
Figure 1.
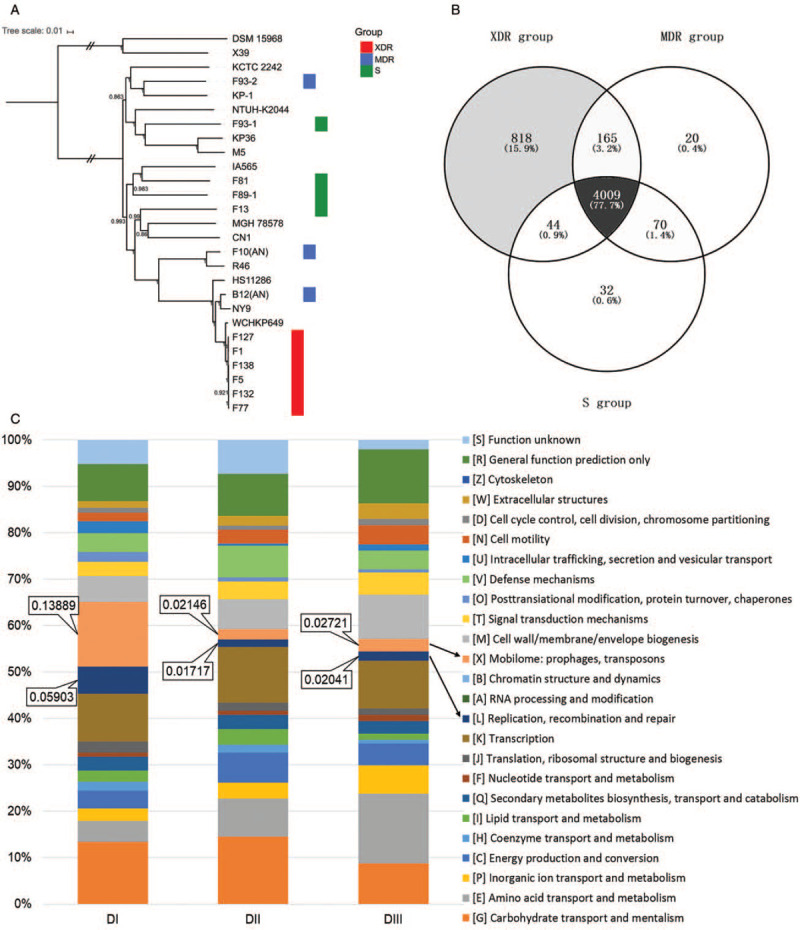
(A) The phylogeny analysis of K. pneumoniae strains. SNPs across the 27 examined genomes were called. The core SNPs were used to construct phylogenetic trees using the ML method based approach FastTreeMP with 1000 bootstraps. K. variicola X39 and DSM 15968 was included as the out-group sequences. The branch bearing double hatch marks indicates that it has been truncated and is not proportional to the rest. (B) Venn diagram of the tally of homologous proteins shared by or unique among K. pneumoniae strains of the XDR, MDR, and S groups. The core genome for XDR, MDR, or S group was determined as homologous proteins presenting in all of the isolates inside the group. The “supercore” genome was determined as the core genes that were present in each of the groups. The dispensable genome of each group were created by subtracting the “supercore” from each group's core genome. (C) Distribution of COG functional categories among DI, DII, and DIII. All proteins in DI, DII, and DIII were aligned against the COG database and comparisons among DI, DII, and DIII were conducted. COG: Clusters of Orthologous Groups; K. pneumoniae: Klebsiella pneumoniae; MDR: Multidrug-resistant; ML: Maximum-likelihood; SNPs: Single-nucleotide polymorphisms; XDR: Extensively drug resistant.