

ABSTRACT
Phosphoinositides (PIPs) are a dynamic family of lipids that execute diverse roles in cell biology. PIP levels are regulated by numerous enzymes, but our understanding of how these enzymes are controlled in space and time is incomplete. One role of the PIP phosphatidylinositol (4,5)-bisphosphate [PI(4,5)P2] is to anchor the cytokinetic ring (CR) to the plasma membrane (PM) in Schizosaccharomyces pombe. While examining potential PI(4,5)P2-binding proteins for roles in CR anchoring, we identified the dual pleckstrin homology (PH) domain-containing protein Opy1. Although related proteins are implicated in PIP regulation, we found no role for S. pombe Opy1 in CR anchoring, which would be expected if it modulated PM PI(4,5)P2 levels. Our data indicate that although Opy1 senses PM PI(4,5)P2 levels and binds to the phosphatidylinositol 4-phosphate 5-kinase (PI5-kinase) Its3, Opy1 does not regulate Its3 kinase activity or PM PI(4,5)P2 levels, a striking difference from its Saccharomyces cerevisiae homolog. However, overexpression of Opy1 resulted in cytokinesis defects, as might be expected if it sequestered PI(4,5)P2. Our results highlight the evolutionary divergence of dual PH domain-containing proteins and the need for caution when interpreting results based on their overexpression.
This article has an associated First Person interview with the first author of the paper.
KEY WORDS: Phosphoinositides, Lipid kinase, Opy1, Fission yeast
Summary: The fission yeast dual PH domain-containing protein Opy1 binds PI(4,5)P2 and senses this lipid in the plasma membrane. Unlike related proteins, Opy1 binds but does not regulate phosphatidylinositol 4-phosphate 5-kinase.
INTRODUCTION
Phosphoinositides (PIPs) are a family of lipids that are essential for signaling, membrane identity and diverse cellular processes involving membrane rearrangements (reviewed by Schink et al., 2016). The different membranes in the cell are each comprised of a unique PIP composition, and these differences are driven and regulated by the activities of numerous PIP kinases, phosphatases and lipases (Dickson and Hille, 2019). Despite decades of study, our understanding of how each of these enzymes is coordinated in space and time to control PIP levels and their downstream effectors remains incomplete.
Phosphatidylinositol (4,5)-bisphosphate [PI(4,5)P2] is the most abundant PIP species in the plasma membrane (PM). Proper PM PI(4,5)P2 promotes cell division in multiple eukaryotic organisms (Field et al., 2005; Snider et al., 2018, 2017; Wong et al., 2005). In Schizosaccharomyces pombe, PI(4,5)P2 is important for anchorage of the cytokinetic ring (CR) in the cell center, which in turn promotes medial division and avoids damage to the dividing genome (Snider et al., 2018, 2017). PM PI(4,5)P2 is produced in S. pombe by the phosphatidylinositol 4-kinase (PI4-kinase) Stt4, which is localized to the PM by its binding partners Efr3 and Ypp1 (Snider et al., 2017). There, it converts phosphatidylinositol (PI) to phosphatidylinositol-4-phosphate (PI4P), which is subsequently phosphorylated by the phosphatidylinositol 4-phosphate 5-kinase (PI5-kinase) Its3 to generate PI(4,5)P2. Loss of function of Stt4 complex members (efr3Δ, GFP–stt4) or Its3 (its3-1) results in reduced PM PI(4,5)P2, CR sliding events and subsequent off-center septation (Snider et al., 2018, 2017), underscoring the importance of PM PI(4,5)P2 for eukaryotic cytokinesis.
In our effort to understand which PM components modulate CR anchoring and are affected by efr3Δ and its3-1, we identified Opy1. Opy1 localizes to the cell cortex in punctae, but upon reduction in PM PI(4,5)P2 in efr3Δ or its3-1, it becomes primarily cytosolic (Snider et al., 2018, 2017). Although Opy1 localization to the PM is sensitive to PIP content, opy1Δ cells do not display CR sliding (Snider et al., 2018). Additionally, we previously demonstrated using lipid biosensors that PI4P and PI(4,5)P2 PM levels in opy1Δ are indistinguishable from those in wild-type cells (Snider et al., 2018). Taken together, these results indicate that S. pombe Opy1 is neither a major CR–PM anchor nor a regulator of PIP composition.
Opy1 is the presumptive ortholog of Saccharomyces cerevisiae Opy1, based on protein sequence similarity. Both S. cerevisiae and S. pombe Opy1 proteins contain one N- and one C-terminal pleckstrin homology (PH) domain (Fig. 1A) (Ling et al., 2012). PH domains are common in eukaryotic proteomes and are well-appreciated to be potential membrane-binding modules. Although once thought to be exclusively PIP-binding domains, only a small fraction of PH domains bind a particular PIP with specificity, and not all PH domains bind membranes (Lemmon, 2007). A function was proposed for each of the two S. cerevisiae Opy1 PH domains in regulation of the PI5-kinase and Its3 ortholog, Mss4. According to this model, Opy1 acts as a coincidence detector by binding PI(4,5)P2 at the PM through its C-terminal PH domain and simultaneously binding Mss4 to inhibit its kinase activity when PI(4,5)P2 levels are high (Ling et al., 2012). Although this is an appealing model, it has not yet been demonstrated that Mss4 and Opy1 interact directly in vitro or that Mss4 kinase activity is diminished in the presence of Opy1.
Fig. 1.

Opy1 binds PI(4,5)P2-containing membranes. (A) Schematic of Opy1. Amino acid numbers are indicated below the schematic. PH1 and PH2 indicate pleckstrin homology domains. (B) Co-pelleting assay with Folch fraction liposomes and full-length Opy1. (C) Co-pelleting assay with GST and GST fusions of PH1 (amino acids 1–128) and PH2 (amino acids 208–340). (D) Co-pelleting assay of full length Opy1 with liposomes of indicated compositions. See Materials and Methods for details. (E) Quantification of the mean fraction of Opy1 bound to liposomes of the indicated compositions from two experiments as shown in D. Data are presented as mean± s.e.m. (F) Live-cell imaging of Opy1–GFP and Opy1(4N)–GFP. GFP signal is shown in inverted grayscale. Scale bar: 5 µm. (G) Western blot showing expression of the indicated proteins in cell lysates. PSTAIRE was used as a loading control. Numbers on the left indicate position of molecular mass markers (kDa). IB, immunoblot. Experiments in B–G were repeated twice with similar results. S, supernatant; P, pellet.
Because deleting S. pombe opy1 does not result in CR sliding or changes to PIP levels (Snider et al., 2018), we set out to compare features of S. pombe Opy1 with those of S. cerevisiae Opy1 (Ling et al., 2012) to better understand the generality of dual PH domain-containing protein function. Our findings suggest that these proteins have evolved unexpectedly distinct properties, suggesting that these proteins may have organism-specific functionality.
RESULTS AND DISCUSSION
We tested whether S. pombe Opy1 binds membrane directly using liposome co-pelleting assays. Opy1, produced and purified from bacteria, co-pelleted with Folch fraction liposomes, indicating that it can directly bind membrane in vitro (Fig. 1B). We then generated GST fusions to Opy1 fragments, including the N-terminal PH domain (amino acids 1–128; PH1) or the C-terminal PH domain (from amino acid 208 to the C-terminal end; PH2). PH1 robustly interacted with liposomes, whereas PH2 showed no significant interaction with liposomes (Fig. 1C). We next tested whether full-length Opy1 binds specific PIPs using co-pelleting assays with liposomes of defined composition. Opy1 interacted with PI(4,5)P2-containing liposomes, but displayed little to no interaction with liposomes containing PI4P or phosphatidylinositol (3,4,5)-trisphosphate [PI(3,4,5)P3] (Fig. 1D,E). The specificity of Opy1 for PI(4,5)P2 can explain the loss of Opy1 PM targeting in both the efr3Δ and its3-1 mutants, in which PM PI(4,5)P2 levels are reduced (Snider et al., 2018, 2017).
We noted significant sequence similarity between Opy1 PH1 and the N-terminal PH domain of the mammalian dual-PH domain protein pleckstrin (Fig. S1A), which has a PI(4,5)P2-binding pocket that confers the specificity of pleckstrin for PI(4,5)P2 in vitro and is required for targeting to the PM in cells (Harlan et al., 1994, 1995; Ma et al., 1997). We mutated the analogous residues in opy1 [K34N, K35N, K42N and K43N; opy1(4N)] and examined the localization of endogenously tagged Opy1(4N)–GFP in cells using fluorescence microscopy. Opy1(4N) did not localize to the PM even though it was expressed at comparable levels to wild-type Opy1–GFP (Fig. 1F,G), indicating that the PH1 domain is required for Opy1 PM targeting, and that it may employ a similar mechanism to that of pleckstrin for binding PI(4,5)P2. We next visualized endogenously produced Opy1 fragments fused to GFP to determine whether membrane-binding capacity in vitro equated with PM localization. The localizations of PH1–GFP and PH2–GFP fusions that either included the linker between the PH domains (PH1, amino acids 1–220; PH2, from amino acid 123 to the C-terminal end; Fig. S1B) or excluded it (PH1, amino acids 1–128; PH2, from amino acid 208 to the C-terminal end; Fig. S1C) were compared with that of full-length Opy1–GFP. We found that none of the truncations localized to the PM, although all of the truncations were produced, as determined using immunoblotting (Fig. S1D). Thus, despite PH1 conferring membrane binding in vitro and being required for PM localization in vivo, neither PH domain is sufficient to target Opy1 to the PM in vivo, indicating that there is another layer of regulation governing stable PM localization of Opy1.
S. cerevisiae Opy1 associates with both PI(4,5)P2 and the PI5-kinase Mss4 (Ling et al., 2012). To test whether S. pombe Opy1 binds the PI5-kinase Its3, we first performed a large-scale purification of TAP-tagged Opy1 (Opy1–TAP) from S. pombe and identified associated proteins using 2D liquid chromatography-tandem mass spectrometry (LC-MS/MS). After background subtraction, Its3 was the top hit and the only PM protein identified in the top ten Opy1-associated proteins, indicating that Opy1 and Its3 associate in vivo (Fig. 2A; Table S2). To test whether Opy1 and Its3 interact directly, we produced and purified both proteins recombinantly and performed in vitro binding assays. We found that MBP–Its3 immobilized on resin directly pulled down GST–Opy1, and that this interaction was mediated by PH1 (Fig. 2B). Thus, S. pombe PH1 binds both PI(4,5)P2 and PI5-kinase. The function of PH2 remains unknown.
Fig. 2.

Opy1 and Its3 directly interact. (A) Summary of data from mass spectrometry analysis of proteins associated with Opy1–TAP. T.S.C., total spectral counts. (B) MBP-Trap resin-bound MBP or MBP–Its3 was incubated with GST, GST–Opy1, GST–PH1 or GST–PH2, washed, and the proteins were separated by SDS-PAGE. Representative immunoblot of the in vitro binding assays are shown. The experiment was performed three times with similar results. PD, pull down; IB, immunoblot.
To determine the functional consequence of the Its3–Opy1 interaction, we first used live-cell imaging to examine whether Opy1 influences Its3 PM localization. The punctate pattern of Its3 at the PM was unaffected by opy1 deletion (Fig. 3A). The PI4-kinase Stt4, which also localizes to PM punctae, was similarly unaffected by Opy1 loss (Fig. S2A). However, Opy1–mNeonGreen (Opy1–mNG) localization to the PM is dramatically reduced in its3-1, a temperature-sensitive allele (Snider et al., 2018). Thus, Its3 is necessary for Opy1 PM localization, possibly through directly binding Opy1, by generating PI(4,5)P2 that recruits Opy1 to the PM, or by a combination of both mechanisms.
Fig. 3.

Opy1 does not regulate Its3 localization or kinase activity. (A) Live-cell imaging of Its3–mNG in the indicated genotypes. mNeonGreen signal is shown in inverted grayscale. Scale bar: 5 µm. (B) Schematic of Its3 lipid kinase assay. Image adapted from Nishimura et al., 2019, where it was published under a CC-BY-4.0 license. (C) ADP-Glo assay measuring Its3 kinase activity toward PI4P-containing liposomes in the presence or absence of Opy1. Control values from liposomes without PI4P have been subtracted from each. Data are presented as the mean±s.e.m. of four experiments. N.S., not significant (unpaired, two-tailed Student's t-test). (D) Liposome co-pelleting assay for Its3 in the presence and absence of Opy1 with PI4P-containing liposomes. The experiment was repeated twice with similar results. S, supernatant; P, pellet. Numbers on the left indicate positions of molecular mass markers (kDa).
We next tested whether S. pombe Opy1 modulates Its3 kinase activity. We performed kinase assays with recombinant MBP–Its3, liposomes containing the Its3 substrate PI4P, and ATP, then used the ADP-Glo assay as a readout for lipid kinase activity, as described previously (Nishimura et al., 2019) (Fig. 3B). Adding Opy1 to the kinase assay did not significantly affect Its3 activity (Fig. 3C). This result is consistent with our previous determination that PM PI4P and PI(4,5)P2 levels are not affected by opy1 deletion in vivo (Snider et al., 2018). Opy1 also had no effect on Its3 membrane binding or access to substrate, based on Its3 co-pelleting with PI4P liposomes in vitro (Fig. 3D), and the observation that Opy1 overproduction did not alter Its3 PM localization in vivo (Fig. S2B). We hypothesized that if Opy1 negatively regulates Its3 in cells, then deletion of opy1 would suppress the growth defects of its3-1, analogously to S. cerevisiae opy1Δ suppressing the growth defects of mss4 temperature-sensitive cells (Ling et al., 2012). However, opy1Δ did not suppress the its3-1 growth phenotype (Fig. S2C). Additionally, one would expect that combining opy1Δ with deletion of phosphatidylinositol 5-phosphatases (PI5-phosphatases) would result in negative genetic interactions if Opy1 also acted to oppose Its3 kinase activity. To the contrary, we found no growth defects when opy1Δ was combined with either single or double deletions of PI5-phosphatases (Fig. S2D,E). Taken together, these results indicate that Opy1 does not regulate Its3 localization or kinase activity.
Given that Opy1 binds PI(4,5)P2 specifically, we hypothesized that high levels of Opy1 in cells would sequester PI(4,5)P2 and result in cytokinesis defects, similar to what we previously observed when a PI(4,5)P2 sensor was overproduced (Snider et al., 2017). In S. pombe, reduced levels of PM PI4P and PI(4,5)P2 cause CR anchoring defects that manifest as sliding of the CR away from the cell center and off-center septation (Snider et al., 2018, 2017). We drove overexpression of opy1 (opy1+) using the nmt1 promoter and first examined changes in PM PIPs using low-level production of biosensors that do not cause any observable growth defect in live cells (Snider et al., 2018). We found that not only the PI(4,5)P2 sensor (GFP–2×PHPlc) but also the PI4P sensor (GFP–P4CSidC) were reduced in abundance at the PM when Opy1 was overproduced (Fig. 4A–D). These results can be explained by Opy1 outcompeting the PI(4,5)P2 sensor. Furthermore, given the importance of PM PI(4,5)P2 for many processes, it is likely that cells expend their PI4P reservoir in an attempt to re-establish the PI(4,5)P2 PM pool. Consistent with the idea that excess Opy1 sequesters PI(4,5)P2, efr3Δ cells exhibited growth defects upon Opy1 overproduction (Fig. S3A), similar to our previous result that efr3Δ and its3-1 are synthetically lethal, presumably because multiple steps in the PI(4,5)P2 synthesis pathway are disrupted (Snider et al., 2018). Also consistent with what would be expected from elevated levels of a PI(4,5)P2-binding protein, Opy1 overproduction resulted in off-center septation (Fig. 4E,F), and overproduced GFP–Opy1 localized in a more homogeneous pattern along the PM compared with the punctate pattern of endogenous Opy1 (compare Fig. S3B with Fig. S1B,C), more closely resembling the localization of a PI(4,5)P2 sensor (Snider et al., 2018). If Opy1 does compete with GFP–2×PHPlc for PI(4,5)P2, we predicted that high levels of the sensor would outcompete Opy1 for PM PI(4,5)P2 binding. To test this, we overproduced the PI(4,5)P2 sensor (2×PHPlc) in Opy1–GFP cells. In control cells, Opy1–GFP localized along the PM and septum, as expected, but upon overexpression of 2×PHPlc, more Opy1 was diffuse in the cytoplasm and less was detected at the cell cortex, as shown by line scans across the cell (Fig. 4G,H). These results further support the conclusion that Opy1 specifically binds PM PI(4,5)P2.
Fig. 4.
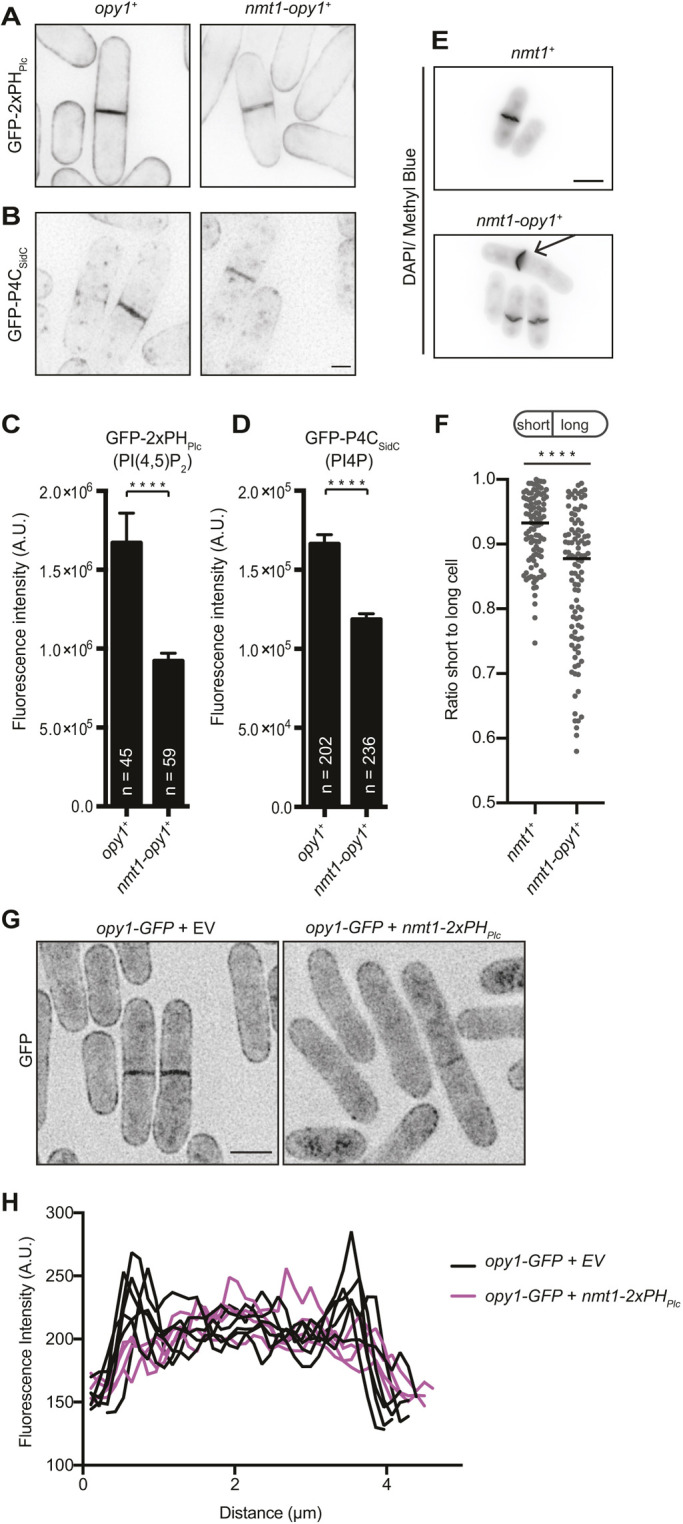
Opy1 overproduction results in cytokinesis defects. (A,B) Representative live-cell images of (A) GFP–2×PHPlc and (B) GFP–P4CSidC PIP biosensors in wild-type cells (opy1+) or cells overproducing opy1 from the nmt1 promoter (nmt1-opy1+). GFP signal is shown in inverted grayscale. Scale bar: 2 µm. (C,D) Quantification of division site intensity of (C) GFP–2×PHPlc and (D) GFP–P4CSidC biosensors, as shown in A and B. Data are presented as the mean±s.e.m. of three experiments. Total numbers of cells analyzed are shown on the graphs. ****P<0.0001 (unpaired, two-tailed Student's t-test). (E) Representative images of fixed cells stained with DAPI and Methyl Blue. Prior to fixation, overexpression was induced in cells carrying either empty vector (nmt1+) or an opy1 overexpression vector (nmt1-opy1+) for 28 h in medium lacking thiamine. Arrow indicates a cell with an off-center septum. DAPI/Methyl Blue signal is shown in inverted grayscale. Scale bar: 5 µm. (F) Quantification of off-center septation as a measure of the ratio of short to long daughter cell length for the strains described in E. Bars represent means. ****P<0.0001(unpaired, two-tailed Student's t-test). (G) Representative live-cell images of opy1–GFP cells expressing either an empty vector (EV) or nmt1-2×PHPlc. GFP signal is shown in inverted grayscale. Scale bar: 5 µm. (H) Five representative line scans across cells as described in G. Line scans were drawn from cortex to cortex in the area between the septum and curved ends of the cell.
We have found that S. pombe Opy1 binds membranes through its N-terminal PH domain. This is similar to some mammalian dual PH-domain proteins (e.g. pleckstrin), which also bind lipids with N-terminal PH domains instead of, or in addition to, C-terminal ones (Bach et al., 2006; Edlich et al., 2005; Ma et al., 1997), but different from S. cerevisiae Opy1, which uses its C-terminal PH domain (Ling et al., 2012). However, full-length S. cerevisiae and S. pombe Opy1 proteins share the property of specifically binding PI(4,5)P2-containing liposomes and show no appreciable interaction with PI4P- or PI(3,4,5)P3-containing membranes. Furthermore, both proteins depend on PI5-kinases for PM localization (Ling et al., 2012; Snider et al., 2018, 2017). Thus, both S. cerevisiae and S. pombe Opy1 have the properties of an endogenous PI(4,5)P2 sensor.
Although a direct interaction between S. cerevisiae Opy1 and Mss4 has not been reported, both PH1 and PH2 of Opy1 can isolate Mss4 from cell lysates (Ling et al., 2012). In contrast, we found that S. pombe Opy1 directly binds both membrane and Its3 through PH1, whereas no interaction was detected between PH2 and Its3. It is somewhat surprising that PH1–GFP did not localize to the PM given that PH1 binds to both Its3 and PI(4,5)P2. However, other PH domain proteins oligomerize, and in some cases oligomerization is required for high-affinity membrane binding (Klein et al., 1998). It is unknown whether S. pombe Opy1 shares this property, but the need for multimerization could explain why PH1–GFP did not localize to the PM (Fig. S1B,C).
Importantly, although we found that Its3 and Opy1 can directly interact, we obtained no evidence that Opy1 negatively regulates Its3 kinase activity. This differs from the model wherein S. cerevisiae Opy1 acts as a coincidence detector for PI(4,5)P2 and Mss4 to inhibit Mss4 in the presence of high levels of its product (Ling et al., 2012). We propose, therefore, that Opy1 evolved to perform different cellular functions in S. cerevisiae and S. pombe. Different functions are somewhat unexpected, considering that there are mammalian proteins with similar domain structure to the yeast Opy1 proteins, and one of these, TAPP1 (also known as PLEKHA1), has similar effects to S. cerevisiae Opy1 when overproduced in S. cerevisiae cells (Ling et al., 2012). Specifically, overexpression of either of these proteins in S. cerevisiae cells deficient in PI5-phosphatase activity restores PI(4,5)P2 levels (Ling et al., 2012). Based on these results, Ling et al. (2012) proposed that the dual PH domain-containing proteins may utilize their PH2 domains to bind specific PIPs and their PH1 domains to bind different PIP kinases (Ling et al., 2012). However, S. pombe Opy1 does not fit this model, despite its similarity in domain layout. Also, the PH2 domains of TAPP1, TAPP2 (PLEKHA2) and pleckstrin specifically bind PI(3,4)P2 (Dowler et al., 2000; Edlich et al., 2005; Thomas et al., 2001; Wullschleger et al., 2011), but the connections between the PH1 domains of these proteins and regulation of PIPs are not fully defined (Abrams et al., 1996; Bach et al., 2006; Ma et al., 1997). As exemplified by S. pombe Opy1, a major complication in the study of these proteins is that they can sequester their lipid binding partners, giving rise to indirect changes in PM composition. Therefore, caution is warranted when interpreting results from overexpression of dual PH domain-containing proteins. Further study of these proteins, particularly at endogenous expression levels, will be required to determine whether there are unifying characteristics amongst this group of proteins beyond a similar domain structure.
MATERIALS AND METHODS
Recombinant protein expression and purification
6×His–Opy1, GST, GST–Opy1, GST–PH1, GST–PH2, MBP and MBP–Its3 were produced in Escherichia coli Rosetta2(DE3)pLysS cells (Novagen; 70954). 6×His–Opy1 was purified on cOmplete His-Tag resin (Roche), according to the manufacturer's protocol. 6×His tags were removed by thrombin digestion at room temperature followed by purification on a benzamidine FF Hitrap column (GE Healthcare) to remove thrombin. GST and GST fusions were purified using GST-Bind resin, according to the manufacturer's protocol (EMD Millipore). MBP and MBP–Its3 were purified using amylose resin, according to the manufacturer's protocol (New England Biolabs). MBP–Its3 was purified further by gel filtration on a 16/600 Superdex 200 column (GE Healthcare).
Liposome methods
Liposome formation and co-pelleting assays were performed as previously described (McDonald et al., 2016). Folch fraction lipids were obtained from Sigma-Aldrich, all others were obtained from Avanti Polar Lipids. CHCl3 lipid stocks were mixed at the desired ratios and dried under N2 gas followed by removal of chloroform under high vacuum. For Fig. 1, liposomes were rehydrated in 50 mM Tris-HCl pH 7.4 and 150 mM NaCl, and for Fig. 3, liposomes were rehydrated in 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 10 mM MgCl2 and 1 mM dithiothreitol (DTT) to a concentration of 1 mg/ml. Liposomes were subjected to ten freeze/thaw cycles and extruded through polycarbonate filters of 400 nm pore size (Whatman) using a mini-extruder (Avanti). For Fig. 1E,F, phosphatidic acid (PA, egg, chicken), PI4P (brain), PI(4,5)P2 (brain) and 18:1 PI(3,4,5)P3 were added at 5% where indicated. DOPS (1,2-dioleoyl-sn-glycero-3-phospho-L-serine) was added at 10% (PS), DOPE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine) at 15% (PE), and DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine) was added to make up the total to 100% (PC). For Fig. 3, liposomes contained 70% PC, 15% PE, 10% PS, and 5% PI4P. For control liposomes in the ADP-Glo assay, no PI4P was included in the liposomes.
For co-pelleting assays in Fig. 1, 20 µg of Opy1 was added to 0.5 mg/ml (final concentration) liposomes or buffer for control. For Fig. 3D, MBP–Its3 was added at 150 nM and Opy1 was added at a tenfold molar excess to MBP–Its3. The reaction was incubated for 15 min at room temperature before centrifugation at 150,000 g in an Optima TL ultracentrifuge for 15 min at 25°C. Pellet and supernatant fractions were resuspended in equal volumes and analyzed using SDS-PAGE. Protein bands were visualized using Coomassie staining and quantified using a LI-COR Odyssey CLx.
For the ADP-Glo assay, 400 µM liposomes (either PI4P-containing or control) were incubated in a 50 µl kinase reaction with 1 µM ATP, 50 nM MBP–Its3 and 500 nM Opy1 where added. The reaction was buffered in 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 10 mM MgCl2 and 1 mM DTT. The kinase reactions were incubated for 30 min at 27°C. The kinase reactions were then added to the ADP-Glo assay according to the manufacturer's protocol (Promega). A 25 µl volume from the kinase reaction was transferred to a white 96-well plate. Subsequently, 25 µl of the ADP-Glo reagent was added and allowed to incubate for 40 min, followed by addition of 50 µl of kinase detection reagent for 30 min. Luminescence was detected using a BIO-TEK Synergy HT Microtiter platereader. For the graph in Fig. 3C, four experiments were averaged after subtraction of the luminescence signal resulting from a kinase reaction with control liposomes containing no PI4P.
Yeast methods
S. pombe strains (Table S1) were grown in yeast extract (YE) medium or minimal medium with appropriate supplements (Forsburg and Rhind, 2006). To construct opy1(4N) and opy1 truncation strains, constructs were generated using PCR to amplify full-length opy1 or opy1 fragments from genomic DNA with 300 bp 5′ and 3′ flanking regions, which were then inserted into the BamHI/PstI restriction sites of pSK+ by Gibson assembly. The opy1(4N) mutations were introduced by site-directed mutagenesis using a QuikChange Lightning Multi Site-directed Mutagenesis kit according to the manufacturer's protocol (Agilent Technologies) and were verified by sequencing. 1 µg of the pSK+ opy1 constructs was then linearized by digestion with XhoI and transformed into opy1::ura+ cells. Integrants were selected on YE+5-FOA (1.5 mg/ml) medium. Transformants were verified by PCR and by sequencing. Genes were tagged at the 3′ end of their ORFs with TAP:kanR, GFP:kanR, or mNG:hygR using pFA6 cassettes as previously described (Bähler et al., 1998; Wach et al., 1994), and integration of tags was verified by PCR. A lithium acetate method (Keeney and Boeke, 1994) was used in S. pombe transformations. Introduction of tagged loci into other genetic backgrounds was accomplished using standard S. pombe mating, sporulation, and tetrad dissection techniques (Forsburg and Rhind, 2006).
For spot assays, cells were grown to log phase at 25°C in minimal medium containing thiamine (5 µg/ml), then washed into media lacking thiamine for 24 h prior to plating. 10 million cells were resuspended in 1 ml of water, and tenfold serial dilutions were made. A 2.5 µl volume of each dilution was spotted on plates lacking thiamine (induced) or containing thiamine (repressed) for Fig. S3 and on YE plates for Fig. S2. The plates were incubated at the indicated temperatures.
Overexpression of Opy1 and of 2×PHPlc was accomplished by using the repressible nmt1 or nmt81 promoters in the pREP series of vectors (Basi et al., 1993). For Fig. 4E,F, 1 µg of pREP1 or pREP1-opy1+ plasmids, for Fig. 4G,H 1 µg of pREP1 or pREP1-2×PHPlc plasmids and for Fig. S3B, 1 µg of pREP81-GFP or pREP81-GFP–opy1 were transformed into wild-type cells by electroporation and plated on medium containing 5 µg/ml thiamine. Cells were then grown in liquid culture in the presence of thiamine before being washed into medium lacking thiamine for 28 h (Fig. 4E,F), 26 h (Fig. 4G,H) or 24 h (Fig. S3B) to induce overproduction. For Fig. 4A–D, Fig. S2B and Fig. S3A, a strain was constructed in which nmt1-opy1+ was integrated into the genome. This was accomplished by subcloning nmt1-opy1+ from the pREP1-opy1+ plasmid into pJK210 (Keeney and Boeke, 1994). The resulting plasmid was then linearized with NruI and integrated into the ura4-294 locus. Integration was verified by PCR. To induce overproduction of Opy1 in the resulting strain, cells were grown in medium lacking thiamine for 24 h.
In vitro binding assay
For the binding assay in Fig. 2B, all proteins were combined at 1 µM in 50 mM Tris-HCl pH 7.4, 150 mM NaCl and 0.1% NP-40 with 10 µl MBP–Trap agarose (Chromotek) to pull down MBP or MBP–Its3. The reactions were incubated for 1.5 h at 4°C, then the beads were washed four times in binding buffer. Associated proteins were analyzed using SDS-PAGE followed by transfer to PVDF and immunoblotting with anti-MBP antibody at a dilution of 1:10,000 (E8032S; New England Biolabs) and anti-GST antibody at 1:10,000 (VU160, rabbit polyclonal; Cocalico Biologicals). Secondary antibodies were conjugated to IRDye680LT or IRDye800 (LI-COR Biosciences). Blotted proteins were detected using an Odyssey CLx (LI-COR Biosciences).
TAP purification and MS analysis
Tandem affinity purification of Opy1–TAP and subsequent identification of interacting proteins by mass spectrometry were performed as previously described (Chen et al., 2013; Elmore et al., 2014; Gould et al., 2004) with the following changes: a newer version of Scaffold (Scaffold v4.4.1.1; Proteome Software) was used, and the minimum peptide identification probability was changed to 95.0%. Proteins identified in a mock tandem affinity purification and in an Ada1–TAP purification (Ada1 is a nuclear protein) were subtracted from the Opy1–TAP results.
Microscopy
Yeast for live-cell imaging were grown at 25°C. Live-cell and fixed-cell images of S. pombe cells were acquired using a Personal DeltaVision (Cytiva) that includes a microscope (IX71; Olympus), 60× NA 1.42 Plan Apochromat and 100× NA 1.40 U Plan S Apochromat objectives, fixed and live-cell filter wheels, a camera (CoolSNAP HQ2; Photometrics) and softWoRx imaging software (Cytiva). Z-sections were spaced at 0.5 μm. Images for quantification were not deconvolved. Images used for representative images were deconvolved with ten iterations. For all representative images, the middle z-slice is shown.
Intensity measurements in Fig. 4 were made using FIJI (Schindelin et al., 2012). A region of interest (ROI) was created to measure the intensity around the membranes lining the division septum, and background was subtracted by creating a ROI in the same image in an area containing no cells (Snider et al., 2017; Waters, 2009). For intensity measurements, sum intensity projections were analyzed.
For fixed-cell imaging, cells were grown to log phase at 32°C and then fixed in 70% ethanol for 30 min. Cells were then washed three times with phosphate-buffered saline before DAPI and Methyl Blue staining. To quantify off-center septation, fixed cells stained for nuclei and cell wall were imaged. The coordinates of the cell tips and septum were logged. Lengths of the shorter and longer cell were calculated from these coordinates and reported as a ratio (Snider et al., 2017).
Western blotting of cell lysates
Cell pellets were snap frozen in dry ice-ethanol baths. Lysates were prepared using a Fastprep cell homogenizer (MP Biomedicals). Cells were extracted with 8 M urea, 50 mM Tris, 1 mM PMSF and 1.3 mM benzamidine. Cleared lysate was then resolved using SDS-PAGE and transferred to PVDF membrane using an iBlot (Invitrogen). The membrane was then probed with an anti-GFP antibody at a dilution of 1:1000 (118144600001; Roche) or an anti-PSTAIRE antibody at a dilution of 1:10,000 (Cdc2; P7962; Sigma-Aldrich). Secondary antibodies were conjugated to IRDye680LT or IRDye800 (LI-COR Biosciences). Blotted proteins were detected using an Odyssey CLx (LI-COR Biosciences).
Statistical analysis
All statistical analyses were performed in Prism 8 (GraphPad Software).
Supplementary Material
Acknowledgements
We thank S. Cullati, J. Bisht, R. Bhattacharjee and A. Rossi for critical reading of this manuscript, and Liping Ren and Seth Drey for technical assistance.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: C.E.S., A.H.W., K.L.G.; Methodology: C.E.S., A.H.W., J.-S.C., K.L.G.; Validation: C.E.S., A.H.W., K.L.G.; Formal analysis: C.E.S., A.H.W., H.T.B., J.-S.C., J.M.E.; Investigation: C.E.S., A.H.W., H.T.B., J.-S.C., J.M.E.; Resources: K.L.G.; Data curation: C.E.S., A.H.W., J.-S.C., J.M.E., K.L.G.; Writing - original draft: C.E.S., A.H.W., K.L.G.; Writing - review & editing: C.E.S., A.H.W., H.T.B., J.-S.C., J.M.E., K.L.G.; Visualization: C.E.S., A.H.W., H.T.B., J.-S.C., J.M.E.; Supervision: K.L.G.; Project administration: C.E.S., A.H.W., K.L.G.; Funding acquisition: K.L.G.
Funding
We are grateful for the following support: National Institutes of Health grants R01GM101035 and R35GM131799 (to K.L.G.) and T32GM008554-21 (to C.E.S.); and American Heart Association grants 14PRE19740000 (to A.H.W.) and 17PRE33410245 (to C.E.S.). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at https://jcs.biologists.org/lookup/doi/10.1242/jcs.247973.supplemental
References
- Abrams C. S., Zhang J., Downes C. P., Tang X.-W., Zhao W. and Rittenhouse S. E. (1996). Phosphopleckstrin Inhibits Gβγ-activable platelet phosphatidylinositol-4,5-bisphosphate 3-Kinase. J. Biol. Chem. 271, 25192-25197. 10.1074/jbc.271.41.25192 [DOI] [PubMed] [Google Scholar]
- Bach T. L., Kerr W. T., Wang Y., Bauman E. M., Kine P., Whiteman E. L., Morgan R. S., Williamson E. K., Ostap E. M., Burkhardt J. K. et al. (2006). PI3K regulates pleckstrin-2 in T-cell cytoskeletal reorganization. Blood 109, 1147-1155. 10.1182/blood-2006-02-001339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bähler J., Wu J.-Q., Longtine M. S., Shah N. G., McKenzie A. III, Steever A. B., Wach A., Philippsen P. and Pringle J. R. (1998). Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast 14, 943-951. [DOI] [PubMed] [Google Scholar]
- Basi G., Schmid E. and Maundrell K. (1993). TATA box mutations in the Schizosaccharomyces pombe nmt1 promoter affect transcription efficiency but not the transcription start point or thiamine repressibility. Gene 123, 131-136. 10.1016/0378-1119(93)90552-E [DOI] [PubMed] [Google Scholar]
- Chen J.-S., Broadus M. R., McLean J. R., Feoktistova A., Ren L. and Gould K. L. (2013). Comprehensive proteomics analysis reveals new substrates and regulators of the fission yeast clp1/cdc14 phosphatase. Mol. Cell. Proteomics 12, 1074-1086. 10.1074/mcp.M112.025924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson E. J. and Hille B. (2019). Understanding phosphoinositides: rare, dynamic, and essential membrane phospholipids. Biochem. J. 476, 1-23. 10.1042/BCJ20180022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowler S., Currie R. A., Campbell D. G., Deak M., Kular G., Downes C. P. and Alessi D. R. (2000). Identification of pleckstrin-homology-domain-containing proteins with novel phosphoinositide-binding specificities. Biochem. J. 351, 19-31. 10.1042/bj3510019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edlich C., Stier G., Simon B., Sattler M. and Muhle-Goll C. (2005). Structure and Phosphatidylinositol-(3,4)- bisphosphate binding of the C-terminal PH domain of human pleckstrin. Structure 13, 277-286. 10.1016/j.str.2004.11.012 [DOI] [PubMed] [Google Scholar]
- Elmore Z. C., Beckley J. R., Chen J.-S. and Gould K. L. (2014). Histone H2B ubiquitination promotes the function of the anaphase-promoting complex/cyclosome in Schizosaccharomyces pombe. G3 4, 1529-1538. 10.1534/g3.114.012625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field S. J., Madson N., Kerr M. L., Galbraith K. A. A., Kennedy C. E., Tahiliani M., Wilkins A. and Cantley L. C. (2005). PtdIns(4,5)P2 functions at the cleavage furrow during Cytokinesis. Curr. Biol. 15, 1407-1412. 10.1016/j.cub.2005.06.059 [DOI] [PubMed] [Google Scholar]
- Forsburg S. L. and Rhind N. (2006), Basic methods for fission yeast. Yeast 23, 173-183. 10.1002/yea.1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould K. L., Ren L., Feoktistova A. S., Jennings J. L. and Link A. J. (2004). Tandem affinity purification and identification of protein complex components. Methods 33, 239-244. 10.1016/j.ymeth.2003.11.019 [DOI] [PubMed] [Google Scholar]
- Harlan J. E., Hajduk P. J., Yoon H. S. and Fesik S. W. (1994). Pleckstrin homology domains bind to phosphatidylinositol-4, 5-bisphosphate. Nature 371, 168-170. 10.1038/371168a0 [DOI] [PubMed] [Google Scholar]
- Harlan J. E., Yoon H. S., Hajduk P. J. and Fesik S. W. (1995). Structural characterization of the interaction between a pleckstrin homology domain and phosphatidylinositol 4, 5-bisphosphate. Biochemistry 34, 9859-9864. 10.1021/bi00031a006 [DOI] [PubMed] [Google Scholar]
- Keeney J. B. and Boeke J. D. (1994). Efficient targeted integration at leu1-32 and ura4-294 in Schizosaccharomyces pombe. Genetics 136, 849-856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein D. E., Lee A., Frank D. W., Marks M. S. and Lemmon M. A. (1998). The pleckstrin homology domains of dynamin isoforms require oligomerization for high affinity phosphoinositide binding. J. Biol. Chem. 273, 27725-27733. 10.1074/jbc.273.42.27725 [DOI] [PubMed] [Google Scholar]
- Lemmon M. A. (2007). Pleckstrin homology (PH) domains and phosphoinositides. Biochem. Soc. Symp. 74, 81-93. 10.1042/BSS0740081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling Y., Stefan C. J., MacGurn J. A., Audhya A. and Emr S. D. (2012). The dual PH domain protein Opy1 functions as a sensor and modulator of PtdIns(4,5)P2 synthesis. EMBO J. 31, 2882-2894. 10.1038/emboj.2012.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma A. D., Brass L. F. and Abrams C. S. (1997). Pleckstrin associates with plasma membranes and induces the formation of membrane projections: requirements for phosphorylation and the NH2-terminal PH domain. J. Cell Biol. 136, 1071-1079. 10.1083/jcb.136.5.1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald N. A., Takizawa Y., Feoktistova A., Xu P., Ohi M. D., Vander Kooi C. W. and Gould K. L. (2016). The tubulation activity of a fission yeast F-BAR protein is dispensable for its function in Cytokinesis. Cell Rep. 14, 534-546. 10.1016/j.celrep.2015.12.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T., Gecht M., Covino R., Hummer G., Surma M. A., Klose C., Arai H., Kono N. and Stefan C. J. (2019). Osh proteins control nanoscale lipid organization necessary for PI(4,5)P2 synthesis. Mol. Cell 75, 1043-1057.e8. 10.1016/j.molcel.2019.06.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schink K. O., Tan K.-W. and Stenmark H. (2016). Phosphoinositides in control of membrane dynamics. Annu. Rev. Cell Dev. Biol. 32, 143-171. 10.1146/annurev-cellbio-111315-125349 [DOI] [PubMed] [Google Scholar]
- Snider C. E., Willet A. H., Chen J.-S., Arpağ G., Zanic M. and Gould K. L. (2017). Phosphoinositide-mediated ring anchoring resists perpendicular forces to promote medial cytokinesis. J. Cell Biol. 216, 3041-3050. 10.1083/jcb.201705070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider C. E., Willet A. H., Brown H. S. T. and Gould K. L. (2018). Analysis of the contribution of phosphoinositides to medial septation in fission yeast highlights the importance of PI(4,5)P2 for medial contractile ring anchoring. Mol. Biol. Cell 29, 2148-2155. 10.1091/mbc.E18-03-0179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C. C., Dowler S., Deak M., Alessi D. R. and van Aalten D. M. F. (2001). Crystal structure of the phosphatidylinositol 3,4-bisphosphate-binding pleckstrin homology (PH) domain of tandem PH-domain-containing protein 1 (TAPP1): molecular basis of lipid specificity. Biochem. J. 358, 287-294. 10.1042/bj3580287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wach A., Brachat A., Pöhlmann R. and Philippsen P. (1994). New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 10, 1793-1808. 10.1002/yea.320101310 [DOI] [PubMed] [Google Scholar]
- Waters J. C. (2009). Accuracy and precision in quantitative fluorescence microscopy. J. Cell Biol. 185, 1135-1148. 10.1083/jcb.200903097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong R., Hadjiyanni I., Wei H.-C., Polevoy G., McBride R., Sem K.-P. and Brill J. A. (2005). PIP2 hydrolysis and calcium release are required for cytokinesis in drosophila spermatocytes. Curr. Biol. 15, 1401-1406. 10.1016/j.cub.2005.06.060 [DOI] [PubMed] [Google Scholar]
- Wullschleger S., Wasserman D. H., Gray A., Sakamoto K. and Alessi D. R. (2011). Role of TAPP1 and TAPP2 adaptor binding to PtdIns(3,4)P2 in regulating insulin sensitivity defined by knock-in analysis. Biochem. J. 434, 265-274. 10.1042/BJ20102012 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.