

Summary
Boron-containing compounds represent a promising class of molecules with proven efficacy against a wide range of pathogens, including apicomplexan parasites. Following lead optimization, the benzoxaborole AN13762 was identified as a preclinical candidate against the human malaria parasite, yet the molecular target remained uncertain. Here, we uncovered the parasiticidal mechanisms of AN13762, by combining forward genetics with transcriptome sequencing and computational mutation discovery and using Toxoplasma gondii as a relevant model for Apicomplexa. AN13762 was shown to target TgCPSF3, the catalytic subunit of the pre-mRNA cleavage and polyadenylation complex, as the anti-pan-apicomplexan benzoxaborole compound, AN3661. However, unique mutations within the TgCPSF3 catalytic site conferring resistance to AN13762 do not confer cross-protection against AN3661, suggesting a divergent resistance mechanism. Finally, in agreement with the high sequence conservation of CPSF3 between Toxoplasma and Cryptosporidium, AN13762 shows oral efficacy in cryptosporidiosis mouse model, a disease for which new drug development is of high priority.
Subject Areas: Molecular Biology, Molecular Medicine, Microbiology, Microbiology Parasite
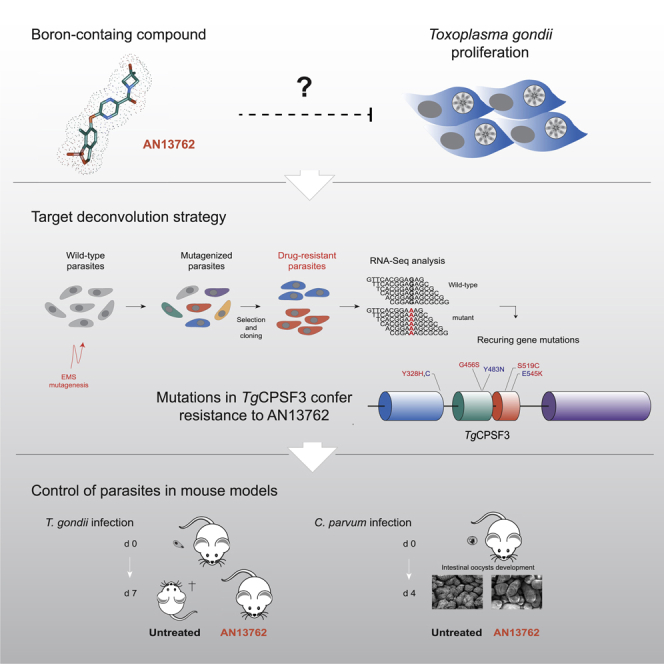
Graphical Abstract
Highlights
-
•
AN13762 is active against T. gondii parasites
-
•
Parasites resistant to AN13762 harbor mutations within TgCPSF3
-
•
Mutations within TgCPSF3 confer resistance to AN13762
-
•
AN13762 offers an alternative for targeting CPSF3 in Toxoplasma and Cryptosporidium
Molecular Biology; Molecular Medicine; Microbiology; Microbiology Parasite
Introduction
The Apicomplexa phylum contains intracellular single-celled parasites several of which are causative agents of animal and human diseases worldwide raising important public health problems (De Rycker et al., 2018). The group comprises important human pathogens such as Plasmodium, Toxoplasma, and Cryptosporidium responsible for malaria, toxoplasmosis, and cryptosporidiosis, respectively. For many of these diseases current treatments are suboptimal, and there are few or no alternatives available for some. Indeed, the current standard of treatment for Cryptosporidium infections, nitazoxanide, shows limited and immune-dependent effectiveness (Manjunatha et al., 2016). Although the current medication against Toxoplasma is quite effective, it has adverse side effects, particularly in immunocompromised patients, such as pyrimethamine-induced hematological toxicity and sulfonamide-induced skin rash, leukopenia, and thrombocytopenia (Dunay et al., 2018). In the case of malaria, emergence and spread of resistance to artemisinin-based combination therapy, the primary form of treatment, poses a constantly growing threat (De Rycker et al., 2018). Therefore, new classes of small-molecule drugs or drugs with novel modes of action are needed to overcome these limitations.
In an effort to optimize the efficacy of a novel class of boron-containing molecules against malarial parasites, the lead candidate AN13762 was identified in a phenotype-based screening (referred to as compound 46 in Zhang et al., 2017). Evaluation of pharmacokinetics showed that AN13762 has improved potency and metabolic stability, is orally bioavailable, and is equally potent across multidrug-resistant strains of Plasmodium falciparum, demonstrating no cross-resistance and a possible new mechanism of action. AN13762 has not exhibited either significant toxicology or cytotoxicity liabilities at any dose tested, and AN13762 was selected for preclinical development by Medicines for Malaria Venture in 2017 (Zhang et al., 2017). Although previous research on parental scaffold AN3661 identified Cleavage and Polyadenylation Specificity Factor 3 (CPSF3) as the direct target (Palencia et al., 2017; Sonoiki et al., 2017; Swale et al., 2019), a recent study investigating the resistance mechanisms of AN13762 in P. falciparum identified multiple components involved in prodrug activation or sumoylation and ubiquitination pathways along with PfCPSF3 suggesting that the latter was not the primary target (Sindhe et al., 2020).
The work described here was undertaken to shed light on the parasiticidal mechanisms of AN13762 using Toxoplasma gondii as a relevant representative of apicomplexan parasites. Here, we present evidence that AN13762 is effective against both T. gondii and Cryptosporidium parvum in vitro at low micromolar concentrations and in vivo in mouse models of toxoplasmosis and cryptosporidiosis, respectively. Using a forward genetic approach based on transcriptome sequencing, we identified its target as CPSF3, a common target of several benzoxaboroles such as AN3661, a compound active against apicomplexan parasites (Palencia et al., 2017; Sonoiki et al., 2017; Swale et al., 2019), or the trypanocidal compounds AN11736 and acoziborole (Wall et al., 2018). Importantly, several point mutations found in T. gondii CPSF3 conferring resistance against AN13762 were not effective against AN3661, suggesting a divergent mode of resistance mechanisms between CPSF3 and benzoxaboroles. Hence this work uncovers the molecular mechanism for the antiparasitic activity of a preclinical antimalarial candidate AN13762 and extends the clinical spectrum of activity of this chemotype to other life-threatening apicomplexan parasites.
Results
AN13762 Is Active against T. gondii In Vitro and In Vivo
To assess the effectiveness of AN13762 against T. gondii parasites, growth of the type I reference RH strain was monitored within human foreskin fibroblasts (HFFs) treated with AN13762; its parental scaffold AN3661 as a positive control (Figure 1A); pyrimethamine, the standard of care for toxoplasmosis; or vehicle (DMSO). Efficient in vitro inhibition of T. gondii growth was repeatedly confirmed, with measured half maximum effective concentration (EC50) of 2.1 μM, which is almost 40 times higher than that of AN3661 (Figures 1B and 1C). Complete and sustained inhibition of growth was observed at 10 μM AN13762 without any adverse effects for the host cells (Figures 1D and S1).
Figure 1.

Activity of AN13762 against Toxoplasma gondii
(A) Chemical structures of benzoxaborole leads AN13762 and AN3661.
(B) Dose-response curves for inhibition of T. gondii growth in vitro in response to increasing concentration of the indicated compounds. Confluent HFF monolayer was infected with tachyzoites of T. gondii RH strain expressing the NanoLuc luciferase (RH Δku80 UPRT::NLuc-P2A-EmGFP). The T. gondii strains used in this study are listed in Table S1. Data are presented as mean ± standard deviation (SD) of at least two independent biological assays, each with 3 technical replicates. Shaded error envelopes depict 95% confidence intervals.
(C) EC50 values of each biological replicate were determined by non-linear regression analysis. EC50 data are presented as mean ± SD from at least 2 independent biological replicates, each with 3 technical replicates.
(D) HFF cells were infected with tachyzoites (RH Δku80 UPRT::NLuc-P2A-EmGFP) and incubated with 10 μM AN13762, 5 μM AN3661, or 0.1% DMSO as control. Cells were fixed 24 h post-infection and then stained with antibodies against the T. gondii inner membrane complex protein GAP45 (magenta). The cytosolic GFP is shown in green. Scale bars, 10 μm. A complete dataset can be found in Figure S1.
(E) Acute toxoplasmosis: timeline of mouse infections and treatments. Untreated mice succumbed to infection, and thus a new group of healthy CBA/JRj mice was used for the second challenge (n = 3 in each group).
(F) Survival curves of the CBA/JRj mice infected intraperitoneally (i.p.) with 103 tachyzoites of type I RH wild-type (WT) or the indicated CPSF3 mutant strains (E545K or G456S). During the first challenge, mice were treated orally with 40 mg/kg AN13762 or 200 mg/kg sulfadiazine once daily beginning 1 day post-infection (n = 6 for each condition; two independent experiments, each with three mice per experimental group). Mice surviving the primary infection were challenged a second time with the T. gondii WT strain (second challenge). A new group of naive mice was used as control (n = 3 in each group).
When AN13762 was administered orally for 7 days to T. gondii-infected mice, beginning on the first day following intraperitoneal injection of parasites, 100% of the animals survived the lethal infection by the highly virulent type I RH strain in contrast to untreated controls (Figures 1E and 1F). Second lethal challenges to the mice that survived the first infection confirmed that the initial 7-day treatment with AN13762 resulted in a protective immune response to subsequent T. gondii infection (Figures 1E and 1F), thus strengthening the biological and pharmacokinetic profile of AN13762 in animal efficacy studies. Altogether, these results indicate that AN13762 is effective against T. gondii both in vitro and in vivo allowing long-term cures in mouse model of acute toxoplasmosis with comparable efficacy to current treatment.
Selection of T. gondii Parasites Resistant to AN13762
In an attempt to shed light on the mechanism of action of AN13762, we performed a forward genetic screen combining chemical mutagenesis to isolate AN13762-resistant parasites and next-generation sequencing analysis to map mutations conferring drug resistance (Figure 2A). Central to our approach, we reasoned that the gene(s) that would be mutated in more than one independently mutagenized resistant clone might be relevant to the drug resistance mechanism and by this means alleviating the notoriously difficult molecular mapping of point mutations induced by mutagens. For this purpose, seven independent ethyl methanesulphonate (EMS) mutagenesis experiments were performed and the resulting mutagenized parasites were selected in the presence of 10 μM AN13762 (Figure 2B), which corresponds to approximately 5-fold the EC50 value. Resistant parasites were obtained from each of the seven mutagenesis experiments, whereas none of the non-mutagenized parasites survived the selection at 10 μM AN13762, attesting once more to the parasiticidal efficacy of this compound (Figure 2B). The resistant parasite lines were then cloned by limited dilution, and we selected a single clone from each mutagenesis experiment (named A1 to G1) for transcriptome sequencing by RNA sequencing (RNA-seq). All the resistant clones were able to grow and formed plaques when grown in the presence of 10 μM AN13762 (Figures 2C–2E and S2). In parallel, the parental strain was analyzed by RNA-seq and used as a reference to identify EMS-induced mutations. We use transcriptome sequencing as most drugs target expressed proteins, with levels of gene expression and mutations being part of the sequencing results.
Figure 2.

Strategy for AN13762 Target Deconvolution
(A) Diagram of key steps of the forward genetic screen to map mutations conferring drug resistance in T. gondii parasites. EMS-mutagenized population of T. gondii tachyzoites was selected in the presence of a lethal concentration of AN13762 to isolate drug-resistant parasites. Analysis of the parental wild-type strain and multiple resistant clones by variant-calling of sequencing reads generated by RNA-seq to identify EMS-induced mutations in coding sequences conferring drug resistance.
(B) Schematic of the strategy used to obtain T. gondii-resistant parasites to AN13762. From each mutagenesis experiment a single clone (A1 to F1) was isolated and analyzed by RNA-seq.
(C) AN13762-resistant parasites form plaques after 7 days of growth in the presence of 10 μM AN13762. Complete dataset in shown in Figure S2A.
(D) Quantification of plaque sizes of wild-type parasites and resistant lines (A1 to G1) when cultured in the presence of AN13762. n.d., not detected. Associated data are shown in Figure S2C.
(E) Fluorescence microscopy showing intracellular growth of T. gondii AN13762-resistant lines. HFF cells were infected by the indicated T. gondii strains in the presence or absence of 10 μM AN13762. At 24 h post-infection, cells were fixed and stained with antibodies against GAP45 (magenta) and Hoechst (blue) to detect the inner membrane complex (IMC) of parasites and nuclei, respectively.
(F) Circos plot summarizing single nucleotide variants (SNVs), insertions, and deletions detected by transcriptomic analysis of the T. gondii AN13762-resistant lines, grouped by chromosome (numbered in Roman numerals with size intervals given outside). Each dot in the seven innermost gray tracks corresponds to a scatterplot of the mutations identified in the coding regions of the seven drug-resistant strains, with each ring representing one of the seven drug-resistant lines (A1 to G1). In the second outermost track, lines depicting whole-genome RNA-seq data of the T. gondii parental strain (RPKM values of genes are shown). Each bar in the outermost track represents locations of selected archetypal essential genes. See Table S2 and Figure S4 for transcriptomic analysis.
(G) TgCPSF3 domain architecture as predicted from PFAM databases and crystal structures of Cryptosporidium CPSF3 (Swale et al., 2019). Positioning of residues that were mutated in parasites resistant to AN13762 (Y328H, G465S, S519C, and E545K, in red) or AN3661 (Y328C, Y483N, and E545K, in blue) are indicated.
Parasites Resistant to AN13762 Harbor Mutations within TgCPSF3
To map the EMS-induced mutations that confer drug resistance, the Illumina sequencing reads were aligned to the ~65-Mb T. gondii GT1 reference genome. The assembled sequences were analyzed to identify single nucleotide variations (SNVs), small insertions, or short deletions using the parental strain as a reference (see Transparent Methods). By focusing on mutations present in coding sequences, we identified a single gene, CPSF3 (Cleavage and Polyadenylation Specific Factor 3, TGGT1_285200), that harbored SNVs leading to amino acid substitutions in each of the seven drug-resistant lines that were not present in the parental strain (Figure 2F and Table 1). CPSF3 encodes a nuclear mRNA-processing endonuclease that functions in pre-mRNA maturation (Ryan, 2004), which has been previously identified as the target of several benzoxaborole compounds active against distantly related pathogens (Lunde et al., 2019; Sonoiki et al., 2017; Swale et al., 2019; Wall et al., 2018), including T. gondii (Palencia et al., 2017). Importantly, four different mutations were identified (G456S, E545K, Y328H, and S519C; Figure 2G; Table 1), among which E545K conferred resistance against AN3661 in T. gondii (Palencia et al., 2017). Mutations span from the metallo-β-lactamase domain to the RNA specificity domain of CPSF3 (Figure 2G). Therefore, these data suggest that mutations in CPSF3 were responsible for resistance against AN13762.
Table 1.
Mutations Found in Candidate Gene Identified by RNA-Seq Transcriptome Analysis
| Chr. | Gene | Annotation | Position | Variant Calling |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Parental Strain |
Resistant Mutants |
||||||||||
| WT | A1 | B1 | C1 | D1 | E1 | F1 | G1 | ||||
| V | TGGT1_285200 | CPSF3 | 2395898 | E545K (GAG to AAG) | E545K (GAG to AAG) | E545K (GAG to AAG) | |||||
| V | TGGT1_285200 | CPSF3 | 2395976 | S519C (AGC to TGC) | |||||||
| V | TGGT1_285200 | CPSF3 | 2396165 | G456S (GGC to AGC) | G456S (GGC to AGC) | ||||||
| V | TGGT1_285200 | CPSF3 | 2396549 | Y328H (TAC to CAC) | |||||||
Amino acid substitutions with the corresponding codons shown in parentheses are indicated for each mutagenized T. gondii-resistant mutant strain.
Mutations within CPSF3 Confer Resistance to AN13762
To confirm that the CPSF3 mutations were sufficient to confer resistance to AN13762, we reconstructed each of the mutations identified in AN13762-resistant parasites into the sensitive parental wild-type strain using CRISPR/Cas9 system coupled to homology-directed repair for gene editing in T. gondii (Figure 3A) (Palencia et al., 2017). Thus, RHΔku80 parasites were co-transfected with a vector expressing the Cas9 endonuclease and synthetic guide RNA (sgRNA) and the corresponding homologous single-stranded donor oligonucleotides as repair template. After selection with AN13762, emerging resistant parasites were cloned, and DNA sequencing established that the mutations have been correctly inserted at CPSF3 locus (Figures 3B and S3A). Transfections with the Cas9 control vectors alone produced no surviving parasites. In the engineered parasites, we observed that the CPSF3 mutations E545K, G456S, S519C, and Y328H substantially decreased the sensitivity against AN13762 when compared with wild-type parasites (Figures 3C–3E and S3B–3D). It is noteworthy that Y328H mutation had a significant effect on parasite growth in the absence of drug (Figure 3C, upper panel), suggesting that this mutation might affect basal activity of CPSF3 in tachyzoites, which is in line with CPSF3 being essential to parasite growth (Palencia et al., 2017; Sidik et al., 2016). In addition, the CPSF3-edited parasites harboring the mutations E545K or G456S were also resistant to AN13762 treatment in mice (Figure 1F). Altogether, these data confirm the primary role of CPSF3 mutations in conferring resistance to AN13762 and indicate that AN13762, in a similar fashion to AN3661, targets CPSF3 (Palencia et al., 2017; Swale et al., 2019).
Figure 3.

Validation of T. gondii CPSF3 as the Benzoxaborole Target
(A) Schematic of the CPSF3 gene editing strategy in T. gondii parasites. Detailed view of CPSF3 locus and CRISPR/Cas9-mediated homology-directed repair with single-stranded oligo DNA nucleotides (ssODNs) carrying nucleotide substitutions (orange letters). After homologous recombination (HR) events with ssODNs, CPSF3 recombinants were selected with AN13762.
(B) Sanger sequencing validation of CPSF3 editing. Chromatograms of CPSF3 DNA sequences from parental and engineered parasites are shown. Nucleotide positions relative to the ATG start codon on genomic DNA are indicated. A complete dataset can be found in Figure S3A.
(C) Effects of CPSF3 mutations on T. gondii lytic cycle as determined by plaque assay. Plaque sizes were measured for WT and the engineered CPSF3 mutant strains (G456S, S519C, Y328H, E545K, Y483N, Y328C) after 7 days of growth in the absence or presence of 10 μM AN13762 or 5 μM AN3661. n.d., not detected. p values corresponding to Kruskal-Wallis test with Dunn's multiple comparisons with the wild-type (WT) strain are indicated. ns, not significant. Associated data are shown in Figure S3B.
(D) EC50 values for pyrimethamine (Pyr), AN13762, and AN3661 were determined for WT and the engineered CPSF3 mutant strains (G456S, S519C, Y328H, E545K, Y483N, Y328C). Data are mean from at least 2 independent biological replicates, each with 3 technical replicates. Associated dose-response curves are shown in Figure S3C. Mean EC50 values ± SD with fold changes (FC) in EC50 relative to that of the WT parasites are indicated.
(E) Fluorescence microscopy showing intracellular growth of WT and the CPSF3-edited parasites (G456S and E545K). HFF cells were infected with tachyzoites of the indicated T. gondii strains expressing the NLuc-P2A-EmGFP reporter gene and incubated with 10 μM AN13762, 5 μM AN3661, or 0.1% DMSO as control. Cells were fixed 24 h post-infection and then stained with antibodies against the T. gondii inner membrane complex protein GAP45 (magenta). The cytosolic GFP is shown in green. Scale bars, 10 μm. Complete dataset in shown in Figure S3D.
AN13762-Resistant Mutations G456S and S519C Do Not Confer Cross-Resistance to AN3661
We had previously found that mutations in CPSF3 were conferring resistance to another oxaborole compound, AN3661 (Figure 2G, mutations Y328C, Y483N, and E545K, Palencia et al., 2017). To examine whether the AN13762-resistant mutations in CPSF3 confer cross-resistance to AN3661, we assayed AN3661 against reconstructed parasites harboring CPSF3 mutations E545K, G456S, S519C, and Y328H. As expected, the most prevalent mutation E545K that was identified in the mutagenesis experiments conducted against either AN13762 or AN3661 conferred resistance to both compounds (Figures 3C–3E and S3B–S3D). Note that the increase in resistance to AN3661 was more dramatic than for AN13762 (~100- and ~3-fold increase in EC50, respectively; Figure 3D). Very different results were obtained for G456S and S519C mutations, which did not allow parasite growth when exposed to 5 μM AN3661 (Figure 3C). The CPSF3G456S mutation conferred the strongest resistance phenotype to AN13762 with a ~42-fold increase in AN13762 EC50 when compared with wild-type parasites, whereas sensitivity to AN3661 remained unaffected (Figures 3D and 3E). Of note, the latter mutations were not identified in the AN3661 screen, presumably reflecting their inability to protect against AN3661 at 5 μM. Conversely, the Y483N mutation identified in AN3661-resistant parasites conferred cross-resistance to AN13762. Similarly, mutations affecting the Y328 residue of CPSF3 decreased sensitivity to both compounds (Figures 3C and 3D). Altogether, these results further confirm the role of CPSF3 mutations in drug resistance and indicate a divergent mode of resistance between AN13762 and AN3661.
Molecular Docking Suggests a Divergent Resistance Mechanism between Oxaboroles
Multiple sequence alignments show a high overall sequence conservation within the metallo-β-lactamase (MBL), Beta-Casp, and RNA specificity domains of CPSF3 within apicomplexan parasites and humans (Figure 4A). One notable difference between the apicomplexan and human enzyme is the presence of an extended loop or “apicomplexan specific insert” whose length varies from 20 to 59 residues. However, conservation of the generated resistant SNVs to AN13762 within T. gondii CPSF3 coding sequence is absolute across species and appears close to the catalytic residues but is never directly involved in the coordination of the catalytic zinc atoms. Next, we visualized the resistance-conferring mutations within the recently obtained structure of Cryptosporidium CPSF3 (ChCPSF3) in co-crystal with AN3661 (pdb id 6Q55) (Swale et al., 2019). With the assumption that AN13762 interacts with a comparable geometry as the AN3661 benzoxaborole group, notably through the boron-driven octahedral coordination of the two catalytic zinc ions, we placed the AN13762 derivative in the same plane as AN3661 (Figure 4B–4D). Through this modeling, we did not generate any clashes with CPSF3, despite the much bigger size of AN13762 (13.4 Å in length against 7 Å for AN3661). When visualizing both AN3661 and AN13762 placement with regard to the resistance-conferring mutations, two important features can be noted. First, most of the mutations found (Y328C/H, E545K, S519C, and Y483N) that rescue parasites from both compounds are not directly observed in contact with the compound-binding site. Instead, the mutated residues are generally placed on loop regions lining the interfacial cavity between the RNA specificity domain and Beta-Casp domain. These resistance-conferring mutations probably act indirectly on the compound activity through either an allosteric mechanism preventing compound binding or by modifying RNA recognition by CPSF3 as these loop regions are believed to regulate RNA access and recognition (Sun et al., 2020). Second, the G456S mutation, which exclusively rescues T. gondii parasites from AN13762, is observed separated to the other resistance-conferring mutations. Because of its close proximity with the AN13762 pyrazine ring and methylazetidine (2.2 Å distance), the G456S mutant probably introduces an important steric hindrance to AN13762 binding. AN3661, with a much shorter organic extension, does not come close enough for the mutation to have an effect on its binding and activity. As a result, the G456S mutant remains sensitive to AN3661 (Figure 3E).
Figure 4.

Docking Studies for Chemotypes AN13762 and AN3661
(A) Multiple sequence alignment of CPSF3 proteins from T. gondii (Tg), C. hominis (Ch), P. falciparum (Pf), and CPSF73 of H. sapiens (Hs). The domain architecture is indicated as follows: blue, metallo-β-lactamase; green, β-CASP; orange, RNA specificity domain; magenta, the insertion within the MBL domain of apicomplexan parasites. Residues mutated in drug-resistant parasites are indicated by asterisks. The highly conserved residues involved in the coordination the zinc atoms Zn1 or Zn2 are indicated in gray. Mutations identified in parasites resistant to AN13762 or AN3661 are indicated in red and blue text, respectively.
(B) Schematic of Cryptosporidium hominis CPSF3/AN3661 co-crystal structure and modeling with AN13762. CPSF3 is displayed in a cartoon fashion with the same domain color code as in (A) surrounded by a light gray surface representation. Catalytic zinc atoms and coordinating residues are shown in gray sticks, whereas resistant mutations are shown in yellow and pink spheres.
(C) Zoom into the catalytic pocket with AN13762 manually placed colored in cyan.
(D) Zoom into the catalytic pocket binding AN3661 colored in dark purple.
AN13762 Is Active against Cryptosporidium In Vitro and In Vivo
The aforementioned data provide evidence that AN13762 targets CPSF3 enzyme. Given that it has been shown that CPSF3 is a bona fide target for inhibiting Cryptosporidium development (Swale et al., 2019), we assessed the anticryptosporidial activity of AN13762 in vitro and in vivo. The ability of AN13762 to inhibit C. parvum INRAE Nluc fast-growing strain in human ileocecal HCT-8 was assessed with its parental scaffold AN3661 as a positive control. Although less potent than AN3661, an efficient in vitro inhibition of C. parvum growth was repeatedly observed with AN13762 (EC50 13 ± 9 μM) (Figures 5A, 5B and S5). AN13762 presented no detectable toxicity for the host cells, even at 100 μM (Figure 5C). AN13762 activity was therefore assessed in vivo in a neonatal mouse model. Seven-day-old neonates were orally treated with AN13762 mixed in carboxymethyl cellulose (CMC) 4 h after C. parvum infection and daily until 3 days post-infection (dpi). Parasite load was assessed in the intestine at 4 dpi by oocyst count and measuring Nluc activity representing transgenic expression by the INRAE Nluc strain. Both methods revealed an impressive and significant inhibition of parasite development as illustrated in Figure 5D and by scanning electron microscopy where only very scarce parasites can occasionally be found on the intestinal villi of treated mice. Remarkably, the enzymatic assay revealed a 4-log reduction in luminescence signals in treated mice, and oocysts were not detected by coproscopic intestinal material examination, which is much less sensitive than the former method. Altogether, these results indicate that AN13762 is effective against C. parvum both in vitro and in vivo and provide an additional drug presumably acting by a different mode of action than AN3661 to block CPSF3 activity.
Figure 5.

Efficacy Against C. parvum in Cell Culture and Neonatal Mouse Model
(A) Comparative inhibitory activity of AN13762 and AN3661 against C. parvum INRAE Nluc strain in human ileocecal HCT-8 cells. The effect of both drugs in reducing parasitic load in epithelial cells was monitored by the luminescence signal of transgenic Nluc parasites (each concentration point represents the average of six measurements ±SD.) Curves corresponding to AN13762 and AN3661 are in magenta and blue, respectively. Corresponding fluorescence microscopy images showing intracellular growth of C. parvum parasites can be found in Figure S5.
(B) Calculated EC50 measurements are shown for AN13762 and AN3661 (n = 4 for each drug). Mean EC50 values ±SD from 4 independent biological replicates are indicated.
(C) HCT-8 cell viability assay performed 48 h with increasing concentration of AN13762. Percent viability compared with the untreated control is displayed as a function of compound concentration in micromolar concentrations. Data are presented as mean ± SD of at least two independent biological assays. Dotted line represents 100% viability.
(D) Schematic representation of the 4-day oral dosage of AN13762 (40 mg/kg) in CMC from day 0 (4 h post-infection) in 7-day-old neonatal mice previously infected with 5 × 105 oocysts. The degree of infection was monitored by counting the oocysts in the small intestine of the animals at 4 dpi (D.L., detection limit = 6 × 104 oocysts/intestine) and by monitoring Nluc activity on a small piece of ileum of each neonatal mouse (n = 14 animals per group). n.d., not detected. Mann-Whitney test, ∗∗∗∗P < 0.0001. Scanning electron microscopic imaging of neonatal mice ileum was performed at the end of the experiment on treated (AN13762) and mock treated (control) animals.
Discussion
Whole- cell phenotypic screening is an efficient approach in drug discovery that has led to the identification of numerous antimicrobial lead compounds, although the targets and mode of action remain unknown and challenging to determine. Although clinical development remains possible without this knowledge, lack of insight into the mechanism of action is one of the biggest obstacles for further medicinal chemistry optimization or to predict and track drug resistance. Fortunately, a large variety of target deconvolution technologies are currently available. The approach developed here takes advantage of all the benefits of the EMS mutagenesis method, including its wide and mostly unbiased coverage of the genome with virtually all types of mutations (Farrell et al., 2014). In this work, by combining cost-effective RNA-seq based variant calling, computational mutation discovery and CRISPR/Cas9 genome editing, we identified CPSF3, the catalytic subunit of the pre-mRNA cleavage and polyadenylation complex, as the target of AN13762 in T. gondii parasites.
In eukaryotes, CPSF3 is key to the 3′ end processing of both polyadenylated and replication-dependent histone precursor mRNAs (Shi and Manley, 2015). These distinct 3′ ends are generated co-transcriptionally by specialized 3′ end processing machineries that recognize a conserved hexanucleotide AAUAAA and a downstream G/U-rich sequence on the 3′end of nascent pre-mRNAs destined for polyadenylation or cleave histone mRNA precursors few nucleotides downstream of a highly conserved stem-loop structure (Marzluff et al., 2008). As a result, the majority of histone genes are expressed as nonpolyadenylated transcripts that are poorly detected by poly(A) purification-based RNA-seq (Lyons et al., 2016; Zhao et al., 2018). Despite this technical bias, our transcriptomic data indicate that histone mRNAs (e.g., H2Ba, H4, H2Ax, H2A1, and H2Bb; Table S2 and Figure S4) were dramatically enriched in drug-resistant lines harboring CPSF3 mutations Y328H, E545K, and S519C, but not in those mutants containing the CPSF3G456S allele (strains A1 and F1 in Table 1). This suggests hypomorphic mutations of CPSF3 that retain sufficient activity to overcome lethality but somehow favor histone pre-mRNA processing toward polyadenylation of transcripts that were otherwise barely detected using our poly(A)-selected transcript experiment settings. Interestingly, the mutations Y328H, E545K, and S519C are lining the channel accommodating the RNA substrate on CPSF3 (Figure S6), whereas the G456S mutation that is observed distant from the other mutations did not affect histone mRNA accumulation. It is noteworthy that the Y328 mutations significantly impacted the overall growth fitness (Figure 3C), suggesting a default in TgCPSF3Y328H/C activity. As the G456S mutation in T. gondii is equivalent to the G330S mutation found in the human CPSF3 counterpart conferring resistance against the anti-cancer agent JTE-607 (Ross et al., 2020), it is likely that the mechanism of resistance is shared. Possibly, the G330S and G456S mutations can only be effective for elongated molecules to clash with the compound thereby impeding binding without affecting recognition of the substrate. Yet further studies are required to determine whether the mutations in CPSF3 affect the access of the substrate to the catalytic site, complex assembly, or its conformational dynamics as shown recently by Sun et al. (2020). Altogether, these results underscore the advantage of using transcriptome sequencing to investigate mechanisms of drug action and to provide functional insight into the molecular biology of the target protein.
In mammalian cells, CPSF3 is embedded in a large multisubunit complex including CPSF1, CPSF2, CPSF4, CPSF7, cleavage stimulatory factor 1 (CSTF1), CSTF2, CSTF3, symplekin, and WDR33 (Dominski and Marzluff, 2007; Ryan, 2004). A quite similar complex was purified in T. gondii (Table S2, Swale et al., manuscript in preparation), and the identified subunits were all predicted to be essential for tachyzoite growth in vitro (Sidik et al., 2016). No mutations with significant enrichment were found in the CPSF3 protein partners in the resistant strains, which is in agreement with our docking model based on Cryptosporidium hominis CPSF3 structural data where the oxaboroles are enfolded within the CPSF3 scaffold, presumably precluding any interaction with other components.
The mutations conferring resistance to AN13762 target TgCPSF3 catalytic site, a gold standard evidence for target confirmation of a bioactive small molecule. In the published structure of AN3661 bound to ChCPSF3, the oxaborole competes with the catalytic water molecules for zinc atoms, hence blocking the phosphate bond cleavage of the pre-mRNA substrate (Swale et al., 2019). Given the overall conservation of CPSF3 catalytic core in Apicomplexa and the high conservation of the residues involved in drug resistance, it is likely that AN13762 binds to this site and disrupts the pre-mRNA processing activity of TgCPSF3 that is essential for parasite growth.
Although it is clear that AN13762 targets CPSF3 in T. gondii, different results were observed in P. falciparum where the mechanism of resistance is plural (Sindhe et al., 2020). In fact, while we were investigating the mechanism of action of AN13762 in T. gondii, Sindhe and colleagues have shown that P. falciparum resistance depends not only on the activity of Prodrug Activation and Resistance Esterase (PfPARE), an enzyme responsible for AN13762 processing, but also on enzymes involved in ubiquitination and SUMOylation pathways or PfCPSF3. The latter is responsible for the high level of resistance, thus suggesting that AN13762 or its refined derivative theoretically targets CPSF3 in malaria parasites as well. Whether AN13762 is processed in T. gondii is not known. However, as TgCPSF3G456S selectivity toward AN13762 is based on steric hindrance over the methylazetidine group, which is cleaved off upon processing by the esterase, it seems unlikely that such a modification occurs in T. gondii. Note that no mutations with significant enrichment were found in TGGT1_306330, the closest homolog to PfPARE in T. gondii (Table S2). Furthermore, as AN13762 processing is required for full antimalarial activity, it is tempting to speculate that the lack of intracellular activation explains the decreased sensitivity observed in T. gondii and Cryptosporidium (EC50 values are in the μM range, Figures 1C and 5B) relative to P. falciparum (EC50 values ranging from 18 to 118 nM, Sindhe et al., 2020).
Based on the catalytic core sequence homology between TgCPSF3 and CpCPSF3, both previously chemically validated targets for Toxoplasma and Cryptosporidium (Palencia et al., 2017; Swale et al., 2019), we successfully laid the groundwork for pathogen hopping. In this respect, AN13762 efficiently inhibits C. parvum, a species relevant to human health, in vitro and in vivo in mouse model of infection. These results appear to be even more important for the treatment of cryptosporidiosis, where druggable targets are scarce and there is a high demand for more efficient therapies. However, further work will be needed to demonstrate that AN13762 acts as a direct binder of the CpCPSF3 and inhibits its mRNA processing activity, thereby restricting the growth of parasites. The recent discovery of benzoxaborole-based chemistry has given rise to a series of compounds with great potential against various infectious agents, including trypanosomatids and apicomplexan parasites by targeting different molecular targets (De Rycker et al., 2018). Remarkably, multiple compounds with known or suspected anti-CPSF3 activity across different organisms share a similar benzoxaborole scaffold that could be a prerequisite to CPSF3 binding (Begolo et al., 2018; Lunde et al., 2019; Palencia et al., 2017; Wall et al., 2018). Interestingly, the oxaborole acoziborole can cross the blood-brain barrier (Nare et al., 2010), offering a therapeutic option to eradicate persistent Toxoplasma cysts that are resistant to most, if not all, medications currently prescribed.
Limitations of the Study
Although our study is reasonably clear about AN13762 targeting CPSF3 in Toxoplasma and its activity against Cryptosporidium parasites, it remains possible that the mechanism of action in the latter is different and depends on prodrug-activating enzyme(s) such as PfPARE as described in Plasmodium species. Hopefully, recent advances in Cryptosporidium genetics will make it possible to carry out such investigations and genetically validate the CpCPSF3 molecular target in this organism (Vinayak et al., 2020).
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Alexandre Bougdour (alexandre.bougdour@inserm.fr).
Materials Availability
All unique materials generated in this study are available from the Lead Contact upon request.
Data and Code Availability
This study did not generate/analyze code.
The Illumina RNA-seq dataset generated during this study is available at NCBI GEO: GSE156685.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We acknowledge A. Palencia for initial fruitful discussions. We thank D. Cannella and C. Corrao for technical assistance. This research was supported by funds from the Agence Nationale pour la Recherche Project ToxoP53 (grant no. ANR-19-CE15-0026) to A.B., Project HostQuest (grant no. ANR-18-CE15-0023), and the European Research Council (ERC Consolidator grant no. 614880 Hosting TOXO) to M.-A.H.
Author Contributions
F.L., M.-A.H., and A.B. conceptualized the research. A.B. supervised the research. V.B. designed and conducted the in vitro studies performed in T. gondii. C.S. performed structural modelings. M.-P.B.-P. and V.B. designed and conducted the in vivo experiments with T. gondii. A.B. computed and analyzed the RNA-seq data. F.L. supervised the work performed on Cryptosporidium. T.P. realized the in vitro and in vivo studies performed with Cryptosporidium. S.G. performed the electron microscopy study. V.B., C.S., and A.B. wrote the manuscript. Funding Acquisition, M.-A.H. and A.B. All the authors contributed to the editing of the final version of manuscript, discussed, and approved the results.
Declaration of Interests
The authors declare no competing interests.
Published: December 18, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101871.
Contributor Information
Mohamed-Ali Hakimi, Email: mohamed-ali.hakimi@inserm.fr.
Alexandre Bougdour, Email: alexandre.bougdour@inserm.fr.
Supplemental Information
RNA-seq report, T. gondii transcripts (RPKM), transcriptomic histones, transcriptomic CPSF complex, all variants, coding region variants, filtered variants in clones A1 to G1, data used in Circos plot.
References
- Begolo D., Vincent I.M., Giordani F., Pöhner I., Witty M.J., Rowan T.G., Bengaly Z., Gillingwater K., Freund Y., Wade R.C. The trypanocidal benzoxaborole AN7973 inhibits trypanosome mRNA processing. PLoS Pathog. 2018;14:e1007315. doi: 10.1371/journal.ppat.1007315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rycker M., Baragaña B., Duce S.L., Gilbert I.H. Challenges and recent progress in drug discovery for tropical diseases. Nature. 2018;559:498–506. doi: 10.1038/s41586-018-0327-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominski Z., Marzluff W.F. Formation of the 3′ end of histone mRNA: getting closer to the end. Gene. 2007;396:373–390. doi: 10.1016/j.gene.2007.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunay I.R., Gajurel K., Dhakal R., Liesenfeld O., Montoya J.G. Treatment of toxoplasmosis: historical perspective, animal models, and current clinical practice. Clin. Microbiol. Rev. 2018;31:e00057-17. doi: 10.1128/CMR.00057-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell A., Coleman B.I., Benenati B., Brown K.M., Blader I.J., Marth G.T., Gubbels M.-J. Whole genome profiling of spontaneous and chemically induced mutations in Toxoplasma gondii. BMC Genomics. 2014;15:354. doi: 10.1186/1471-2164-15-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunde C.S., Stebbins E.E., Jumani R.S., Hasan M.M., Miller P., Barlow J., Freund Y.R., Berry P., Stefanakis R., Gut J. Identification of a potent benzoxaborole drug candidate for treating cryptosporidiosis. Nat. Commun. 2019;10:2816. doi: 10.1038/s41467-019-10687-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons S.M., Cunningham C.H., Welch J.D., Groh B., Guo A.Y., Wei B., Whitfield M.L., Xiong Y., Marzluff W.F. A subset of replication-dependent histone mRNAs are expressed as polyadenylated RNAs in terminally differentiated tissues. Nucleic Acids Res. 2016;44:9190–9205. doi: 10.1093/nar/gkw620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjunatha U.H., Chao A.T., Leong F.J., Diagana T.T. Cryptosporidiosis drug discovery: opportunities and challenges. ACS Infect. Dis. 2016;2:530–537. doi: 10.1021/acsinfecdis.6b00094. [DOI] [PubMed] [Google Scholar]
- Marzluff W.F., Wagner E.J., Duronio R.J. Metabolism and regulation of canonical histone mRNAs: life without a poly(A) tail. Nat. Rev. Genet. 2008;9:843–854. doi: 10.1038/nrg2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nare B., Wring S., Bacchi C., Beaudet B., Bowling T., Brun R., Chen D., Ding C., Freund Y., Gaukel E. Discovery of novel orally bioavailable oxaborole 6-carboxamides that demonstrate cure in a murine model of late-stage central nervous system African Trypanosomiasis. Antimicrob. Agents Chemother. 2010;54:4379–4388. doi: 10.1128/AAC.00498-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palencia A., Bougdour A., Brenier-Pinchart M.-P., Touquet B., Bertini R.-L., Sensi C., Gay G., Vollaire J., Josserand V., Easom E. Targeting toxoplasma gondii CPSF3 as a new approach to control toxoplasmosis. EMBO Mol. Med. 2017;9:385–394. doi: 10.15252/emmm.201607370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross N.T., Lohmann F., Carbonneau S., Fazal A., Weihofen W.A., Gleim S., Salcius M., Sigoillot F., Henault M., Carl S.H. CPSF3-dependent pre-mRNA processing as a druggable node in AML and Ewing’s sarcoma. Nat. Chem. Biol. 2020;16:50–59. doi: 10.1038/s41589-019-0424-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan K. Evidence that polyadenylation factor CPSF-73 is the mRNA 3’ processing endonuclease. RNA. 2004;10:565–573. doi: 10.1261/rna.5214404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Manley J.L. The end of the message: multiple protein–RNA interactions define the mRNA polyadenylation site. Genes Dev. 2015;29:889–897. doi: 10.1101/gad.261974.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidik S.M., Huet D., Ganesan S.M., Huynh M.-H., Wang T., Nasamu A.S., Thiru P., Saeij J.P.J., Carruthers V.B., Niles J.C., Lourido S. A genome-wide CRISPR screen in toxoplasma identifies essential apicomplexan genes. Cell. 2016;166:1423–1435.e12. doi: 10.1016/j.cell.2016.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindhe K.M.V., Wu W., Legac J., Zhang Y.-K., Easom E.E., Cooper R.A., Plattner J.J., Freund Y.R., DeRisi J.L., Rosenthal P.J. Plasmodium falciparum resistance to a lead benzoxaborole due to blocked compound activation and altered ubiquitination or sumoylation. mBio. 2020;11:e02640-19. doi: 10.1128/mBio.02640-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoiki E., Ng C.L., Lee M.C.S., Guo D., Zhang Y.-K., Zhou Y., Alley M.R.K., Ahyong V., Sanz L.M., Lafuente-Monasterio M.J. A potent antimalarial benzoxaborole targets a Plasmodium falciparum cleavage and polyadenylation specificity factor homologue. Nat. Commun. 2017;8:14574. doi: 10.1038/ncomms14574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Zhang Y., Aik W.S., Yang X.-C., Marzluff W.F., Walz T., Dominski Z., Tong L. Structure of an active human histone pre-mRNA 3′-end processing machinery. Science. 2020;367:700–703. doi: 10.1126/science.aaz7758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swale C., Bougdour A., Gnahoui-David A., Tottey J., Georgeault S., Laurent F., Palencia A., Hakimi M.-A. Metal-captured inhibition of pre-mRNA processing activity by CPSF3 controls cryptosporidium infection. Sci. Transl. Med. 2019;11:eaax7161. doi: 10.1126/scitranslmed.aax7161. [DOI] [PubMed] [Google Scholar]
- Vinayak S., Jumani R.S., Miller P., Hasan M.M., McLeod B.I., Tandel J., Stebbins E.E., Teixeira J.E., Borrel J., Gonse A. Bicyclic azetidines kill the diarrheal pathogen cryptosporidium in mice by inhibiting parasite phenylalanyl-tRNA synthetase. Sci. Transl. Med. 2020;12:eaba8412. doi: 10.1126/scitranslmed.aba8412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall R.J., Rico E., Lukac I., Zuccotto F., Elg S., Gilbert I.H., Freund Y., Alley M.R.K., Field M.C., Wyllie S., Horn D. Clinical and veterinary trypanocidal benzoxaboroles target CPSF3. Proc. Natl. Acad. Sci. U. S. A. 2018;115:9616–9621. doi: 10.1073/pnas.1807915115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.-K., Plattner J.J., Easom E.E., Jacobs R.T., Guo D., Freund Y.R., Berry P., Ciaravino V., Erve J.C.L., Rosenthal P.J. Benzoxaborole antimalarial agents. Part 5. Lead optimization of novel amide pyrazinyloxy benzoxaboroles and identification of a preclinical candidate. J. Med. Chem. 2017;60:5889–5908. doi: 10.1021/acs.jmedchem.7b00621. [DOI] [PubMed] [Google Scholar]
- Zhao S., Zhang Y., Gamini R., Zhang B., von Schack D. Evaluation of two main RNA-seq approaches for gene quantification in clinical RNA sequencing: polyA+ selection versus rRNA depletion. Sci. Rep. 2018;8:4781. doi: 10.1038/s41598-018-23226-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
RNA-seq report, T. gondii transcripts (RPKM), transcriptomic histones, transcriptomic CPSF complex, all variants, coding region variants, filtered variants in clones A1 to G1, data used in Circos plot.
Data Availability Statement
This study did not generate/analyze code.
The Illumina RNA-seq dataset generated during this study is available at NCBI GEO: GSE156685.