beads, due to bonded forces, enhances the assembly process; longer chains lead to denser coacervates. On the basis of these observations, we anticipate that the effects of chain length will saturate at some point, with polymer concentration in the coacervate becoming independent of molecular mass. The overall agreement with experiment is satisfactory, particularly given the difficulty of performing such measurements and the large error bars that are associated with the corresponding experimental data. For completeness, we also show results of calculations with the Voorn–Overbeek model in Figure 3 as presented in work by Spruijt et al.,17 with model parameters: α = 0.9, σ = 0.95; Np is equal to number of monomers. Note that several parameters in the model were adjusted by those authors to fit the data, leading to a concentration of polymer that reproduces experiments and that captures at a qualitative level the general trends observed in experiments (dependence on molecular mass and salt concentration).
Figure 2.

Prediction of composition of the supernatant and the coacervate and comparison with experimental data by Spruijt et al.17 Here cAA is monomer concentration of polyanion (poly(acrylic acid)), and csalt is concentration of added salt ions. Filled color symbols are coacervate predictions of the model, filled black symbols correspond to experimental data for coacervate, and empty color symbols are supernatant predictions of the model; experimental data for supernatant is not shown. For all predictions of supernatant the polyelectrolyte concentration is 0. Predictions are obtained with Gibbs ensemble calculations. Dashed lines show corresponding supernatant and coacervate phases. See Spruijit et al.17 for a discussion of the determination of the uncertainty in the measurements.
Figure 3.

Prediction by the Voorn–Overbeek model and comparison with experimental data as presented by Spruijt et al.17 Model parameters: α = 0.9, σ = 0.95; Np is equal to the number of monomers.
Having shown that the coarse-grained model introduced here is capable of describing the equilibrium phase behavior, we turn our attention to the dynamic response of the model in the linear regime and refer to later experimental work of Spruijt et al.47 on the same system. In that study, the authors characterized the effect of salt concentration on relaxation times. Interestingly, upon addition of salt, the shape of the modulus curve over a wide range of frequencies is unaffected, but relaxation happens at shorter time scales. In other words, the whole dynamic modulus is shifted to higher frequencies, leading to a “time–salt superposition” behavior that was reminiscent of that observed in neutral polymer solutions as temperature is changed. More specifically, the relaxation curves for different salt concentrations could be shifted and superimposed on each other, leading to a universal curve. Spruijt et al.47 exploited that feature to arrive at a complete mechanical relaxation spectrum spanning several orders of magnitude in the frequency domain. Figure 4 shows the mean-squared displacement (MSD) of the polymers’ center of mass for concentrations corresponding to the equilibrium coacervate phase. The results are consistent with fluid-like behavior and exhibit a diffusive regime at long times. However, the electrostatic forces lead to an intermediate subdiffusive region that is particularly pronounced in the N50 and N140 systems. For reference, Figure 4 also shows the MSD for electrostatically neutral systems having the same composition as the coacervate. The neutral system does not exhibit a subdiffusive region, and the corresponding diffusion coefficient is larger by a factor of ≈2.4 for N20 and N50 and by a factor of ≈1.7 for N140. These results show that electrostatic attractions between beads of different polymers and ions slow down motion and change the overall dynamics of the coacervate system. However, in contrast to the experimental observations mentioned above, we do not observe the relaxation time dependence on salt content (see the insets of Figure 4). Instead, we observe a slight, insignificant change in the MSD with added salt. The changes are systematic with higher salt concentration, resulting in larger MSD. Importantly, the changes in diffusion coefficient are on the order of 1% at most, orders of magnitude smaller than in experiments.
Figure 4.

Soft-core model prediction for polymer center-of-mass mean-square displacement. Also shown is the mean-square displacement for the neutral polymer at the same concentration as coacervate. Insets highlight the small effect of salt concentration on MSD.
Figure 5 shows the dynamic modulus G* of the coacervate as a function of molecular weight. As expected from the MSD results, G* exhibits liquid-like behavior, with a loss modulus that is higher than the storage modulus everywhere except in the high-frequency region. Compared to the dynamic modulus of neutral polymers, which can be described by the Rouse model, the coacervate has a more slowly decaying response at intermediate frequencies and a stronger elastic component. Electrostatic interactions in the coacervate phase shift the terminal relaxation region to much lower frequencies. It is important to mention that these extensive relaxation calculations are only accessible through the use of a soft-core model implemented on GPUs. In agreement with the MSD results, we observe only slight changes in G* with salt content, as shown in Figure S4. The Supporting Information also provides details pertaining to the dynamic modulus G*. The shape of the dynamic modulus is similar for all salt concentrations considered here; changes in the height, however, are proportional to polymer concentration in the coacervate. Rather than presenting the dependence of G* on salt content, we focus on a comparison of the dynamic modulus of our model to that reported in the measurements of Spruijt et al.47 The model predictions were individually shifted to superimpose them on the experimental data. The frequency shifts factors are 0.0006 s, 0.012 s, and 0.12 s for N20, N50, and N140, respectively. The vertical shift is 500 kPa. The predictions of the model can be seen to capture both the length and slope of the intermediate relaxation region for all molecular weights. A comparison to results for neutral polymers shows that electrostatic interactions play a key role in leading to agreement with such experimentally observed features and shapes. Unfortunately, however, the model cannot simultaneously capture the experimental time scales for all the molecular weights as well as the effect of salt concentration. Specifically, it does not reproduce the time–salt superposition reported by Spruijt et al. It is important to point out that the estimated overlap concentrations58 for the polymers considered here are c* = (7.6 mol L−1, 4.8 mol L−1, and 2.8 mol L−1) for N20, N50, and N140, respectively. The highest concentration considered in our rheological calculations of coacervates is 2.2 mol L−1, thereby suggesting that overlap concentration or entanglement effects play a negligible role in the effects outlined above. Given the overall agreement with the experimental phase composition and the shape of the dynamic modulus curves, we anticipate that we should be able to capture the reported salt–time superposition behavior by modifying ion mobility–water dispersion interaction strength, and thus the relative ion mobility. A feature that was not included in our simulations is the effect of ion hydration as a function of salt concentration; note that experimental data indicate that ion hydration plays an important role in the dynamic properties (e.g., viscosity) of salt solutions.59–61 We therefore speculate that such effects could be partly responsible for deviations in the absolute frequency observed in experiments and simulations—a feature that we plan to address in future work.62
Figure 5.

Prediction for dynamic modulus superimposed on experimental data.47 Filled symbols show the G′ elastic component of the modulus, and empty symbols show the G′ viscous component of the modulus. Predictions are scaled by 500 kPa vertically and by {0.0006 s, 0.012 s, and 0.12 s} for N20, N50, and N140 horizontally. Also shown is the dynamic modulus of the neutral N50 polymer at the same concentration as coacervate.
CONCLUSION
We conclude our observations by pointing out that a simple model consisting for charged bead–spring polymers is capable of capturing the complex coacervation behavior of polyelectrolytes. A first important prediction of the model is that in a good solvent the concentration of salt in the coacervate phase is slightly larger than in the supernatant phase. This result is contrary to the predictions of the VO model, where the concentration of salt is higher in the supernatant than in the coacervate. Note that past analysis of experimental data assumed that the salt concentration is comparable in the two coexisting phases—a feature that is not generally valid. A second important prediction of the model is that the concentration of polymer in the coacervate phase increases with molecular weight only up to a point after which it appears to become saturated. A simulation of the thermodynamic and transport properties of polyelectrolytes has been proposed. The model consists of an extension of previous models shown to provide excellent agreement with experiment in studies of neutral polymers, block polymers, and their mixtures. The predictions of the model are shown to be in good agreement with experimental measurements of polymer coacervation and capture the effects of salt concentration and polymer length. Salt concentration is found to be lower in the coacervate phase than in the supernatant phase. Precise data on salt concentration are not available, and it will be important to address and verify this prediction in future experimental and theoretical work. We note that the model relies on soft potentials to attain long time scales. As presented here, the model is able to describe the general effects of molecular mass and salt on the dynamic modulus, but it is unable to describe the salt–temperature superposition behavior that has been observed in recent experiments. We suggest that this discrepancy is due to the hydration of the ions, but at the same time we stress that additional experiments and more detailed models should be used to address this issue. Moving forward, it will also be of interest to examine how chain stiffness and ion valency influence not only phase behavior, but also the rheology of coacervates.63
Supplementary Material
ACKNOWLEDGMENTS
This work was performed under the following financial assistance award 70NANB14H012 from U.S. Department of Commerce, National Institute of Standards and Technology as part of the Center for Hierarchical Materials Design (CHiMaD).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.macromol.8b00556 .
Details of the integration method; details of the phase diagram calculation, including typical pathway of the iterative method, representative simulation snapshots and comparison of the phase diagrams for the iterative and Gibbs ensemble methods; dynamic modulus in simulation units as a function of the salt concentration; table for conversion between the simulation units and the real units (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Capito RM; Azevedo HS; Velichko YS; Mata A; Stupp SI. Self-Assembly of Large and Small Molecules into Hierarchically Ordered Sacs and Membranes. Science 2008, 319, 1812–1816. [DOI] [PubMed] [Google Scholar]
- (2).Cohen Stuart MA; Besseling NAM; Fokkink RG. Formation of Micelles with Complex Coacervate Cores. Langmuir 1998, 14, 6846–6849. [Google Scholar]
- (3).Penchev H; Paneva D; Manolova N; Rashkov I. Novel Electrospun Nanofibers Composed of Polyelectrolyte Complexes. Macromol. Rapid Commun 2008, 29, 677–681. [Google Scholar]
- (4).Hunt JN; Feldman KE; Lynd NA; Deek J; Campos LM; Spruell JM; Hernandez BM; Kramer EJ; Hawker CJ. Tunable, High Modulus Hydrogels Driven by Ionic Coacervation. Adv. Mater 2011, 23, 2327–2331. [DOI] [PubMed] [Google Scholar]
- (5).Srivastava S; Andreev M; Levi AE; Goldfeld DJ; Mao J; Heller WT; Prabhu VM; de Pablo JJ; Tirrell MV. Gel Phase Formation in Dilute Triblock Copolyelectrolyte Complexes. Nat. Commun 2017, 8, 14131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Biesheuvel PM; Cohen Stuart MA. Electrostatic Free Energy of Weakly Charged Macromolecules in Solution and Intermacromolecular Complexes Consisting of Oppositely Charged Polymers. Langmuir 2004, 20, 2785–2791. [DOI] [PubMed] [Google Scholar]
- (7).Bucur CB; Sui Z; Schlenoff JB. Ideal Mixing in Polyelectrolyte Complexes and Multilayers: Entropy Driven Assembly. J. Am. Chem. Soc 2006, 128, 13690–13691. [DOI] [PubMed] [Google Scholar]
- (8).Priftis D; Laugel N; Tirrell M. Thermodynamic Characterization of Polypeptide Complex Coacervation. Langmuir 2012, 28, 15947–15957. [DOI] [PubMed] [Google Scholar]
- (9).Lemmers M; Sprakel J; Voets I; van der Gucht J; Cohen Stuart M. Multiresponsive Reversible Gels Based on Charge-Driven Assembly. Angew. Chem 2010, 122, 720–723. [DOI] [PubMed] [Google Scholar]
- (10).Wang X; Lee J; Wang Y-W; Huang Q. Composition and Rheological Properties of β-Lactoglobulin/Pectin Coacervates: Effects of Salt Concentration and Initial Protein/Polysaccharide Ratio. Biomacromolecules 2007, 8, 992–997. [DOI] [PubMed] [Google Scholar]
- (11).Cirulis JT; Keeley FW; James DF. Viscoelastic Properties and Gelation of an Elastin-Like Polypeptide. J. Rheol 2009, 53, 1215–1228. [Google Scholar]
- (12).Winslow BD; Shao H; Stewart RJ; Tresco PA. Biocompatibility of Adhesive Complex Coacervates Modeled after the Sandcastle Glue of Phragmatopoma Californica for Craniofacial Reconstruction. Biomaterials 2010, 31, 9373–9381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Stewart RJ; Wang CS; Shao H. Complex Coacervates as a Foundation for Synthetic Underwater Adhesives. Adv. Colloid Interface Sci 2011, 167, 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Thomasin C; Nam-Trân H; Merkle HP; Gander B. Drug Microencapsulation by PLA/PLGA Coacervation in the Light of Thermodynamics. 1. Overview and Theoretical Considerations. J. Pharm. Sci 1998, 87, 259–268. [DOI] [PubMed] [Google Scholar]
- (15).Luzzi LA. Microencapsulation. J. Pharm. Sci 1970, 59, 1367–1376. [DOI] [PubMed] [Google Scholar]
- (16).Jong H. G. B. d.; Kruyt HR. Coacervation. Proc. Koninkl. Med. Akad. Wetershap 1929, 32, 849–856. [Google Scholar]
- (17).Spruijt E; Westphal AH; Borst JW; Cohen Stuart MA; van der Gucht J. Binodal Compositions of Polyelectrolyte Complexes. Macromolecules 2010, 43, 6476–6484. [Google Scholar]
- (18).Priftis D; Tirrell M. Phase Behaviour and Complex Coacervation of Aqueous Polypeptide Solutions. Soft Matter 2012, 8, 9396–9405. [Google Scholar]
- (19).Priftis D; Farina R; Tirrell M. Interfacial Energy of Polypeptide Complex Coacervates Measured via Capillary Adhesion. Langmuir 2012, 28, 8721–8729. [DOI] [PubMed] [Google Scholar]
- (20).Priftis D; Xia X; Margossian KO; Perry SL; Leon L; Qin J; de Pablo JJ; Tirrell M. Ternary, Tunable Polyelectrolyte Complex Fluids Driven by Complex Coacervation. Macromolecules 2014, 47, 3076–3085. [Google Scholar]
- (21).Hwang DS; Zeng H; Srivastava A; Krogstad DV; Tirrell M; Israelachvili JN; Waite JH. Viscosity and Interfacial Properties in a Mussel-Inspired Adhesive Coacervate. Soft Matter 2010, 6, 3232–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Chollakup R; Beck JB; Dirnberger K; Tirrell M; Eisenbach CD. Polyelectrolyte Molecular Weight and Salt Effects on the Phase Behavior and Coacervation of Aqueous Solutions of Poly(acrylic acid) Sodium Salt and Poly(allylamine) Hydrochloride. Macromolecules 2013, 46, 2376–2390. [Google Scholar]
- (23).Perry SL; Li Y; Priftis D; Leon L; Tirrell M. The Effect of Salt on the Complex Coacervation of Vinyl Polyelectrolytes. Polymers 2014, 6, 1756. [Google Scholar]
- (24).Jha PK; Desai PS; Li J; Larson RG. pH and Salt Effects on the Associative Phase Separation of Oppositely Charged Polyelectrolytes. Polymers 2014, 6, 1414. [Google Scholar]
- (25).Perry SL; Leon L; Hoffmann KQ; Kade MJ; Priftis D; Black KA; Wong D; Klein RA; Pierce CF III; Margossian KO; et al. Chirality-Selected Phase Behaviour in Ionic Polypeptide Complexes. Nat. Commun 2015, 6, 6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Hoda N; Larson RG. Explicit- and Implicit-Solvent Molecular Dynamics Simulations of Complex Formation between Polycations and Polyanions. Macromolecules 2009, 42, 8851–8863. [Google Scholar]
- (27).Overbeek JTG; Voorn MJ. Phase Separation in Polyelectrolyte Solutions. Theory of Complex Coacervation. J. Cell. Comp. Physiol 1957, 49, 7–26. [PubMed] [Google Scholar]
- (28).Borue VY; Erukhimovich IY. A Statistical Theory of Weakly Charged Polyelectrolytes: Fluctuations, Equation of State and Microphase Separation. Macromolecules 1988, 21, 3240–3249. [Google Scholar]
- (29).Castelnovo M; Joanny JF. Phase Diagram of Diblock Polyampholyte Solutions. Macromolecules 2002, 35, 4531–4538. [Google Scholar]
- (30).Kudlay A; Ermoshkin AV; Olvera de la Cruz M. Complexation of Oppositely Charged Polyelectrolytes: Effect of Ion Pair Formation. Macromolecules 2004, 37, 9231–9241. [Google Scholar]
- (31).Mahdi KA; Olvera de la Cruz M. Phase Diagrams of Salt-Free Polyelectrolyte Semidilute Solutions. Macromolecules 2000, 33, 7649–7654. [Google Scholar]
- (32).Perry SL; Sing CE. PRISM-Based Theory of Complex Coacervation: Excluded Volume versus Chain Correlation. Macromolecules 2015, 48, 5040–5053. [Google Scholar]
- (33).Kayitmazer AB; Seeman D; Minsky BB; Dubin PL; Xu Y. Protein-Polyelectrolyte Interactions. Soft Matter 2013, 9, 2553–2583. [Google Scholar]
- (34).Jeon J; Dobrynin AV. Monte Carlo Simulations of Polyampholyte-Polyelectrolyte Complexes: Effect of Charge Sequence and Strength of Electrostatic Interactions. Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top 2003, 67, 061803. [DOI] [PubMed] [Google Scholar]
- (35).Jeon J; Dobrynin AV. Molecular Dynamics Simulations of Polyampholyte-Polyelectrolyte Complexes in Solutions. Macromolecules 2005, 38, 5300–5312. [Google Scholar]
- (36).Qin J; de Pablo JJ. Criticality and Connectivity in Macromolecular Charge Complexation. Macromolecules 2016, 49, 8789–8800. [Google Scholar]
- (37).Shen K; Wang Z-G. Electrostatic Correlations and the Polyelectrolyte Self Energy. J. Chem. Phys 2017, 146, 084901. [DOI] [PubMed] [Google Scholar]
- (38).Popov YO; Lee J; Fredrickson GH. Field-Theoretic Simulations of Polyelectrolyte Complexation. J. Polym. Sci., Part B: Polym. Phys 2007, 45, 3223–3230. [Google Scholar]
- (39).Lee J; Popov YO; Fredrickson GH. Complex Coacervation: A Field Theoretic Simulation Study of Polyelectrolyte Complexation. J. Chem. Phys 2008, 128, 224908. [DOI] [PubMed] [Google Scholar]
- (40).Riggleman RA; Kumar R; Fredrickson GH. Investigation of the Interfacial Tension of Complex Coacervates Using Field-Theoretic Simulations. J. Chem. Phys 2012, 136, 024903. [DOI] [PubMed] [Google Scholar]
- (41).Audus DJ; Gopez JD; Krogstad DV; Lynd NA; Kramer EJ; Hawker CJ; Fredrickson GH. Phase Behavior of Electrostatically Complexed Polyelectrolyte Gels Using an Embedded Fluctuation Model. Soft Matter 2015, 11, 1214–1225. [DOI] [PubMed] [Google Scholar]
- (42).Delaney KT; Fredrickson GH. Theory of Polyelectrolyte Complexation-Complex Coacervates are Self-Coacervates. J. Chem. Phys 2017, 146, 224902. [DOI] [PubMed] [Google Scholar]
- (43).Lytle TK; Sing CE. Transfer Matrix Theory of Polymer Complex Coacervation. Soft Matter 2017, 13, 7001–7012. [DOI] [PubMed] [Google Scholar]
- (44).Muthukumar M. 50th Anniversary Perspective: A Perspective on Polyelectrolyte Solutions. Macromolecules 2017, 50, 9528–9560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Orkoulas G; Kumar SK; Panagiotopoulos AZ. Monte Carlo Study of Coulombic Criticality in Polyelectrolytes. Phys. Rev. Lett 2003, 90, 048303. [DOI] [PubMed] [Google Scholar]
- (46).Ou Z; Muthukumar M. Entropy and Enthalpy of Polyelectrolyte Complexation: Langevin Dynamics Simulations. J. Chem. Phys 2006, 124, 154902. [DOI] [PubMed] [Google Scholar]
- (47).Spruijt E; Cohen Stuart MA; van der Gucht J. Linear Viscoelasticity of Polyelectrolyte Complex Coacervates. Macromolecules 2013, 46, 1633–1641. [Google Scholar]
- (48).Pike DQ; Detcheverry FA; Müller M; de Pablo JJ. Theoretically Informed Coarse Grain Simulations of Polymeric Systems. J. Chem. Phys 2009, 131, 084903. [DOI] [PubMed] [Google Scholar]
- (49).Detcheverry FA; Kang H; Daoulas KC; Müller M; Nealey PF; de Pablo JJ. Monte Carlo Simulations of a Coarse Grain Model for Block Copolymers and Nanocomposites. Macromolecules 2008, 41, 4989–5001. [Google Scholar]
- (50).Pike DQ; Müller M; de Pablo JJ. Monte-Carlo simulation of ternary blends of block copolymers and homopolymers. J. Chem. Phys 2011, 135, 114904. [DOI] [PubMed] [Google Scholar]
- (51).Ramírez-Hernández A; Peters BL; Andreev M; Schieber JD; de Pablo JJ. A Multichain Polymer Slip-Spring Model with Fluctuating Number of Entanglements for Linear and Nonlinear Rheology. J. Chem. Phys 2015, 143, 243147. [DOI] [PubMed] [Google Scholar]
- (52).Ewald PP. Die Berechnung Optischer und Elektrostatischer Gitterpotentiale. Ann. Phys. (Berlin, Ger.) 1921, 369, 253–287. [Google Scholar]
- (53).Hockney R; Goel S; Eastwood J. Quiet High-Resolution Computer Models of a Plasma. J. Comput. Phys 1974, 14, 148–158. [Google Scholar]
- (54).LeBard DN; Levine BG; Mertmann P; Barr SA; Jusufi A; Sanders S; Klein ML; Panagiotopoulos AZ. Self-Assembly of Coarse-Grained Ionic Surfactants Accelerated by Graphics Processing Units. Soft Matter 2012, 8, 2385–2397. [Google Scholar]
- (55).Miller TF; Eleftheriou M; Pattnaik P; Ndirango A; Newns D; Martyna GJ. Symplectic Quaternion Scheme for Biophysical Molecular Dynamics. J. Chem. Phys 2002, 116, 8649–8659. [Google Scholar]
- (56).Cranford SW; Buehler MJ. Variation of Weak Polyelectrolyte Persistence Length through an Electrostatic Contour Length. Macromolecules 2012, 45, 8067–8082. [Google Scholar]
- (57).Widom B. Some Topics in the Theory of Fluids. J. Chem. Phys 1963, 39, 2808–2812. [Google Scholar]
- (58).Graessley WW. Polymer Chain Dimensions and the Dependence of Viscoelastic Properties on Concentration, Molecular Weight and Solvent Power. Polymer 1980, 21, 258–262. [Google Scholar]
- (59).Jenkins HDB; Marcus Y. Viscosity B-Coefficients of Ions in Solution. Chem. Rev 1995, 95, 2695–2724. [Google Scholar]
- (60).Collins KD. Sticky Ions in Biological Systems. Proc. Natl. Acad. Sci. U. S. A 1995, 92, 5553–5557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Kim JS; Wu Z; Morrow AR; Yethiraj A; Yethiraj A. Self-Diffusion and Viscosity in Electrolyte Solutions. J. Phys. Chem. B 2012, 116, 12007–12013. [DOI] [PubMed] [Google Scholar]
- (62).Andreev M; Chremos A; de Pablo J; Douglas JF. Coarse-Grained Model of the Dynamics of Electrolyte Solutions. J. Phys. Chem. B 2017, 121, 8195–8202. [DOI] [PubMed] [Google Scholar]
- (63).Lytle TK; Sing CE. Tuning chain interaction entropy in complex coacervation using polymer stiffness, architecture, and salt valency. Mol. Syst. Des. Eng 2018, 3, 183–196. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.