

Abstract
Objective
To determine the ordering of changes in Alzheimer disease (AD) biomarkers among cognitively normal individuals.
Methods
Cross-sectional data, including CSF analytes, molecular imaging of cerebral fibrillar β-amyloid (Aβ) with PET using the [11C] benzothiazole tracer Pittsburgh compound B (PiB), MRI-based brain structures, and clinical/cognitive outcomes harmonized from 8 studies, collectively involving 3,284 cognitively normal individuals 18 to 101 years of age, were analyzed. The age at which each marker exhibited an accelerated change (called the change point) was estimated and compared across the markers.
Results
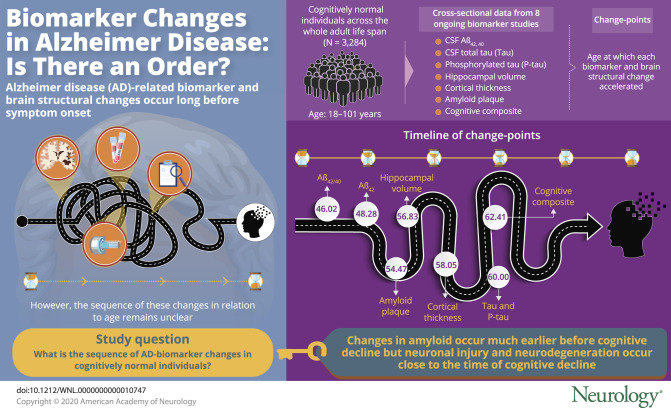
Accelerated changes in CSF Aβ1-42 (Aβ42) occurred at 48.28 years of age and in Aβ42/Aβ40 ratio at 46.02 years, followed by PiB mean cortical standardized uptake value ratio (SUVR) with a change point at 54.47 years. CSF total tau (Tau) and tau phosphorylated at threonine 181 (Ptau) had a change point at ≈60 years, similar to those for MRI hippocampal volume and cortical thickness. The change point for a cognitive composite occurred at 62.41 years. The change points for CSF Aβ42 and Aβ42/Aβ40 ratio, albeit not significantly different from that for PiB SUVR, occurred significantly earlier than that for CSF Tau, Ptau, MRI markers, and the cognitive composite. Adjusted analyses confirmed that accelerated changes in CSF Tau, Ptau, MRI markers, and the cognitive composite occurred at ages not significantly different from each other.
Conclusions
Our findings support the hypothesized early changes of amyloid in preclinical AD and suggest that changes in neuronal injury and neurodegeneration markers occur close in time to cognitive decline.
The neuropathologic course of Alzheimer disease (AD) is decades long and dynamic and begins years before symptom onset.1–4 Many clinicopathologic studies have demonstrated that asymptomatic individuals can manifest the neuropathologic changes of AD, notably senile plaques and neurofibrillary tangles.1,5,6 Major biomarker studies, including the Alzheimer’s Disease Neuroimaging Initiative and the Dominantly Inherited Alzheimer Network (DIAN), converge to suggest that Aβ accumulation and deposition in the brain is a very early pathologic process in AD, detectable by PET imaging of amyloid plaques and the CSF β-amyloid (Aβ)42 concentration.7–9 Neurofibrillary tangles and neuronal death appear to begin during the preclinical phase of AD, and by the time of early symptoms, neuronal cell death is already significant in CA1 of the hippocampus and layer II of the entorhinal cortex.10 The acceleration of tau aggregation and neurodegeneration, detectable by CSF total tau (Tau) and phosphorylated tau (Ptau11) and tau PET tracer uptake,12 may mark the transition just before symptom onset. By symptom onset, brain structural changes are also observed.5,10,13 The cascade of AD biomarker changes during preclinical AD has been hypothesized graphically,14,15 incorporated into the diagnostic criteria of preclinical AD,16 and further extended to the A/T/N (A = amyloid, T = tau, N = neurodegeneration or neuronal injury) framework.17
The objective of this study is to infer the ordering of AD biomarker changes at the mean level as a function of age in cognitively normal individuals by using a large, harmonized cross-sectional database across 8 biomarker studies and an age span of 18 to 101 years.
Methods
Participants
Participants are cognitively normal individuals from 8 ongoing biomarker studies of AD: (1) Washington University (WU) Adult Children Study (ACS); (2) Johns Hopkins University Biomarkers for Older Controls at Risk for Dementia Study; (3) Wisconsin Registry for Alzheimer's Prevention; (4) Australian Imaging, Biomarkers and Lifestyle (AIBL) Study; (5) WU DIAN; (6) WU Healthy Aging and Senile Dementia (HASD) study; (7) WU Knight Alzheimer's Disease Research Center (ADRC); and (8) Wisconsin ADRC. All studies have focused primarily on the asymptomatic phase of AD and/or recruited young and middle-aged participants at risk for AD and followed them up longitudinally with assessments of AD biomarkers, cognition, and everyday function. All individuals have a Clinical Dementia Rating18 global score of 0 (indicating cognitively normal) and have data on at least 1 of the following modalities: CSF biomarker concentrations, PET amyloid standardized uptake value ratio (SUVR), MRI structural measures (hippocampal volume and cortical thickness), and cognition. All 8 studies also collect data on APOE genotypes, obtained through standard techniques with either a blood draw or buccal swab and subsequent genotyping. In this study, APOE status is classified by the presence or absence of ε4, denoted by the terms APOE ε4 positive and negative, respectively. For participants from the DIAN, only those who are noncarriers of a highly penetrant mutation for AD (in the gene encoding amyloid precursor protein, presenilin 1 or 22) are included in the analyses. Details of the assessment protocols for each of the 8 studies have been described previously.19 Symptomatic individuals are excluded from the study because of our focus on the relative ordering of changes for AD biomarkers before symptom onset that may help improve the design (i.e., biomarker targets) and analysis of future prevention trials on AD.
Standard protocol approvals, registrations, and patient consents
All participants have given written informed consent and agreed to data sharing under the 8 ongoing AD studies. The protocol of the current study is approved by the Institutional Review Board of the WU School of Medicine.
Clinical and cognitive assessments
Details of the clinical and cognitive assessment protocols from each of the 8 studies and the harmonization of clinical and cognitive databases across these studies are described previously.19 In brief, the clinical assessment protocols are largely consistent with that of the National Alzheimer Coordinating Center Uniform Data Set,20 which includes standard diagnostic criteria for detection of dementia and its differential diagnoses.21 The presence or absence of dementia and, when present, its severity are operationalized with the Clinical Dementia Rating by all studies. Five cognitive tests are shared by almost all studies: the Mini-Mental State Examination,22 Boston Naming Test,23 Animal Naming (60 seconds),24 Wechsler25 Adult Intelligence Scale Digit Symbol,25 and Logical Memory Delayed Recall. A cognitive composite is calculated by averaging the individual z scores of the 5 psychometric tests, using the overall mean and SD of each test. If a participant has missing data from 1 or 2 cognitive tests, the cognitive composite is averaged over the number of tests for which data are available. Only those with data from at least 3 of the 5 tests are included in the cognitive analyses.
CSF and imaging biomarkers
Four well-established CSF biomarkers for AD are analyzed: CSF Aβ42, Aβ42/Aβ40 ratio, Tau, and Ptau. Both PET imaging of amyloid plaques with a small molecule radiotracer, the [11C] benzothiazole tracer Pittsburgh compound B (PiB7), and MRI imaging of brain structures are analyzed, including PET PiB mean cortical SUVR (obtained by averaging SUVRs over FreeSurfer regions within the prefrontal cortex, precuneus,26 and temporal cortex27), PET PiB SUVR in the precuneus (PiB precuneus), MRI total hippocampal volume (MRI hippocampal volume), and the MRI cortical thickness. PET and MRI data are obtained after centrally reprocessing scans across studies at the WU NeuroImaging Lab using a standard protocol. The cerebellum (gray matter) is chosen as the reference region. Details of the imaging protocol have been described previously.19 Because absolute values for a given CSF analyte differ as a function of collection protocol and assay platform,28,29 only data from the samples in the 4 studies (WU ACS, DIAN, WU ADRC, WU HASD) that shared similar CSF collection protocols and used the Roche Elecsys immunoassay30 are included in the statistical analyses. Details of the CSF processing have been previously described.19
Statistical analyses
The statistical analyses follow an intuitive conceptualization (figure 1) that each AD biomarker, as a function of age and at the mean level, initially reflects no or minimal effect due to AD neuropathology at young ages because very few individuals have preclinical AD and then, after a certain age (called a change point), an additional effect of AD neuropathology and neurodegeneration due to the fact that a significant portion of the older population has preclinical AD. This conceptualization implies a cross-sectional acceleration on the age-related change in the older population that represents a combination of (pure) age effect and the effect of AD neuropathology and other possible pathologies (i.e., vascular) after the change point compared to individuals younger than the change point. Hence, a piecewise linear regression model is used to describe the mean pattern of the biomarker as a function of age. Statistically, the expected biomarker change follows the first linear trend for individuals younger than the change point, with a positive slope indicating an increasing trend and a negative slope indicated a decreasing trend. When individuals are older than the change point, the expected biomarker change follows, in a continuous manner, another (the second) linear trend that describes the accelerated age-related change, with a larger slope (in magnitude) than the first linear trend. The change points in age across all major AD markers where the accelerated changes occur are of central interest in ordering the AD markers at the mean level (but not at the individual level). For example, biomarker 1 with a cross-sectional change point at age 50 years has an accelerated relationship with age among individuals >50 years of age (by virtue of how much the biomarker differs at the mean level between 2 independent subcohorts of individuals whose ages differ by 1 year) compared to the relationship of the marker with age among individuals <50 years old. Compared to biomarker 2 with a change point at age 60 years, biomarker 1 started to have a stronger relationship with age earlier than biomarker 2. Given that age is the most important risk factor of AD, we use this comparison to infer the ordering of biomarkers 1 and 2 at the mean level.
Figure 1. Cross-sectional conceptualization of an AD biomarker as a function of age at the mean level.

The 2 piecewise linear lines represent the average level of the marker across all individuals of the same ages. A change point (in the unit of age) connecting the 2 linear lines is conceptualized to reflect the higher prevalence of preclinical Alzheimer disease (AD) in the older population.
Change point regression modeling31 is applied to fit data from each AD biomarker as a piecewise linear function of age, initially allowing only 1 change point. Given that biomarker changes in cognitively normal individuals are correlated with major AD risk factors,32 adjusted analysis for the effects of important covariates (study cohort, APOE ε4 status, race, sex, family history of dementia, and education) is further implemented. Maximum likelihood estimator to the change point is obtained, along with its 95% confidence interval (CI).33 Maximum likelihood estimators to both slopes of the biomarker against age (for individuals younger than and older than the change point) are also obtained simultaneously. By comparing these 2 slopes, we can test and confirm the existence of the change point.33 Once a change point is confirmed, the same procedure is applied to each of the subcohorts of individuals (with age either younger or older than the initially identified change point) to further explore the possibility of another distinctive change point within each subinterval of age. Sensitivity analyses are conducted by treating study cohorts as both fixed and random effects. No missing data are imputed. Further analyses are conducted to identify and compare change points by APOE ε4 status (positive vs negative).
To test the ordering of the markers at the mean level, the bootstrapping technique34 is used to generate 1,000 bootstrapped datasets by sampling with replacement from the original dataset so that the change points of all markers can be simultaneously estimated with the same bootstrapped datasets. Subsequently, for each pair of 2 markers, the paired difference in change points is calculated, and the 95% bias-corrected bootstrap quantiles-based CI is derived for the difference. If a 95% CI does not include zero, it provides statistical support (at the 5% significance level) that 1 of the 2 markers in the pair changes earlier than the other at the mean level.
All computations are performed with R (version 3.3.1, R Foundation for Statistical Computing, Vienna, Austria). The R package segmented (version 0.5-1.4) is used for estimating the change point and testing its existence for each marker. All statistical tests and CIs are 2 sided.
Data sharing
Requests for deidentified data can be sent to the corresponding author.
Results
Participant characteristics are summarized in table 1 overall for all participants with data from at least 1 of the 4 modalities (CSF, amyloid PET, MRI, and cognition) and separately for each modality-specific cohort. In total, data from 3,284 cognitively normal participants were analyzed: 3,102 participants were included for the analysis of the cognitive composite, 807 participants for the analysis of the CSF biomarkers, 830 participants for the analysis of the PET PiB SUVRs, and 1,489 participants for the analysis of the MRI structural measures. The 4 cohorts across modalities overlapped significantly: 1,467 participants had data from at least 2 of the 4 modalities; 407 participants had data from 3 of the 4 modalities; and 535 had data from all 4 of the modalities. As a result, these cohorts shared largely similar characteristics. The median ages of participants across the 4 cohorts were between 65 and 67 years. Across all the modality-specific cohorts, participants were predominantly White and female (≈60%). The median education was 16 years, and ≈30% of all participants were APOE ε4 positive.
Table 1.
Demographic and APOE ε4 characteristics of the entire cohort and the modality-specific cohorts

The unadjusted analyses confirmed that for each marker under analyses, a change point exists in age when age-related change started to accelerate. Table 2 presents the estimated change points along with their 95% CIs across the markers, the estimated slopes against age younger or older than the change point, and the associated p values for comparing the 2 slopes. The earliest change points were observed for CSF Aβ42 at the age of 48.28 years (95% CI 39.97–56.60) and Aβ42/Aβ40 ratio at 46.02 years (95% CI 38.54–53.51). Before the change point, CSF Aβ42 increased slightly with age as indicated by a positive slope (estimate/standard error [SE] 15.23/6.23 pg/mL) but decreased significantly with age after the change point (slope estimate/SE −10.58/2.80 pg/mL). The PiB PET mean cortical SUVR (and the SUVR in the precuneus) initially showed very minimal change with age (slope estimate/SE −0.0013/0.0035 and −0.0007/0.0044 for cortical mean and precuneus SUVRs, respectively), but both significantly increased with age after the estimated change point at ≈54 years of age. A change point at 56.83 years (95% CI 51.80–61.86) and at 58.05 years (95% CI 51.75–64.35) was detected for the MRI hippocampal volume and cortical thickness, respectively, with an accelerated decrease of hippocampal volume and thinning of the cortical thickness after these ages. CSF Tau, Ptau, and the cognitive composite all had a change point detected at ≈60 years of age, indicating a faster accumulation of measurable soluble Tau and Ptau proteins in the CSF and more rapid deterioration of cognition at about the same age. Figure 2 provides a visualization of the change points overlaid on the scatterplots of all 9 markers as functions of age.
Table 2.
Individual change point estimate without covariate adjustment

Figure 2. Scatterplots of AD markers against age overlaid with the estimated piecewise linear lines and change points.

Each Alzheimer disease (AD) marker (as indicated by y-axis labels) is plotted against age. Estimated piecewise linear lines are overlaid over the data points and connected at the change point (indicated by the blue dot at top) with associated 95% confidence interval (blue line at top). Aβ = β-amyloid; MR = magnetic resonance; p-tau = phosphorylated tau; PiB = Pittsburgh compound B.
No additional change points were found for CST Tau, Ptau, PiB PET SUVRs, or MRI hippocampal volume. For MRI cortical thickness, we identified a second change point at the age of 39.91 years (p = 0.0372). For CSF Aβ42, a change point at 34.97 years (p = 0.0104) and another change point at 87.26 years (p = 0.0162) were detected. However, none of these additional change points for the MRI cortical thickness or CSF Aβ42 were statistically significant after multiplicity adjustments.
For each pair of markers, the bias-corrected percentile bootstrapping 95% CI for the difference of the 2 change points is presented in table 3. Age-related accelerated change in CSF Aβ42/Aβ40 ratio and Aβ42 occurred significantly earlier (nearly 12 or >12 years) than that for CSF Tau and Ptau, ≈10 years earlier than that for cortical thickness, and >14 years earlier than that for the cognitive composite. In addition, accelerated age-related changes in PiB PET SUVR (both cortical mean and in the precuneus) occurred ≈8 years earlier than that for the cognitive composite. The 95% CIs for the differences of the change points in every other pair crossed zero; thus, no statistically significant evidence existed to indicate an ordering between the markers within these pairs from the unadjusted analyses (table 3).
Table 3.
95% Bias-corrected bootstrap CI for pairwise difference in change points without adjustment for covariates

The adjusted analyses after accounting for the effect of covariates are displayed in table 4. The adjusted estimates to change points varied only slightly from the unadjusted estimates in table 2. The 2 exceptions were the MRI cortical thickness and the cognitive composite. The MRI cortical thickness had an adjusted change point at 62.00 years (95% CI 56.44–67.55), and the cognitive composite had an adjusted change point at 55.02 years (95% CI 51.42–58.61). Further adjusted analyses after identifying the initial change point for each marker resulted in no additional change points, with the possible exception of CSF Aβ42, in which an additional change point at 35.00 years (p = 0.01) was observed but was not statistically significant after multiplicity adjustments. The bootstrap bias-corrected 95% CIs for the pairwise difference of change points from each pair of markers after accounting for the covariates are presented in table 5. These results further confirmed that the age-related changes in CSF Aβ42/Aβ40 ratio with an estimated change point of 45.98 years and in CSF Aβ42 with an estimated change point of 47.55 years, albeit not significantly different from the change point in PiB PET SUVRs at the age of 54.52 years, were significantly earlier than those for CSF Ptau, MRI-based cortical thickness, and the cognitive composite. Furthermore, there were no statistically significant differences in change points among CSF Tau, Ptau, MRI hippocampal volume and cortical thickness, and the cognitive composite. Sensitivity analyses (by treating the study cohorts as a random effect) resulted in largely consistent results.
Table 4.
Individual change point estimate with covariate adjustment

Table 5.
95% Bias-corrected bootstrap CI for pairwise differences of change points, accounting for covariates

Table e-1 (available on Dryad, doi.org/10.5061/dryad.x69p8czfs) presents the estimated change points for each marker that are further stratified by APOE ε4 status (positive vs negative). The estimated change points were not significantly different between APOE ε4–positive and –negative individuals and were largely consistent with the overall estimates among all participants (table 2). However, likely due to much smaller sample sizes (only 137 participants had data on CSF Aβ42/Aβ40 ratio), the estimated change points were not statistically significant among APOE ε4–positive individuals for CSF Aβ42/Aβ40 ratio, Tau, and Ptau and among APOE ε4–negative individuals for CSF Aβ42/Aβ40 ratio.
Discussion
Given that the neuropathologic change of AD begins many years before symptom onset,1–4 the temporal ordering of the neuropathologic and neurodegenerative events during the preclinical phase of AD provides critical information for designing prevention trials for AD. The cascade of biomarker changes in preclinical late-onset AD has been hypothesized16,17 to follow specific orderings from amyloid to tau, then to brain structure, and finally to cognition. This hypothesis, if proven correct, suggests possibly different targets for preventive interventions during different stages of preclinical AD, namely that primary prevention trials may target the change in amyloid or tau in the brain, whereas the secondary prevention trials may target cognitive changes.
The hypothesis of the temporal ordering of biomarker changes in preclinical AD, however, remains to be statistically tested. The reason in part is the lack of cross-sectional and longitudinal biomarker data that can capture the very early biomarker changes, which may occur decades earlier than symptom onset, in addition to the analytic challenges that these biomarkers are from different modalities with different measurement units and different distributions, which make direct comparisons across markers meaningless. Leveraging a large and harmonized biomarker database across 8 biomarker studies (3,284 cognitively normal individuals whose ages span from 18 to 101 years), we assessed the relative ordering of changes for AD biomarkers in cognitively normal individuals by conceptualizing a cross-sectional acceleration in the age-related changes at a latent age (the change point) due to the fact that a significant portion of older population have preclinical AD. Our cross-sectional piecewise linear regression analyses searched for the change point for each marker, tested the existence of the change point by comparing the age-related changes between individuals younger and older than the change point, and finally compared biomarkers across modalities on the same scale, namely, their change points in age, with a computationally intensive bootstrapping technique to infer the ordering of biomarker changes in cognitively normal individuals.
Our results from the unadjusted analyses largely support the hypothesized orderings of biomarker changes during the preclinical stage. Specifically, we confirm that CSF Aβ42/Aβ40 ratio and Aβ42 showed the earliest change points in age, as young as 46.02 and 48.28 years, respectively, which are not statistically different from the change point (≈54 years) for PiB PET mean cortical SUVR or the precuneus PiB PET SUVR but are significantly earlier than the change points for CSF Tau and Ptau, MRI cortical thickness, and the cognitive composite. The unexpected positive slope of 15.2 pg/mL for CSF Aβ42 observed before the change point of 48.28 years is interesting but consistent with several published studies.35,36 Future studies on this are needed. Our analyses did not find a significant difference among the change points for the markers presumed to represent neurodegeneration and neuronal injury (CSF Tau, Ptau, MRI-based hippocampal volume and cortical thickness) and that of the cognitive composite. In fact, the estimated change points from the unadjusted analyses for these markers are all around the age of 60 years, suggesting almost simultaneous acceleration of change for these markers among cognitively normal individuals. These results, albeit not perfectly consistent with the hypothesized ordering of biomarker changes during preclinical stage, are nonetheless supported by multiple studies reporting that CSF Tau, Ptau, and MRI structural changes predict each other's change, and most importantly, the cognitive change. For example, the Harvard Aging Brain Study recently reported that cognitive decline was most closely associated with tau change, beyond baseline Aβ and tau.12 Furthermore, we have previously found that baseline values of CSF Tau, Ptau, and MRI hippocampal volume all predicted the rate of longitudinal change in cognition among cognitively normal individuals and, more importantly, that the longitudinal rate of change in CSF Tau (Ptau), but not CSF Aβ42 or PiB PET SUVR, was correlated negatively with longitudinal rate of cognitive change over the same windows of longitudinal follow-up.11 The longitudinal rate of change in hippocampal volume was also positively correlated with the longitudinal rate of change in cognition over the same window of follow-up. Recently, it was reported that increasing levels of tau most consistently relate to declines in cognition preceding biomarker collection and suggested that elevated Aβ alone may be insufficient to produce cognitive change in individuals at risk for AD dementia.37
Our findings have important implications for the design and analysis of future prevention trials in AD. First, because the change points in biomarkers for brain amyloid occur at least a decade earlier than the change point for cognition, drugs targeting amyloid may have limited chance to demonstrate cognitive benefit if the duration of the prevention trial is not long enough. The ongoing and future secondary prevention trials of AD may need to consider much longer follow-up, especially given the absence of sensitive cognitive tests that can detect subtle cognitive changes when amyloid buildup initiates. Second, if, as our results indicate, change points in CSF Tau and Ptau occur almost simultaneously with the change point in cognition, prevention trials targeting tau may have a better chance to demonstrate cognitive benefit with a relatively short follow-up. Furthermore, because factors other than AD (e.g., vascular insults38) could result in change in brain structures such as hippocampal volume and cortical thickness, compounds that help preserve structural integrity of the brain may provide another channel to slow cognitive decline, highlighting the importance of simultaneously targeting tau and other comorbid conditions or mixed pathologies in preventing dementia due to AD, perhaps through combinations of different compounds.
Our findings from the adjusted analyses, albeit largely consistent with those from the unadjusted analyses, suggest that accelerated changes in cognition may occur as young as 55 years, right after the accelerated change in CSF Aβ42 and PiB PET SUVRs. The surprisingly early estimate of the cognitive change point differs from other published studies of cognitive changes.39 The most likely reason behind this discrepancy is that our harmonized database included cognitively normal individuals from almost the entire adult lifespan from 18 to 101 years in age, whereas most of the previous studies were based on cohorts of much older ages (e.g., >76 years39) and hence tended to overestimate the change points. Our estimated early change point for cognition, on the other hand, is supported by findings from a large study of aging (The Whitehall II study with 5,198 men and 2,192 women over a 10-year period from 199740) that reported that cognition can start to deteriorate as early as 45 years of age after adjustment for the effect of education. Another longitudinal observational study in 2,124 participants from the Study of Women's Health Across the Nation also provides strong, longitudinal evidence of cognitive aging in midlife women, with substantial within-woman declines in processing speed and memory.41 Findings from the Interdisciplinary Study on Adult Development (n = 346) further suggest that cognitive changes may occur among middle-aged individuals (mean 43.8 years).42 A subsequent and fundamental question is, what are the causes behind this early cognitive change? On the one hand, early cognitive decline may be accompanied by the accelerated age-related change in CSF Aβ42 and PiB PET SUVR even when cognitively normal individuals are Aβ negative.43 On the other hand, given that the estimated cognitive change point is numerically earlier than those for some of the AD biomarkers (CSF Tau, Ptau, MRI hippocampal volume and cortical thickness) and that most of these biomarkers do not show appreciable age-related changes during this young age window (table 4), the early cognitive change may be unrelated to the preclinical changes in some of these biomarkers. Furthermore, the fact that the differences between the adjusted cognitive change point and the change points from CSF Tau, Ptau, and MRI hippocampal volume and cortical thickness are not statistically significant suggests that these biomarkers and cognition start to show accelerated age-related changes at similar ages. Hence, soon after these change points (55–62 years), there may exist a bidirectional relationship between the early cognitive change and the early biomarker changes.44 Further large-scale and longitudinal studies are needed to fully appreciate the complexity of preclinical cognitive changes and their relationship with changes in these biomarkers.
Our study has several major strengths. First, this study represents our great efforts of rigorous statistical testing of the hypothesized cascade of changes for AD biomarkers on one of the largest biomarker and cognitive cohorts (n = 3,284) of cognitively normal individuals covering almost the entire adulthood from 18 to 101 years. Second, data from all major AD biomarkers across the modalities of CSF, amyloid PET, and MRI were available on a large overlapping subset of participants and jointly analyzed to estimate and compare the change points across the markers. Third, the CSF biomarker measures were obtained with the same high-performance automated assay platform (Elecsys),30,45 and all PET and MRI imaging data were obtained after centrally reprocessing raw imaging scans across the studies. This study also has limitations. First, it is a cross-sectional study with findings that may not generalize to longitudinal data, which are necessary to fully test the hypothesized ordering of biomarker changes during the preclinical stage of AD. Specifically, as demonstrated previously,46 it is dangerous to extrapolate our estimated cross-sectional change at the mean level as a function of age to longitudinal and within-participant change. Furthermore, it is not possible to fully differentiate (pure) age-related changes from AD-specific changes in a cross-sectional analysis because older ages are confounded with higher prevalence of preclinical AD. Hence, although all markers we analyzed are well-established AD biomarkers, the reported slopes can be interpreted only as a combined effect of age, AD neuropathology, and even other possible pathologies (i.e., vascular) for which the database has no or limited information. Second, although there was a large subset of participants with biomarker data on all modalities, not all participants had data on all biomarker modalities. Third, the entire study cohort may not represent the general population, and selection bias may exist. Finally, the cognitive data were restricted to a relatively small number of tests shared by all the studies, which may miss cognitive domains that may be particularly affected during the preclinical stage of AD. The difference between unadjusted and adjusted cognitive analyses suggests the difficulty in estimating the ordering of preclinical changes in cognitively normal individuals that may be sensitive to the study cohort and assessment method. Despite these potential shortcomings, our study represents the appreciable effort to rigorously and statistically test the hypothesized ordering of changes for all major AD biomarkers using a large sample of cognitively normal individuals from 18 to 101 years of age. Our findings corroborate the hypothesis that amyloid deposition occurs early and that changes in cognition and other biomarkers thought to represent degeneration (CSF tau, Ptau, volumetric MRI) occur years later, either simultaneously or in close succession. Further studies using longitudinal data and analyses will be required to refine this understanding.
This study largely confirms the hypothesized cascade of biomarker changes in preclinical late-onset AD and moreover provides substantiated and precise knowledge of their relationships with age. This may facilitate more expedient study designs and enhance the probability of success in future prevention trials of AD.
Acknowledgment
The authors acknowledge the altruism of the participants and their families and contributions of the DIAN research and support staff at each of the participating sites for their contributions to this study.
Glossary
- Aβ
β-amyloid
- ACS
Adult Children Study
- AD
Alzheimer disease
- ADRC
Alzheimer's Disease Research Center
- AIBL
Australian Imaging, Biomarkers and Lifestyle
- CI
confidence interval
- DIAN
Dominantly Inherited Alzheimer Network
- HASD
Healthy Aging and Senile Dementia
- PiB
Pittsburgh compound B
- Ptau
phosphorylated tau
- SE
standard error
- SUVR
standardized uptake value ratio
- Tau
total tau
- WU
Washington University
Appendix. Authors
Study funding
This study was supported by National Institute on Aging (NIA) grant R01 AG053550 (Dr. Xiong) and NIA grants P50 AG005681, P01AG026276, and P01 AG0399131 (Dr. Morris), UF1AG032438 (Dr. Bateman), U19-AGO33655 and R01 AG059869 (Dr. Albert), R01 AG027161 and R01 AG021155 (Dr. Johnson), and Australian Commonwealth Scientific Industrial Research Organization (Dr. Masters).The AIBL study (AIBL.csiro.au) was supported by the Alzheimer's Association (United States), the Alzheimer's Drug Discovery Foundation, an anonymous foundation, the Science and Industry Endowment Fund, the Dementia Collaborative Research Centres, the Victorian Government's Operational Infrastructure Support program, the McCusker Alzheimer's Research Foundation, the National Health and Medical Research Council, and the Yulgilbar Foundation, plus numerous commercial interactions that supported data collection and analysis. Image processing was supported in part by the Neuroimaging Informatics and Analysis Center (1P30NS098577) and R01 EB009352. Data collection and sharing for this project were supported by DIAN (UF1AG032438) funded by the NIA, the German Center for Neurodegenerative Diseases, and Raul Carrea Institute for Neurological Research. Partial support was provided by the Research and Development Grants for Dementia from Japan Agency for Medical Research and Development, and the Korea Health Technology R&D Project through the Korea Health Industry Development Institute. This manuscript has been reviewed by DIAN Study investigators for scientific content and consistency of data interpretation with previous DIAN Study publications.
Disclosure
J. Luo, F. Agboola, E. Grant, C.L. Masters, and M.S. Albert report no disclosures relevant to the manuscript. S.C. Johnson has served on a scientific advisory board for Roche Diagnostics and has received research funding from the NIH and from Cerveau Technologies. E.M. McDade and J. Vöglein report no disclosures relevant to the manuscript. A.M. Fagan has received research funding from the NIA of the NIH, Biogen, Centene, Fujirebio, and Roche Diagnostics; she is a member of the scientific advisory boards for Roche Diagnostics, Genentech and AbbVie and consults for Araclon/Grifols, Diadem, and DiamiR. T. Benzinger, P. Massoumzadeh, J. Hassenstab, R.J. Bateman, J.C. Morris, and R.J. Perrin report no disclosures relevant to the manuscript. J. Chhatwal has served on a medical advisory board for Otsuka Pharmaceuticals. M. Jucker, B. Ghetti, C. Cruchaga, N.R Graff-Radford, P.R. Schofield, H. Mori, and C. Xiong report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Morris JC, Price JL. Pathologic correlates of nondemented aging, mild cognitive impairment, and early-stage Alzheimer's disease. J Mol Neurosci 2001;17:101–118. [DOI] [PubMed] [Google Scholar]
- 2.Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med 2012;367:795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Katzman R. Editorial: the prevalence and malignancy of Alzheimer disease: a major killer. Arch Neurol 1976;33:217–218. [DOI] [PubMed] [Google Scholar]
- 4.Vos SJB, Xiong CJ, Visser PJ, et al. Preclinical Alzheimer's disease and its outcome: a longitudinal cohort study. Lancet Neurol 2013;12:957–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Price JL, McKeel DW Jr, Buckles VD, et al. Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol Aging 2009;30:1026–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 2006;66:1837–1844. [DOI] [PubMed] [Google Scholar]
- 7.Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer's disease with Pittsburgh compound-B. Ann Neurol 2004;55:306–319. [DOI] [PubMed] [Google Scholar]
- 8.Fagan AM, Xiong C, Jasielec MS, et al. Longitudinal change in CSF biomarkers in autosomal-dominant Alzheimer's disease. Sci Transl Med 2014;6:226ra230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jagust W. Mapping brain beta-amyloid. Curr Opin Neurol 2009;22:356–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gomez-Isla T, Price JL, McKeel DW, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J Neurosci 1996;16:4491–4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiong CJ, Jasielec MS, Weng H, et al. Longitudinal relationships among biomarkers for Alzheimer disease in the Adult Children Study. Neurology 2016;86:1499–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hanseeuw BJ, Betensky RA, Jacobs HIL, et al. Association of amyloid and tau with cognition in preclinical Alzheimer disease: a longitudinal study. JAMA Neurol 2019;76:915–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Price JL, Ko AI, Wade MJ, Tsou SK, McKeel DW, Morris JC. Neuron number in the entorhinal cortex and CA1 in preclinical Alzheimer disease. Arch Neurol 2001;58:1395–1402. [DOI] [PubMed] [Google Scholar]
- 14.Jack CR, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol 2010;9:119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jack CR, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 2013;12:207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jack CR, Bennett DA, Blennow K, et al. NIA-AA research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement 2018;14:535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43:2412–2414. [DOI] [PubMed] [Google Scholar]
- 19.Xiong C, Luo J, Agboola F, et al. A harmonized longitudinal biomarkers and cognition database for assessing the natural history of preclinical Alzheimer's disease from young adulthood and for designing prevention trials. Alzheimers Dement 2019;15:1448–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morris JC, Weintraub S, Chui HC, et al. The Uniform Data Set (UDS): clinical and cognitive variables and descriptive data from Alzheimer Disease Centers. Alz Dis Assoc Dis 2006;20:210–216. [DOI] [PubMed] [Google Scholar]
- 21.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Folstein MF, Folstein SE, McHugh PR. “Mini-Mental State”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 23.Goodglass H, Kaplan E. Boston Diagnostic Aphasia Examination Booklet. 3rd ed. Philadelphia: Lea & Febiger; 1983. [Google Scholar]
- 24.Rosen WG. Verbal fluency in aging and dementia. J Clin Neuropsychol 1980;2:135–146. [Google Scholar]
- 25.Wechsler D. Wechsler Adult Intelligence Scale. New York: Psychological Corp; 1955. [Google Scholar]
- 26.Sperling RA, LaViolette PS, O'Keefe K, et al. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron 2009;63:178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gordon BA, Blazey T, Su Y, et al. Longitudinal beta-amyloid deposition and hippocampal volume in preclinical Alzheimer disease and suspected non-Alzheimer disease pathophysiology. JAMA Neurol 2016;73:1192–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mattsson N, Andreasson U, Persson S, et al. The Alzheimer's Association external quality control program for cerebrospinal fluid biomarkers. Alzheimers Dement 2011;7:386–395 e386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mattsson N, Andreasson U, Persson S, et al. CSF biomarker variability in the Alzheimer's Association quality control program. Alzheimers Dement 2013;9:251–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bittner T, Zetterberg H, Teunissen CE, et al. Technical performance of a novel, fully automated electrochemiluminescence immunoassay for the quantitation of beta-amyloid (1-42) in human cerebrospinal fluid. Alzheimers Dement 2016;12:517–526. [DOI] [PubMed] [Google Scholar]
- 31.Muggeo VM. Estimating regression models with unknown break-points. Stat Med 2003;22:3055–3071. [DOI] [PubMed] [Google Scholar]
- 32.Morris JC, Roe CM, Xiong C, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol 2010;67:122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muggeo VMR. Testing with a nuisance parameter present only under the alternative: a score-based approach with application to segmented modelling. J Stat Comput Sim 2016;86:3059–3067. [Google Scholar]
- 34.Efron B. 1977 Rietz Lecture: bootstrap methods—another look at the Jackknife. Ann Stat 1979;7:1–26. [Google Scholar]
- 35.Schupf N, Tang MX, Fukuyama H, et al. Peripheral Aβ subspecies as risk biomarkers of Alzheimer's disease. Proc Natl Acad Sci USA 2008;105:14052–14057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cirrito RJ, Yamada AK, Finn MB, et al. Synaptic activity regulates interstitial fluid amyloid-B levels in vivo. Neuron 2005;48:913–922. [DOI] [PubMed] [Google Scholar]
- 37.Aschenbrenner AJ, Gordon BA, Benzinger TLS, Morris JC, Hassenstab JJ. Influence of tau PET, amyloid PET, and hippocampal volume on cognition in Alzheimer disease. Neurology 2018;91:e859–e866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DeCarli C. Clinically asymptomatic vascular brain injury: a potent cause of cognitive impairment among older individuals. J Alzheimers Dis 2013;33(suppl 1):S417–S426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilson RS, Leurgans SE, Boyle PA, Bennett DA. Cognitive decline in prodromal Alzheimer disease and mild cognitive impairment. Arch Neurol 2011;68:351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singh-Manoux A, Kivimaki M, Glymour MM, et al. Timing of onset of cognitive decline: results from Whitehall II prospective cohort study. BMJ 2012;344:d7622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karlamangla AS, Lachman ME, Han W, Huang M, Greendale GA. Evidence for cognitive aging in midlife women: study of women's health across the nation. PLoS One 2017;12:e0169008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zimprich D, Mascherek A. Five views of a secret: does cognition change during middle adulthood? Eur J Ageing 2010;7:135–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Landau SM, Horng A, Jagust WJ; Alzheimer's Disease Neuroimaging Initiative. Memory decline accompanies subthreshold amyloid accumulation. Neurology 2018;90:e1452–e1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Younes L, Albert M, Moghekar A, Soldan A, Pettigrew C, Miller MI. Identifying changepoints in biomarkers during the preclinical phase of Alzheimer's disease. Front Aging Neurosci 2019;11:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kollmorgen G, Manuilova E, Oelschlaegel T, et al. Elecsys® total-tau and phospho-tau (181P) CSF assays: analytical performance of the novel, fully automated immunoassays for quantification of tau proteins in human cerebrospinal fluid. Clin Biochem 2019;72:30–38. [DOI] [PubMed] [Google Scholar]
- 46.McDade E, Wang G, Gordon BA, et al. Longitudinal cognitive and biomarker changes in dominantly inherited Alzheimer disease. Neurology 2018;91:e1295–e1306. [DOI] [PMC free article] [PubMed] [Google Scholar]