

Abstract
Bioadhesive membranes with controllable and reversible underwater adhesion are desirable for several biomedical applications ranging from biosensing, drug/therapeutic delivery, and tissue regeneration. Here, we present dual soft mucosal and hard bone/enamel tissue adhesive nanofiber membranes composed of chitosan and pectin derivatives for pH-controlled delivery of antimicrobial peptides (AMPs) in the oral cavity. Ex vivo testing with porcine esophagus (soft mucosal mimic) indicated a 2-fold increase in the mucoadhesion of chitosan membranes with 0.05 wt % oxidized pectin coating, while the uncoated membranes exhibited 3–4-fold stronger adhesion to hydroxyapatite discs (enamel/hard bone mimic) compared to the coated membranes. The former is attributed to a synergistic interaction of surface nanofiber topography, intermolecular hydrogen bonding, and aldehyde–amine chemistry between surface polar groups and mucosal proteins, while the latter may arise from electrostatic interactions between cationic amines (─NH3+) in chitosan and anionic phosphates (─PO43─) in hydroxyapatite. Further, the dual hard–soft oral tissue adhesive nanofiber membranes loaded with cationic amphipathic AMPs (D-GL13K and IDR-1018) elicited pH-responsive AMP delivery and antimicrobial action comparable to chlorhexidine (CHX) against oral streptococci. Concurrently, the AMP loaded membranes were cytocompatible to both soft epithelial tissue-derived human oral keratinocytes and hard calvarial murine pre-osteoblast cells. We envision these membranes to function as adhesive gingival grafts and guided bone regeneration (GBR) membranes at the hard–soft tissue interface while simultaneously protecting against oral infections.
Graphical Abstract
1. INTRODUCTION
Bioadhesives are becoming common in clinical use as tissue adhesives, hemostatic agents, and tissue sealants.1 While the majority of bioadhesives are applied externally on the skin as wound dressings or sensors, the need for bioadhesives with underwater or wet adhesion capability is also being recognized.2 Furthermore, reversible and controllable wet adhesives with temporary or permanent tissue bonding capabilities present greater flexibility in a myriad of biomedical applications.3 However, several of the popular wet bioadhesives based on naturally derived fibrin glue, semisynthetic gelatin-resorcinol-formaldehyde/glutaraldehyde (GRFG), or synthetic cyanoacrylate glue are irreversible and used for permanent closure/sealing of surgical wounds.4 Irreversible wet bioadhesives elicit polymerization and cross-linking networks between adjoining tissues or covalent bonding such as that between an aldehyde-based adhesive and tissue collagen/glycoproteins based on aldehyde–amine (─CHO/─NH2) chemistry.4 The strong adhesion of marine mussels on wet inorganic rock surfaces mediated by DOPA (3,4-dihydroxyphenylalanine) or catechol chemistries is also being used to develop another class of bioinspired wet irreversible adhesives.5 On the contrary, reversible underwater adhesives have been designed based on supramolecular host–guest noncovalent interactions between the two surfaces.6 Reversible wet bioadhesives function via weak noncovalent interactions such as intermolecular hydrogen bonding and van der Waals interactions leading to gelation and temporary bonding such as tannic acid/polyphenol-based adhesives for tissue patches and flexible electronics.7 The different forces governing mucosal tissue adhesion such as noncovalent interactions (hydrogen bonding, electrostatic, hydrophobic, and van der Waals), covalent interactions (disulfide links, imine formation), and polymer chain entanglements with mucosal surfaces have been extensively reviewed.8
Most of the adhesives and sealants used in oral and maxillofacial procedures are based on cyanoacrylates or photocurable resin formulations for permanent bonding of tissues.9 Here, we have developed oral tissue adhesive nanofiber membranes with reversible/temporary underwater adhesion properties for biosensing, tissue regeneration, and drug/therapeutic delivery in the oral cavity. The application of nanofiber membranes as bioadhesives has been biologically inspired by nanoscale “setae” structures on gecko-lizard feet that confer adhesion to dry surfaces.10 Nanofiber topography and fiber alignment were demonstrated to be critical factors governing the adhesion strength of electrospun polycaprolactone membranes.11 Notably, detachable mucoadhesive bilayer nanofiber patches have been previously evaluated for residence time in the oral cavity and drug delivery to the oral mucosa.12 Thus, nanofiber membranes with moderate underwater adhesion properties that would allow effective attachment and easy detachment from the wet tissues can be useful for temporary adhesion in the oral cavity.
For this study, we chose chitosan as the base material for our membranes, as it is a naturally derived mucoadhesive pH-responsive polysaccharide that can be exploited toward pH-controlled antibiotic/antimicrobial delivery.13 However, the mucoadhesion properties of chitosan were greatly improved in combination with pectin when casted as free films.14 On similar lines, a combination of nanotopography and optimal coating of oxidized dextran was applied to enhance the mucoadhesion of gecko-inspired synthetic elastomer nanopatterns.15 Taking cues from the aforementioned studies, we aimed to obtain electrospun chitosan nanofiber membranes modified by surface coating with oxidized sugars/polysaccharides for oral tissue adhesion. Pectin has been vastly explored in oral drug delivery,16 and thus, we selected pectin for providing mucoadhesive properties and oral therapeutic delivery capability to our membranes. The selective modification of chitosan membranes with a mucoadhesive polysaccharide coating on only one side of the membrane and/or specific area will enable the manufacturing and application of membranes with dual tissue adhesive properties, i.e., adhesion to soft tissues and hard tissues. This dual adhesive function of the membranes coupled with the delivery of oral therapeutics can be exploited for oral tissue regeneration and protection from oral infections. Moreover, considering that the oral pH can vary from 4.5 to 6.7 for diseased/caries versus healthy individuals,17-19 the tissue adhesive nanofiber membranes incorporated with antimicrobial peptides (AMPs) or chlorhexidine are investigated here for pH-responsive delivery to address prevalent oral mucosal tissue and dental infections, such as periodontitis, peri-implantitis, and caries.20
2. MATERIALS AND METHODS
2.1. Materials.
Chitosan (low mol. wt. 50–190 kDa, 75–85% deacetylation degree), low methyl pectin (from citrus peel, 74% galacturonic acid), trifluoroacetic acid (TFA, 99%), dichloromethane (DCM), glutaraldehyde (GA at 25 wt % in water), sodium periodate (NaIO4), and chlorhexidine (CHX) were purchased from Sigma-Aldrich (St. Louis, MO, USA). The antimicrobial peptides IDR-1018 (VRLIVAVRIWRR-NH2, mol. wt. 1536 g/mol), D-GL13K (gkiiklkaslkll-NH2, mol. wt. 1424 g/mol), and its fluorescent carboxytetramethylrhodamine (TAMRA) analogue TAMRA-GL13K (GKIIKLKASLKLL-Lys(5-TAMRA)-NH2, mol. wt. 1964.5 g/mol) were produced by solid-phase peptide synthesis in >98% purity by AAPPTec (Lousiville, KY, USA).
2.2. Fabrication of Uncoated and Oxidized Pectin Coated Chitosan Nanofiber Membranes.
Chitosan (CS) nanofiber membranes were fabricated by the electrospinning technique following a previously reported protocol with minor modifications.21 CS (5 wt %) was dissolved in a solvent mixture of TFA:DCM = 70:30 (v/v) and fed into a syringe with a 25G needle. Electrospinning was performed horizontally onto a metallic collector using the following parameters: applied DC voltage = 15 kV, flow rate = 0.5–0.6 mL/h, distance between the syringe needle tip and collector = 10–15 cm. The electrospun membrane was peeled off from the collector and cross-linked with GA vapors overnight for ~18 h. The GA-cross-linked chitosan was neutralized in a saturated carbonate solution (7 g of Na2CO3 + 6 g K2CO3 dissolved in 25 mL of DI water) for ~3 h, followed by washing the membrane three times in DI water. The resultant chitosan membrane was vacuum-dried overnight.
Pectin (PE) was oxidized to pectin dialdehyde or oxidized pectin by the sodium periodate oxidation method following a previous protocol.22 Low methyl pectin (from citrus peel) dissolved at 2 wt % in DI water was oxidized with NaIO4 at a 1:10 molar ratio of NaIO4:galacturonic acid by stirring at room temperature for 24 h in the dark. The reaction mixture was dialyzed against water and freezedried to obtain oxidized pectin (Oxi-PE). The chitosan nanofiber membranes were spin coated with oxidized pectin at 0.05, 0.5, and 2.0 wt % using a rotation speed of 4000 rpm for 1 min. The concentrations of the spin-coating solutions were adapted from a previous report.15
2.3. Morphological and Spectroscopic Characterization of Uncoated and Oxidized Pectin Coated Chitosan Nanofiber Membranes.
The surface morphologies and membrane thicknesses of the uncoated and oxidized pectin coated chitosan nanofiber membranes were visualized under a high-resolution scanning electron microscope (SEM, Hitachi SU8230). The membranes were mounted on aluminum stubs using a double-sided carbon tape, followed by sputter coating with 5 nm of iridium (Leica). The images were acquired using an accelerating voltage of 3 kV. The mean, median, and diameter distribution for the different nanofiber compositions were analyzed using the DiameterJ plugin in ImageJ.23 The two-step image analysis consisted of image segmentation into a binary image (black and white) followed by analysis of the segmented images.
The surface chemistry and composition of the membranes were characterized by Fourier-transform infrared (FT-IR) spectroscopy, UV-visible spectrophotometry, X-ray photoelectron spectroscopy (XPS), and water contact angle measurements. The FT-IR spectrometer (Nicolet iS50, Thermo Fisher Scientific) recorded the spectra of the membranes in the attenuated reflectance (ATR) mode from 400 to 4000 cm−1 using an incremental step size of 2 cm−1, and spectra were signal-averaged from 32 scans. The surface aldehydes from the oxidized pectin coatings on the chitosan membranes were characterized by reaction with 2,4-dinitrophenylhydrazine (2,4-DNP: 0.1% w/v) followed by UV–vis spectrophotometry of the reaction adduct.24 Briefly, oxidized pectin (0.05, 0.5, and 2.0 wt %) was reacted with 2,4-DNP in 1:5 (v/v) ratios under acidic pH for 1 h in the dark followed by recording the absorbance of the reaction product—hydrazone Schiff’s base at 412 nm. The aldehyde content (mmol) was calculated using the Beer–Lambert law which postulates a linear relationship between the absorbance and concentration of the chromophore.
For XPS, a PHI 5000 VersaProbe III (ULVAC Inc., Kanagawa, Japan) X-ray photoelectron spectrometer (XPS) was used to determine surface elemental composition and chemistry with a monochromatic Al Kα X-ray source (45°, 1486.6 eV, 50 W, sampling area; 200 μm diameter). Survey spectra were collected using a pass energy of 280 eV with a step size of 1.0 eV. High-resolution C 1s and N 1s spectra were obtained with a pass energy of 190 eV and a step size of 0.1 eV. The surface elemental compositions were determined from the survey scan spectra using CasaXPS software (Casa Software Ltd.). The high-resolution C 1s and N 1s spectra were deconvoluted and peak-fitted using Fityk software.25
The surface hydrophilicity/hydrophobicity of the membranes was determined by measuring dynamic water contact angles using the sessile drop method. 2.0 μL of deionized water was dispensed onto each membrane surface, and water contact angles were recorded for 30 s at 1 Hz frequency using a contact angle meter (DM-CE1, Kyowa Interface Science, Japan). The FAMAS software (Kyowa Interface Science, Japan) was used to capture images of the water droplets and measure the contact angles from the droplet profiles.
2.4. Mucosal and Hard Tissue Adhesion Tests.
The adhesion of uncoated and oxidized pectin (0.05 and 0.5 wt %) spin coated chitosan nanofiber membranes to porcine esophagus as the oral mucosal tissue mimic and hydroxyapatite (9.5 mm discs, 3D Biotek, LLC, Bridgewater, NJ, USA) as the hard tissue/enamel mimic was studied using a texture analyzer (TA.XTPlus, Stable Micro Systems). For the test, the nanofiber membranes glued to a cylindrical probe with double-sided tape were lowered onto the porcine esophagus immersed in DI water until a compressive load of 1 N, held for 60 s, followed by pull-out at a slow rate of 0.01 mm/s. The detailed experimental parameters listed below were adapted from a previous report.26
Temperature: RT
Medium: DI water
Probe contact area: 1 cm2
Pretest velocity: 5 mm/s
Test velocity: 0.1 mm/s
Applied preload: 1 N
Hold time: 60 s
Pull-out velocity: 0.01 mm/s
Trigger force: 0.000 N (smallest detectable force below which the instrument considers the test completed)
Vertical displacement: 50 mm (initial and final position of the probe)
Mucosal soft tissue mimic: porcine esophagus (stored at −80 °C until testing)
Enamel/hard bone tissue mimic: hydroxyapatite discs (9.5 mm)
For the enamel/hard tissue adhesion, hydroxyapatite discs were glued to the upper cylindrical probe lowered onto the nanofiber membrane immersed in DI water at room temperature. From the load–displacement curves recorded during the adhesion tests, the detachment force (Fdet, N) and work of adhesion (Wadh, mJ) were computed.
2.5. Loading and Release of Antimicrobials from Chitosan Nanofiber Membranes.
The d-enantiomer of GL13K derived from the human salivary parotid secretory protein27 and the l-enantiomer of innate defense regulator (IDR)-1018 derived from bovine neutrophil host defense peptide bactenecin28 were the chosen antimicrobial peptides for the current study. The antimicrobial drug chlorhexidine (CHX) and antimicrobial peptides (D-GL13K and 1018) or the analogue TAMRA-GL13K fluorescent tagged peptide were incorporated into chitosan membranes either by (i) co-electrospinning drug:chitosan or peptide:chitosan at 1:5 wt ratios or (ii) surface absorption of the antimicrobials into 8 mm diameter discs punched out from the membrane. The co-electrospun membranes were GA-cross-linked and neutralized similar to pure chitosan membrane. For surface absorption, 100 μL of CHX/D-GL13K/ 1018 dissolved in EtOH/H2O/EtOH:H2O = 1:1(v/v) at ~1 mg/mL was pipetted onto 8 mm chitosan membrane discs and air/vacuum-dried.
Specifically, the TAMRA-GL13K fluorescent-tagged peptide was used for the loading and release studies. Based on the high solubility of the peptides in acidic pH, the loading of TAMRA-GL13K was calculated by repeated extractions of the chitosan absorbed peptide with 30% aq. acetic acid (v/v). The cumulative extracted TAMRA-GL13K peptide is a measure of the antimicrobial peptide loading. The release of TAMRA-GL13K from the chitosan membranes into 0.1 M NaOAc buffers of different pH (4.5, 5.5, and 6.5) was periodically assayed by collecting release solutions for up to 24 h. The fluorescence intensities for all of the release aliquots were recorded using excitation and emission filters of 530 (±25) nm and 590 (±25) nm, respectively, corresponding to the TAMRA fluorophore with a multimode microplate reader (BioTek).
2.6. Antimicrobial Assessment of Chlorhexidine, D-GL13K, and 1018 Loaded Chitosan Nanofiber Membranes against Oral Streptococci.
2.6.1. Zone of Inhibition (ZOI) Assay.
The cryopreserved bacterial stocks were revived by inoculation of Streptococcus gordonii M5 on brain heart infusion (BHI) agar and Streptococcus mutans (ATCC 700610) on Mitis Salivarius Sucrose Bacitracin (MSSB) agar plates, followed by incubation at 37 °C, 5% CO2 for 24–48 h. For the ZOI assay, a few colonies (~5–10) of bacteria were inoculated in 2 mL of phosphate buffer saline (1× PBS) and vortexed for 1 min. A 100 μL portion of the bacterial solution was pipetted on the brain heart infusion (BHI) agar plates and spread with a sterile swab. The UV-sterilized blank chitosan (CS) and peptide/ chlorhexidine loaded chitosan discs (8 mm diameter) were placed on the inoculated plates with sterile forceps, followed by incubation of the BHI agar plates at 37 °C, 5% CO2 for ~24 h. The images of the agar plates were captured with an optical camera, and the diameters of ZOI were measured using ImageJ software. The samples are designated as chitosan (CS), chlorhexidine/peptide encapsulated and co-electrospun chitosan (D-GL13K CS_ES, 1018 CS_ES, and CHX CS_ES), and chlorhexidine/peptide absorbed chitosan (D-GL13K CS_Abs, 1018 CS_Abs, and CHX CS_Abs).
2.6.2. Indirect Antimicrobial Assessment Using Release Aliquots.
For the liquid culture experiments, S. gordonii M5 and S. mutans (ATCC 700610) were cultured separately in Todd Hewitt base (THB) broth at 37 °C, 5% CO2 until they reached logarithmic growth phase (OD600 nm ~ 0.5). The bacteria were diluted to OD600 nm ~ 0.1 and used for further experiments. In order to ascertain pH-controlled antimicrobial peptide (AMP) release and consequent antimicrobial activity, D-GL13K release aliquots under different pH (4.5, 5.5, and 6.5) conditions were cocultured with S. gordonii M5 or S. mutans (ATCC 700610) in 1:1 (v/v) ratio. Similarly, release buffer (pH 4.5, 5.5, and 6.5) aliquots from blank chitosan membranes were cocultured with the oral streptococci as controls along with an additional control of phosphate buffer saline (PBS) so as to account for the dilution of the culture medium with the buffer solutions in 1:1 (v/v) ratio. Growth curves of the bacteria were recorded at intervals of 1 h in a continuous kinetic measurement with the help of a multimode plate reader (Biotek).
2.6.3. Direct Antimicrobial Assessment: ATP Assay, Live/Dead Imaging, and Biomass/Bioburden Determination.
The antimicrobial potency of chitosan-based membranes was assessed by the direct culture of bacteria on membranes loaded with D-GL13K as a representative AMP and compared with chlorhexidine (CHX) at neutral pH. For these experiments, select sample compositions of chitosan (CS), 0.05 wt % oxidized pectin coated chitosan (0.05Oxi-PE_CS), D-GL13K/CHX absorbed chitosan (D-GL13K_CS or CHX_CS), and D-GL13K/CHX absorbed chitosan spin coated with 0.05 wt % oxidized pectin (D-GL13K-0.05Oxi-PE_CS or CHX-0.05Oxi-PE_CS) were evaluated for bacterial adhesion and biofilm formation. The membranes were punched into 8 mm discs, glued onto glass coverslips, and UV-sterilized.
For the ATP assay, the bacteria were diluted to an initial OD600 nm ~ 0.05 and seeded on UV-sterilized chitosan-based membranes. The samples in a 48-well plate were incubated on a rocker at 37 °C, 5% CO2 for ~24 h. Subsequently, the samples were transferred to separate wells and rinsed with PBS to remove loosely attached bacteria. For the quantification of viable bacteria adhered on the membranes via ATP bioluminescence, the BacTiter-Glo Microbial Cell Viability assay kit (Promega) was used. Equal volumes of PBS and BacTiter-Glo reagent (mixture of BacTiter-Glo substrate + BacTiter-Glo buffer) were added to the samples and incubated on a rotary shaker at 37 °C for 5 min. Aliquots of 200 μL were transferred to a 96-well plate and recorded for bioluminescence with a multimode plate reader (Biotek).
For the qualitative assessment of vital bacteria, live dead imaging was performed using the Live/Dead Bac-light Bacterial Viability kit (L7012, ThermoFisher Scientific). The samples were stained with a 1:1 mixture of SYTO 9 and propidium iodide (PI) appropriately diluted in PBS for 20 min in the dark. Excess stain was washed off with PBS, and samples were visualized under a fluorescence microscope at 100× magnification using an oil immersion lens (Leica DM6).
For the estimation of the total biomass/bioburden, S. gordonii M5 and S. mutans (ATCC 700610) were cultured separately on the differently modified chitosan membranes for 24 h. The samples were rinsed in PBS to remove loosely adhered bacteria and stained with 0.1% crystal violet for 15 min on a rotary shaker. The excess crystal violet was aspirated, and the unbound crystal violet was washed off with PBS. The bound crystal violet was solubilized in 30% (v/v) aq. acetic acid for 10 min, and aliquots of the dye were recorded for absorbance at 550 nm. The crystal violet bound to the modified chitosan membranes devoid of bacteria were subtracted from those cultured with bacteria to arrive at the relative amount of biofilm growth.
2.7. Cytocompatibility of Surface Modified Chitosan Nanofiber Membranes.
2.7.1. Cell Proliferation by CCK-8 Assay.
The cytocompatibility of the dual soft and hard tissue adhesive chitosan membranes was evaluated using soft epithelial tissue derived human oral keratinocytes and hard calvarial tissue derived murine pre-osteoblasts. Immortalized human oral keratinocytes (HOK, OKF-6/ TERT2)29 were maintained in the recommended keratinocyte serumfree medium (SFM) supplemented with bovine pituitary extract (BPE) and human recombinant epidermal growth factor (rEGF) at 37 °C, 5% CO2, and 95% relative humidity. Mouse calvaria derived pre-osteoblast cells MC3T3-E1 (ATCC CRL-2593) were maintained in alpha minimal essential medium (α-MEM) supplemented with 10% (v/v) fetal bovine serum (FBS). The UV-sterilized samples were seeded at a density of 10,000 cells/sample and incubated under the aforementioned conditions. After overnight cell attachment, the samples were transferred to new wells and replenished with fresh culture medium. The proliferation of cells on the modified chitosan membranes was evaluated using the Cell Counting Kit 8 (CCK-8, Dojindo Molecular Technologies, Osaka, Japan). At the predetermined time points (1 and 3 days), CCK-8 reagent was added to each well at 10% (v/v) concentration and 90% media. After 2.5 h of incubation, 200 μL aliquots of the supernatant were transferred to a 96-well plate and the absorbance read at 450 nm with a multimode plate reader (Biotek).
2.7.2. Cell Adhesion by Fluorescence Microscopy.
After 3 days of culture on the modified chitosan membranes, the cells were fixed in 4% para-formaldehyde (PFA) overnight at 4 °C, followed by permeabilization with 0.1% Triton X100 for 10 min and blocking with 2 wt % bovine serum albumin (BSA) for 30 min. In the case of HOK, the cells were immunostained for cytokeratin 14 by incubation in primary mouse KRT 14 monoclonal antibody, clone LL002 (Abnova, MAB11337), at 1:100 dilution in 2% BSA in a solution of 0.1% Tween in PBS (PBST) at 4 °C overnight. Subsequently, the samples were washed thrice in PBST and incubated in Alexa Fluor 594 conjugate of goat antimouse IgG (H+L) highly cross-linked secondary antibody (Thermo Fisher Scientific, A11032) at 1:200 dilution in 2% BSA in PBST at room temperature for 1 h. Next, actin was counterstained with Alexa Fluor 488 Phalloidin (Thermo Fisher Scientific, A12379) in 2% BSA in PBST for 30 min and nuclei with DAPI (4′,6-diamidino-2-phenylindole dihydrochloride, D9542, Sigma-Aldrich) for 10 min. A similar protocol was implemented for staining the actin and nuclei of MC3T3-E1 pre-osteoblast cells adhered on the modified chitosan membranes. After washing off excess stain, images were acquired under an upright Leica DM6 fluorescence microscope.
2.8. Statistical Analysis.
The data analysis for statistical significance was performed using the IBM SPSS Statistics 20 software. When data is represented in boxplots, the middle line depicts the median, boxes demark the 25th to 75th percentile, and whiskers show minimum to maximum values of n = 6 replicates. Data shown in the line graphs and tables are means ± standard deviation of n = 3–6 replicates. After validating normality of sample distribution and homogeneity of variance, one-way analysis of variance (ANOVA) with Tukey test was used to assess the statistical significance between groups at p-value <0.05.
3. RESULTS
3.1. Fabrication of Chitosan-Based Nanofiber Membranes.
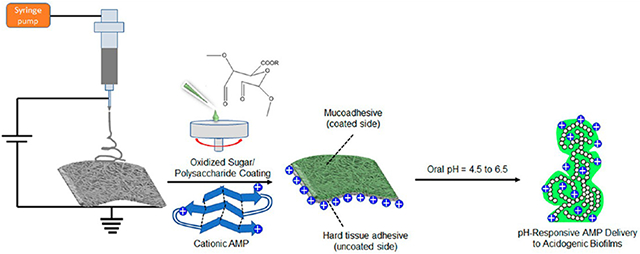
The high surface area to volume ratio and nanotopography of electrospun nanofiber membranes make them apt candidates for membrane-based tissue adhesives. Figure 1 is a schematic illustration of the workflow in the present study. Chitosan was chosen as the base polymer for electrospinning nanofiber membranes, and it was surface coated with oxidized pectin to enhance the mucoadhesion properties. Figure 2A shows the near bead-free nanofiber morphology of glutaraldehyde cross-linked and carbonateneutralized chitosan membrane. Oxidized pectin spin coated chitosan membranes showed diminishing nanofiber topography with increasing concentration of the coating solution (Figure 2B-D). At 0.05 and 0.5 wt % oxidized pectin coating, the surface nanofiber morphology is still apparent, while the chitosan fibrillary structure is completely lost for the 2 wt % oxidized pectin coating. Incidentally, a blend of chitosan and oxidized pectin in different wt. ratios (1:1 and 3:1) did not result in neat fibers but yielded particle morphology (data not shown). Therefore, we chose spin coating of chitosan with oxidized pectin as a viable option to enhance surface mucoadhesivity. ImageJ analysis of the nanofiber diameters using the DiameterJ plugin divulged mean fiber diameters of 150 ± 79, 243 ± 119, and 584 ± 183 nm for uncoated and 0.05 and 0.5 wt % oxidized pectin coated chitosan membranes, respectively (Figure S1A-D). All of the nanofiber diameter distribution plots (Figure S1A-C) were fit with single Gaussian curves with acceptable R2 values of 0.85–0.9. The 2.0 wt % oxidized pectin coated membranes could not be analyzed due to the film-like surface morphology. A statistically significant increase in nanofiber diameter was noted for 0.5 wt % oxidized pectin coated chitosan compared to uncoated and 0.05 wt % oxidized pectin coated chitosan (Figure S1D). This suggests that nanofiber topography was maintained up to an optimal coating of 0.05 wt % oxidized pectin. However, the thicknesses of both the uncoated and oxidized pectin coated chitosan membranes were ~20 μm, as visualized from their cross sections under SEM (Figure S1E,F).
Figure 1.

Schematic illustration of the workflow in the current study: (i) fabrication of oral tissue adhesive nanofiber membranes loaded with cationic AMPs, (ii) pH-responsive delivery of AMPs to acidogenic oral biofilms, (iii) dual hard and soft tissue compatibility of the membranes for oral tissue regeneration. The tooth image in this illustration was adapted and redrawn with permission from ref 30.
Figure 2.

Surface topography and morphologies of (A) uncoated, (B) 0.05 wt %, (C) 0.5 wt %, and (D) 2.0 wt % oxidized pectin coated chitosan nanofiber membranes visualized by SEM. (E) ATR-FT-IR characterization of oxidized pectin (0, 0.05, 0.5, and 2.0 wt % Oxi-PE) spin coated chitosan (CS) nanofiber membranes. (F) Titration of surface aldehyde content (mMoles/membrane) on uncoated and oxidized pectin coated membranes by acid-catalyzed reaction with 2,4-dinitrophenylhydrazine (2,4-DNP) for 1 h. Representative macroscopic images in part F show yellow to orange reaction product for each sample composition. Each data point represents one replicate, and boxplots present statistical values for each group. * p < 0.05 indicates statistical significance between compared groups.
The reaction scheme for the syntheses of oxidized pectin/ pectin dialdehyde by sodium periodate oxidation is depicted in Figure S2A. The formation of pectin dialdehyde was characterized by FT-IR spectroscopy. Figure S2B shows the comparative ATR-FT-IR spectra of pectin and oxidized pectin, distinguished by the presence of aldehyde C─H stretch in ─ CHO at 2850 cm−1 as well as the relative peak intensities of the carbonyl C═O peaks. Figure S2C is a magnified carbonyl ATR-FT-IR spectrum of pectin exhibiting two peaks, an ester─COOR carbonyl at 1740 cm−1, and a carboxylic acid ─ COOH carbonyl at 1614 cm−1.22 In Figure S2D, the newly formed aldehyde ─CHO carbonyl at 1734 cm−1 is masked by the ─COOR ester carbonyl at 1740 cm−1, leading to an increase in the relative intensity of 1740 to 1614 cm−1 IR absorptions in oxidized pectin. Overall, these results demonstrate the successful oxidation of pectin using periodate. In Figure 2E, the ATR-FT-IR spectra of chitosan nanofiber membranes coated with oxidized pectin (0.05, 0.5, and 2.0 wt %) exhibit characteristic carbonyl (─COOR + ─CHO) absorptions at ~1740 cm−1, which is absent in the pure/ uncoated chitosan.
The titration of aldehyde content on the different membrane compositions was determined by the acid-catalyzed reaction of surface aldehydes from oxidized pectin with 2,4-DNP. The reaction scheme for the acid-catalyzed condensation of oxidized pectin with 2,4-DNP to form a hydrazone/Schiff’s base/imine adduct is shown in Figure S3A. UV-visible characterization of the reaction products of oxidized pectin and 2,4-DNP indicates a shift in the absorption maxima (λmax) to longer wavelengths and formation of a shoulder peak with an absorption maximum at ~412 nm (Figure S3B). The titration of aldehyde content in different concentrations of oxidized pectin suggests a concentration dependence of aldehyde content (Figure S3C). The precipitation of the orange reaction product explains the similarity in absorbance values and aldehyde content determined for the 0.5 and 2.0 wt % oxidized pectin (Figure S3C). Figure 2F shows the significant increase in aldehyde content (mmol/membrane) from uncoated chitosan to 0.05, 0.5, and 2.0 wt % oxidized pectin coated membranes. Here too, precipitation of the orange reaction product led to similar absorbance and aldehyde contents determined for 0.5 and 2.0 wt % oxidized pectin coating. The FT-IR and 2,4-DNP titration of aldehydes suggest that we successfully obtained coatings of oxidized pectin on chitosan nanofiber membranes. Further, chitosan and pectin are cationic and anionic polymers, respectively, and their mixtures have been reported to form polyelectrolyte complexes.31 The electrostatic interaction between the two polymers can confer additional stability to the coatings.
3.2. Surface Chemistry, Polarity, and Charge Analysis of Chitosan-Based Nanofiber Membranes.
The presence of hydrophilic macromolecules presenting several hydrogen bond forming groups such as oligo-/polysaccharides on the surface is among the prerequisites for mucoadhesive materials.32 Further, the extent of mechanical interlocking between the free chains of hydrophilic oligo-/polysaccharides with the glycoprotein mucin subunits on mucosal tissues can dictate the strength of mucoadhesion. In this light, the surface chemistry of the chitosan-based nanofiber membranes was characterized in terms of surface polarity and charge by XPS analysis. Figure S4 shows the XPS survey spectra of chitosan coated with 0, 0.05, 0.5, and 2.0 wt % oxidized pectin. The three expected signals of C 1s at ~285 eV, N 1s at ~400 eV, and O 1s at ~531 eV were detected on all of the membrane surfaces. Table 1 is a summary of the surface elemental composition of the uncoated and oxidized pectin coated chitosan nanofiber membranes. An increase in the atomic ratios of O:C and O:N was commensurate with the amount of oxidized pectin (0.05, 0.5, and 2.0 wt %) coated on the chitosan nanofiber membranes (Table 1), which suggests an increase in the density of polar ─OH groups.
Table 1.
Surface Elemental Composition of Uncoated/Oxidized Pectin Coated Chitosan Nanofiber Membranes Determined from XPS Survey Scan Spectra
| sample | C 1s (at. %) | N 1s (at. %) | O 1s (at. %) | O:C | O:N |
|---|---|---|---|---|---|
| CS | 67.68 ± 1.36 | 8.00 ± 1.52 | 24.25 ± 1.97 | 0.35 | 3.03 |
| 0.05 wt % Oxi-PE_CS | 68.82 ± 1.39 | 6.28 ± 2.31 | 24.76 ± 1.65 | 0.36 | 3.94 |
| 0.5 wt % Oxi-PE_CS | 61.52 ± 1.62 | 7.62 ± 1.67 | 30.76 ± 2.59 | 0.5 | 4.03 |
| 2.0 wt % Oxi-PE_CS | 57.46 ± 8.04 | 4.25 ± 2.43 | 38.03 ± 6.69 | 0.66 | 8.93 |
Figure 3 shows deconvoluted high-resolution C 1s peaks. For all of the samples, the C 1s spectra were fit to three peaks corresponding to aliphatic sp3 bonded carbons (C─C) at 284.8 eV, sp3 carbon bonded to electronegative heteroatoms (C─O, C─N) at ~286.5 eV, and sp2 carbon bonded to heteroatoms (C═O, C═N) at ~289.1 eV. Further analysis of the C 1s peak-fit XPS data is shown in Table 2. Surface polarity determined as the Cpolar/Cnonpolar ratio increased with increasing oxidized pectin concentration (0.05, 0.5, and 2.0 wt %) coating due to the greater abundance of carbon—heteroatom bonded polar functional groups (C─O, C─N, C═O, C═N) in comparison to C─C nonpolar bonds. Lastly, the N 1s spectra were deconvoluted and peak-fitted into two peaks corresponding to deprotonated N at a lower binding energy of ~400 eV and protonated N+ at a higher binding energy of ~402 eV (Figure S5). Both the deprotonated and protonated N peaks arise cumulatively from amine (─NH2), amide (─NH─C═O), and imine (─N═C) in their deprotonated and protonated states, respectively. The surface charge determined as the ratio of protonated to deprotonated (N+/N) was highest for chitosan and decreased marginally with oxidized pectin coating (Table S1). Thus, on the one hand, the analysis of surface polarity from the C 1s spectral deconvolution suggested that the notable presence of hydrophilic chains of oxidized pectin on chitosan membranes can contribute to enhanced mucoadhesion. On the other hand, the greater surface positive charge from protonated amine (─NH3+) in chitosan can have greater electrostatic affinity for inorganic phosphates (─PO43−) in hard tissue/enamel, which might confer strong hard tissue adhesion to uncoated chitosan membranes.
Figure 3.

XPS-deconvoluted and peak-fitted high-resolution C 1s spectra of (A) uncoated, (B) 0.05 wt %, (C) 0.5 wt %, and (D) 2.0 wt % oxidized pectin coated chitosan nanofiber membranes. (E) Dynamic water contact angles of uncoated and oxidized pectin coated chitosan nanofiber membranes recorded by the sessile drop method. Data shown are the average ± standard deviation of n = 3–6 replicates. (F) Representative sessile water drop images for the final contact angles shown in part E.
Table 2.
Binding Energies (eV) and Chemical States of Carbon Determined from the Deconvoluted and Peak Fit C 1s Spectra for Uncoated (CS) and Oxidized Pectin Coated Chitosan (Oxi-PE_CS) Nanofiber Membranes
| sample | C─C (eV) |
C─O, C─N (eV) |
C═O, C═N (eV) |
Cpolar/Cnonpolar |
|---|---|---|---|---|
| CS | 284.8 | 286.5 | 289.1 | 1.537 ± 0.338 |
| 0.05 wt % Oxi-PE_CS | 284.8 | 286.6 | 289.0 | 1.951 ± 0.278 |
| 0.5 wt % Oxi-PE_CS | 284.8 | 286.7 | 289.2 | 2.374 ± 0.434 |
| 2.0 wt % Oxi-PE_CS | 284.8 | 286.8 | 289.2 | 4.648 ± 0.107 |
The abundance of surface polar groups resulting from oxidized pectin coatings was further confirmed by contact angle measurements. The chitosan membrane was hydrophobic with a contact angle of ~115° due to glutaraldehyde cross-linking which consumed polar hydrophilic chemical groups (─NH2 and ─OH).33 Spin coating of chitosan membranes resulted in more hydrophilic membranes with water contact angles dropping to ~85, 83, and 63° for 0.05, 0.5, and 2.0 wt % oxidized pectin, respectively (Figure 3E and F). The increased hydrophilicity can aid in intermolecular hydrogen bonding and interpenetration of polymer chains necessary for mucoadhesion.
3.3. Ex Vivo Mucoadhesion and Hard Tissue Adhesion of Chitosan-Based Nanofiber Membranes.
Porcine esophagus was used as the model/mimic for oral/buccal mucosal tissue34 to study the adhesion of chitosan-based nanofiber membranes ex vivo. Figure S6 is a representative force vs time graph of the mucoadhesion test of chitosan nanofiber membrane to porcine esophagus. For the adhesion test, the nanofiber membrane glued to the probe was pressed onto the mucosa until a compressive load of 1 N was reached, followed by a contact time of 60 s between the nanofiber membrane and mucosa. After this, the probe was slowly retracted away from the mucosa and the force of detachment was recorded. The representative load–displacement curves shown in Figure 4A and B correspond to the tensile/pull-out portion of the adhesion test of the membranes to tissues after the application of compressive load. The adhesion test data were analyzed in terms of the maximum force of separation or detachment force, Fdet [N], and the work of adhesion, Wadh [mJ], determined from the area under the pull-out load–displacement curve (Figure 4C-F).
Figure 4.

Representative force–displacement curves for (A) mucoadhesion and (B) adhesion to hydroxyapatite recorded for uncoated, 0.05 wt %, and 0.5 wt % oxidized pectin coated chitosan nanofiber membranes. Adhesion parameters: detachment force and work of adhesion determined from the force–displacement curves of the (C, E) mucoadhesion and (D, F) adhesion to hydroxyapatite tests. Each data point represents one replicate, and boxplots present statistical values for each group. * p < 0.05 and ** p < 0.005 indicate the statistical significance between compared groups. CS, chitosan; Oxi-PE_CS, oxidized pectin coated chitosan.
The detachment force from the soft mucosal tissue increased ~2-fold for both concentrations of oxidized pectin coating on chitosan nanofibers (Figure 4C) compared to uncoated ones, which we attributed to the greater surface polarity, hydrophilicity, and intermolecular hydrogen bonding capability of oxidized pectin coating on chitosan (determined from XPS). However, the work of adhesion was maximized for the 0.05 wt % oxidized pectin coated chitosan (Figure 4D). This suggests that a combination of nanofiber topography and optimal surface coating of oxidized polysaccharide/sugar (Figure 2B-D) can synergistically enhance mucosal/soft tissue adhesion. In this respect, our findings are consistent with previous reports of a similar soft tissue adhesive strategy.15
However, the maximum detachment force and work of adhesion to hydroxyapatite (enamel/hard tissue mimic) was obtained with the uncoated chitosan membranes (Figure 4E and F). The cationic chitosan nanofibers can have preferential electrostatic interaction with anionic phosphates on the hydroxyapatite surface, and therefore, adhesion between uncoated chitosan to the hard tissue was favored. Taken together, these results indicate that a dual soft–hard tissue adhesive interface can be designed by surface coating of oxidized pectin on one of the two surfaces of chitosan nanofiber membranes.
3.4. pH-Responsive Delivery of Antimicrobial Peptides from Oral Tissue Adhesive Nanofiber Membranes.
To demonstrate the potential application of the oral tissue dual adhesive nanofiber membranes for the delivery of antimicrobials in a pH-responsive manner, TAMRA-tagged antimicrobial GL13K peptides were incorporated into the chitosan nanofiber membrane. In our preliminary experiments, we observed that the FAM (6-carboxyfluorescein)-tagged GL13K peptides exhibited quenching of the green fluorescence in acidic pH (6.5 < 5.5 < 4.5), while the red fluorescence from TAMRA-GL13K was notably stable in the pH range being studied (data not shown). Therefore, TAMRA-GL13K was used for the antimicrobial peptide loading and release experiments. Figure 5A is a fluorescence micrograph of TAMRA-GL13K absorbed into a chitosan nanofiber membrane, as indicated by the red fluorescent emission. Figure 5B shows values of absolute release of TAMRA-GL13K from chitosan membranes. Peptide burst release was maximal during the first 1 h, and ~25 μg (Figure 5C). The cumulative release profiles from chitosan membranes for TAMRA-GL13K (Figure 5C) showed a pH-dependent release of antimicrobial peptide from the nanofiber matrixes. After 24 h, about ~60, ~40, and ~20% of the loaded TAMRA-GL13K was released from the chitosan nanofiber membranes at pH 4.5, 5.5, and 6.5, respectively (Figure 5C). Macroscopic images of the membranes (Figure 5D) after 24 h suggest greater residual TAMRA-GL13K red fluorescent peptide in the order of buffer pH (4.5 < 5.5 < 6.5). The pH dependence of antimicrobial peptide release can be attributed to two factors: (i) increasing peptide solubility with decreasing pH and/or (ii) greater chitosan degradation with decreasing pH, which would enable the release of the absorbed peptides. The degradation of chitosan membranes was determined as weight ratio (Wt/Wo), where Wt and Wo are the weights of the membranes at time t and 0, respectively. Chitosan membranes incubated in 0.1 M NaOAc buffers of pH 4.5, 5.5, and 6.5 recorded 28, 10, and 8% weight loss, respectively, within 3 days of incubation (Figure S7A). Furthermore, a similar pH-dependent peptide release profile was logged for TAMRA-GL13K encapsulated in the chitosan fibers of membranes that were obtained by peptide–chitosan co-electrospinning (Figure S7B). We obtained the burst release profile for defining effective dosage of the AMPs to elicit bactericidal action in vitro (Figure 5C). The antimicrobial potency achieved by the AMPs release against streptococci is presented in the next section.
Figure 5.

(A) Fluorescence microscopy image of GL13K-TAMRA loaded chitosan nanofiber membrane exhibiting a red fluorescence signal due to the presence of the TAMRA fluorophore, (B) Absolute and (C) cumulative release of GL13K-TAMRA peptide in a pH-dependent manner from chitosan nanofiber membranes incubated in 0.1 M NaOAc buffers of different pHs (4.5, 5.5, and 6.5). The data shown above are means ± standard deviation of n = 3 replicates. (D) Macroscopic images of TAMRA-GL13K loaded chitosan membranes showing residual peptide after 24 h release following the order pH = 6.5 > 5.5 > 4.5.
3.5. Antimicrobial Potency of Chlorhexidine/AMP Loaded Chitosan Nanofiber Membranes.
The zone of inhibition (ZOI) assay was employed to assess the antimicrobial efficacy of the chlorhexidine loaded and AMP (D-GL13K and 1018) loaded chitosan nanofiber membranes. Figure S8A-D shows limited but distinct ZOIs around chitosan membranes with absorbed AMPs against both S. gordonii and S. mutans, while more obvious ZOIs are formed around chlorhexidine (CHX) loaded membranes (Figure S8E and F). The diffusion of cationic amphipathic AMPs such as D-GL13K and 1018 was probably hindered at the neutral pH of the nutrient agar, thus the limitation in the potency of the AMPs in this test. Further, no distinct ZOIs were formed around the co-electrospun AMP CS membranes where the AMPs were encapsulated within the membrane fibers. The post-electrospinning chemical treatment needed to manufacture these membranes (glutaraldehyde cross-linking and neutralization in saturated carbonate buffer) can render the AMPs inactive. However, the chlorhexidine (CHX) loaded chitosan elicited large ZOIs against both of the oral streptococci. The membranes with absorbed CHX formed larger ZOIs than membranes with encapsulated CHX. Table 3 summarizes the diameters of ZOIs formed by CHX/AMP loaded chitosan nanofiber membranes against the two oral streptococcal species (S. gordonii M5 and S. mutans ATCC 700610). The diffusion of antimicrobials from the chitosan membranes through the nutrient agar leading to ZOI is reminiscent of the CHX/AMP diffusion through infected oral mucosa.
Table 3.
Diameters of ZOIs Formed around Antimicrobial Loaded Chitosan Nanofiber Membranes against Oral Streptococcia
| diameters of growth inhibitory zones (mm) |
||
|---|---|---|
| sample | S. gordonii M5 | S. mutans ATCC 700610 |
| CS | no inhibition | no inhibition |
| D-GL13K CS_ES | no inhibition | no inhibition |
| D-GL13K CS_Abs | 10.2 ± 0.4 | 9.8 ± 0.3 |
| 1018 CS_ES | no inhibition | no inhibition |
| 1018 CS_Abs | 9.8 ± 0.2 | 11.8 ± 1.8 |
| CHX CS_ES | 17.0 ± 1.8 | 19.5 ± 2.6 |
| CHX CS_Abs | 26.0 ± 0.8 | 29.3 ± 2.3 |
ES, encapsulated; Abs, absorbed. Data shown above are means ± standard deviation of n = 4–6 replicates.
The antibiofilm potency of D-GL13K coating on hydrophilic etched dentin surfaces was demonstrated in a previous study from our group.35 Further, the D-GL13K is less prone to proteolytic degradation in the oral cavity, resulting from d-amino acids in its sequence, whereas IDR-1018 comprising l-amino acids is enzymatically less stable.36 Therefore, subsequent antimicrobial evaluation was restricted to D-GL13K as a representative AMP. The antimicrobial assessments were performed either indirectly with D-GL13K buffer release aliquots or directly by streptococcal culture on D-GL13K/CHX absorbed chitosan membranes followed by ATP bioluminescence assay, live/dead imaging, and crystal violet biomass quantification methods.
In order to demonstrate the pH-dependent AMP release from the chitosan membranes and consequent antimicrobial activity, D-GL13K release aliquots from 0.1 M NaOAc buffers of different pH (4.5, 5.5, and 6.5) were cocultured with the oral streptococci in 1:1 buffer:media volume ratio. The bacterial growth curves in Figure 6A and B show negligible or no growth for D-GL13K release buffer aliquots cocultured with S. gordonii M5 and S. mutans (ATCC 700610), respectively, in comparison to the buffer release aliquots from the blank chitosan membranes. Also, no growth was recorded for both of the streptococci cultured at pH 4.5, which is in agreement with earlier reports suggesting that planktonic streptococci are less acid tolerant compared with their biofilm cells.37 It is also worth noting that the AMPs elicited bacterial growth inhibitory effects even under physiological pH conditions (data not shown) and an acidic pH is not a prerequisite for their antimicrobial behavior.
Figure 6.

Indirect assessment of the antimicrobial potency of D-GL13K released from the chitosan membranes under different pH (4.5, 5.5, and 6.5) conditions. The release aliquots from the blank or D-GL13K absorbed membranes were cultured with bacterial suspensions in a 1:1 (v/v) ratio, and growth curves were recorded in the kinetic mode at 1 h intervals for (A) S. gordonii M5 and (B) S. mutans (ATCC 700610). Data shown in the line graphs are means ± standard deviation of n = 3–6 replicates. Direct assessment of (C, E) S. gordonii or (D, F) S. mutans colonization of chitosan-based nanofiber membranes after 24 h of bacterial culture measured by (C, D) ATP bioluminescence assay and (E, F) crystal violet stained biomass/bioburden. Representative macroscopic images of crystal violet stained membranes are shown above the corresponding experimental groups plotted in parts E and F. Each data point represents one replicate, and boxplots present statistical values for each group. * indicates statistical significance (p < 0.05) between the compared groups. CS, chitosan; 0.05Oxi-PE_CS, 0.05 wt % oxidized pectin coated chitosan; D-GL13K_CS, D-GL13K absorbed CS; D-GL13K-0.05Oxi-PE_CS, 0.05 wt % oxidized pectin coating on D-GL13K absorbed CS.
For the direct antimicrobial assessment, D-GL13K absorbed membranes were compared to blank membranes as negative controls and chlorhexidine (CHX) absorbed membranes as positive controls. In the ATP assay, the bioluminescence signal generated by conversion of luciferin to oxyluciferin by firefly luciferase in the presence of cellular ATP (adenosine triphosphate) derived from viable bacteria is a measure of bacterial colonization. Parts C and D of Figure 6 depict ~5-fold and ~10-fold decrease in ATP bioluminescence, an indication of significant reduction of viable S. gordonii M5 and S. mutans (ATCC 700610), respectively, on D-GL13K and CHX loaded chitosan-based membranes compared to membranes devoid of antimicrobials. The absorption of D-GL13K significantly reduced colonization of the two bacterial strains on both the pure chitosan and the 0.05 wt % oxidized pectin coated chitosan membranes. A similar ~4-fold decrease in ATP bioluminescence was previously reported on D-GL13K coated titanium (Ti) versus etched Ti against P. gingivalis.38 No statistically significant differences were noted between D-GL13K and CHX absorbed membranes within the limits of detection.
The ATP bioluminescence data were commensurate with the total biomass/bacterial bioburden determined and visualized by crystal violet staining of biofilms (Figure 6E and F), as total biomass was significantly reduced when membranes were loaded with D-GL13K or CHX. Further, we hypothesized that the oxidized pectin (polysaccharide/sugar) coated chitosan membranes, although beneficial for improving mucoadhesion, would adversely increase the potential cariogenic effects of streptococci colonization of the chitosan membranes. However, our data in Figure 6 suggested that there was no significant increase in streptococci colonization on oxidized pectin coated chitosan membranes.
Live/dead imaging of S. mutans (Figure 7) on unmodified and D-GL13K or CHX absorbed chitosan nanofiber membranes indicated the antimicrobial potency of D-GL13K and CHX loaded membranes. It is clear from Figure 7 that the S. mutans on chitosan and 0.05 wt % oxidized pectin coated chitosan majorly exhibited a biofilm phenotype with mostly live bacteria stained in green by SYTO 9. The vitality of the bacteria on the membranes with D-GL13K absorbed chitosan without and with oxidized pectin coating was clearly reduced with more bacteria presenting compromised membranes stained in red by PI or colocalization of SYTO 9 and PI, suggesting progressive bacterial injury and death.39 Further, the biofilm phenotype was reduced yet persistent in the case of D-GL13K loaded chitosan membranes. The CHX loaded membranes with and without oxidized pectin coating exhibited maximum red fluorescent bacteria with a more planktonic phenotype or small clusters of streptococci. This suggests the greater bactericidal and antibiofilm potency of CHX in comparison to D-GL13K loaded chitosan membranes. Thus, we have demonstrated the antimicrobial potency of oral dual tissue adhesive nanofiber membranes mediated by delivering antimicrobial peptides/agents in a pH-controlled manner.
Figure 7.

Live/dead imaging of S. mutans (ATCC 700610) on chitosan-based nanofiber membranes. Bacteria with uncompromised membranes were stained by SYTO 9 and exhibited green fluorescence, and bacteria with compromised membranes (an indication of dead bacteria) were counter-stained by PI and exhibited red fluorescence. Scale bar = 25 μm. Sample designations: CS, chitosan; 0.05Oxi-PE_CS, 0.05 wt % oxidized pectin coated CS; D-GL13K_CS, D-GL13K absorbed CS; D-GL13K-0.05Oxi-PE_CS, 0.05 wt % oxidized pectin coating on D-GL13K absorbed CS; CHX_CS, chlorhexidine absorbed CS; CHX-0.05Oxi-PE_CS, 0.05 wt % oxidized pectin coating on CHX absorbed CS.
3.6. Cytocompatibility of Modified Chitosan Nanofiber Membranes with Human Oral Keratinocytes and Murine Pre-Osteoblasts.
In order to demonstrate dual soft and hard tissue compatibility, the pristine and surface modified chitosan nanofiber membranes were assessed for their ability to support the adhesion and proliferation of soft epithelial tissue derived human oral keratinocytes (OKF-6/TERT2) and hard calvarial bone derived murine pre-osteoblasts (MC3T3-E1). Parts A and B of Figure 8 show proliferation of human oral keratinocytes (HOK) and murine pre-osteoblasts (MC3T3-E1) on pristine and surface modified chitosan membranes after 1 and 3 days of culture, respectively. The CCK-8 data demonstrated good proliferation of HOK and MC3T3-E1 cells on pure chitosan (CS), 0.05 wt % oxidized pectin coated chitosan (0.05Oxi-PE_CS), and AMP absorbed chitosan (D-GL13K_CS and 1018_CS). However, the absorbed chlorhexidine in chitosan (CHX_CS) presented cytotoxic effects on both cell types, which was consistent with previous reports.40 Notably, MC3T3-E1 pre-osteoblast proliferation was favored on the chitosan membranes with absorbed AMPs, particularly with D-GL13K. In a previous study from our group, D-GL13K coating on etched titanium (e-Ti) elicited greater proliferation of human gingival fibroblasts compared to e-Ti after 3 days of culture.38 Figure 8C shows immunofluorescent images of adhered HOK on pristine (CS) and modified chitosan membranes with oxidized pectin and AMPs. After 3 days in culture, HOK on all of the modified and unmodified chitosan membranes displayed good cytoskeletal spreading, morphology, and cytokeratin 14 (KRT 14) marker expression, except again for cells on CHX loaded chitosan. KRT 14 are cytoplasmic intermediate filament keratin proteins expressed by mitotically active basal layer keratinocytes whose expression is downregulated upon differentiation into stratified epithelia.41 Figure S9 presents analogous results for MC3T3-E1 pre-osteoblast cells. Taken together, the AMP (D-GL13K and 1018) loaded chitosan membranes exhibited the desirable combination of antimicrobial potency and cytocompatibility. In contrast, chlorhexidine loaded chitosan membranes were highly antimicrobial but also considerably cytotoxic to cells associated with both the hard and soft tissues.
Figure 8.

Proliferation of (A) immortalized human oral keratinocytes (OKF-6/TERT2) and (B) murine pre-osteoblasts (MC3T3-E1) on surface modified chitosan nanofiber membranes. Each data point represents one replicate, and boxplots present statistical values for each group. * indicates statistical significance at p < 0.05 between compared groups. (C) Triple immunofluorescent staining of adhered oral keratinocytes on surface modified chitosan nano fiber membranes after 3 days of culture. Green = actin, red = cytokeratin 14, blue = nuclei, scale bar = 75 μm.
4. DISCUSSION
Tissue adhesive nanofiber membranes are promising multifunctional platforms with potential applications in diagnostic and regenerative dentistry. Moreover, oral pH-responsive adhesive nanofiber membranes developed in the current study can be efficient drug/peptide/therapeutic delivery systems, with a “supply on demand” capability. This can be of particular interest for oral disease conditions such as dental caries and periodontitis as the salivary pH becomes acidic.42,43 In the current study, we demonstrate the pH-responsive delivery of AMPs (D-GL13K) from adhesive nanofiber membranes and the consequent antimicrobial activity against oral streptococci. The pH-controlled release of D-GL13K stems from the affinity of the amines (─NH2) in the lysine rich peptide to the glutaraldehyde (─CHO) cross-linked chitosan membranes. In the case of the oxidized pectin coated membranes, the cationic D-GL13K and anionic pectin can form a polyelectrolyte bilayer complex, which gets destabilized under acid pH due to the conversion of carboxylate (─COO─) to carboxylic acid (─COOH) in the oxidized pectin. Thus, the oxidized pectin not only enhances mucoadhesion but can also aid in the pH-controlled delivery of cationic AMPs. The pH-controlled delivery is expected to be universally applicable to all cationic amphipathic AMPs including IDR-1018, DJK-2, DJK-5, etc. However, the same is probably not true for chlorhexidine (CHX). In our preliminary screening of antimicrobials by the zone of inhibition assay, only the CHX loaded membranes led to large ZOIs even at the neutral pH of nutrient agar. This suggests that the diffusion of CHX from the chitosan membranes is not pH-dependent. On the other hand, the release of AMPs from the tissue adhesive membranes is pHregulated, implying that the AMP_CS can be used as a controlled delivery system, which is of particular interest in the context of the development of antimicrobial resistance caused by the overabuse of antibiotics.44
The mucoadhesion properties exhibited by our oxidized pectin coated chitosan membranes can be predominantly attributed to a combination of physical and chemical characteristics, i.e., surface nanotopography, oxidized sugar/ polysaccharide hydrogen bonding, and surface aldehyde affinity for amines of mucosal tissue proteins. The enamel/hard tissue adhesion of the uncoated chitosan is attributed to electrostatic interaction between the protonated amines (─NH3+) in chitosan and the phosphates (─PO43−) on hydroxyapatite (Figure 4). Thus, surface topography and chemistry can be manipulated to promote the adhesion of membranes with drug/peptide therapeutics for eliciting antimicrobial action in response to multiple oral diseases. Examples of other potential applications in dentistry taking advantage of the dual hard–soft tissue adhesion properties of these membranes are guided bone regeneration (GBR),45 synthetic gingival grafts to treat gingival recession in chronic periodontitis,46 augmentation of peri-implant tissues,47 and temporary toothache relief or tooth numbing.48 The versatility of our membranes is reliant on the easy incorporation in the fibrillary structure of multiple biologicals, drugs, and/or anesthetics depending on the specific application. We further envision that the therapeutic and biosensing applications of the mucoadhesive nanofiber membranes developed in this study can be extended to other mucosal tissue surfaces in the body such as gastrointestinal, vaginal, nasal, and bronchial epithelia.
Table 4 presents a summary of studies reporting novel mucoadhesive materials, the adhesion test configurations, dry/ wet testing, and the adhesion values of the bioadhesive test, predominantly in terms of adhesion strength (N/cm2) and adhesion energy/work of adhesion per unit area (mJ/cm2). The lap shear test and normal/tensile adhesion test are the predominant assessment tests for this purpose. Our chitosan membranes coated with oxidized pectin outperformed earlier reported chitosan–pectin composite films14 and synthetic poly(ethylene oxide) (PEO)–carboxymethyl cellulose (CMC) nanofibers26 for soft tissue adhesion under wet and dry conditions, respectively. Dry adhesion tests and lap shear tests provide disproportionately higher values of detachment forces than tensile adhesion tests under wet conditions. Empirically, the shear adhesion strength of the gecko lizard is ~10 times higher than the normal adhesion strength (1 N/cm2 normal vs 10 N/cm2 shear).49 We believe we report for the first time adhesion values of chitosan membranes to hard tissues, but others have tested adhesion to titanium in the context of dental implants.50
Table 4.
Comparison of the Bioadhesive Performance of Chitosan-Based Nanofiber Membranes with Previous Literature Reportsa
| sample | tissue type | adhesion test type | dry/wet condition |
adhesion strength (N/cm2) |
adhesion energy/area (mJ/cm2) |
reference |
|---|---|---|---|---|---|---|
| oxidized dextran coated nanopatterns | porcine intestine | lap shear test | dry | 0.7 ± 0.1 | ND | Mahdavi et al.15 |
| PNIPAm–alginate hydrogels | porcine skin, rodent skin | 180° peel test | dry | ND | 17.5 ± 2.5 | Blacklow et al.51 |
| PEO–CMC nanofibers | lamb esophagus | normal/tensile adhesion | dry | 0.25 ± 0.05 | ND | Brako et al.26 |
| silk fibroin–tannic acid hydrogels | porcine skin | lap shear test | dry | 6.94 ± 0.53 | ND | Luo et al.7 |
| gelatin methacryloyl photocure | porcine gingiva | lap shear test | dry | 5.53 ± 0.67 | ND | Shirzaei Sani et al.50 |
| chitosan–pectin composite films | porcine intestine | normal/tensile adhesion | wet | 0.27 ± 0.05 | ND | Hagesaether et al.14 |
| oxidized pectin coated chitosan nanofibers | porcine esophagus | normal/tensile adhesion | wet | 0.55 ± 0.04 | 0.4 ± 0.05 | current study |
| chitosan nanofibers | hydroxyapatite | normal/tensile adhesion | wet | 0.96 ± 0.2 | 0.13 ± 0.01 | current study |
ND: Not determined.
One of the limitations of our work is that we did not assess the effect of pH on the adhesion properties of our membranes. This is of further relevance, as our membranes contain and can release therapeutics (antimicrobials, anti-inflammatory, and antifibrotics) in response to environmental pH changes. Indeed, the therapeutics, their release, and the pH of the tissue at the site of placement of the adhesive can affect the performance of the adhesive. Therefore, we discuss here the effects of acidic pH on mucoadhesion. The adsorption of mucin glycoproteins on synthetic polymers is a well-known measure of mucoadhesion property.52 This can occur via hydrogen bonding and aldehyde–amine chemistry. A spectroscopic investigation of mucin/poly(acrylic acid) interactions revealed better mucoadhesion at slightly acidic pH ≤ 4.5 compared to neutral pH due to enhanced hydrogen bonding between the un-ionized carboxylic acid (─COOH) groups of poly(acrylic acid) and the deprotonated amines (─NH2) groups of mucin glycoproteins.53 In our oxidized pectin coated membranes too, the ─COOH groups from the D-galacturonic acid units of oxidized pectin can form more intermolecular hydrogen bonding networks with mucin glycoproteins at a mildly acidic pH of 4.5. The other mechanism of enhanced mucoadhesion is the aldehyde–amine chemistry leading to imine formation between oxidized pectin and mucin glycoproteins. Imine formation is an acid-catalyzed reversible reaction with an optimal reaction pH of ~5, above or below which imine formation is reduced. Based on this, we envisage that mucoadhesion of the oxidized pectin coated membranes will be promoted under a mildly acidic pH of 4.5. Summarizing, we expect that the mucoadhesion properties of our membranes will be retained or enhanced in the oral pH (4.5–6.5) range, which will be assessed in future studies. The hard tissue adhesion is mostly electrostatic charge related and therefore will not deteriorate under acidic pH. Next, we discuss the effect of cationic AMPs on the adhesion behavior of our membranes. A careful observation of the adhesive membrane configuration will reveal that the cationic AMP absorbed chitosan faces the hard bone tissue, while the oxidized pectin mucoadhesive coating faces the soft gingival tissue (Figure 1). This positioning of our membranes preserves mucoadhesion, while hard tissue adhesion is favored by the electrostatic attraction between the cationic AMPs and anionic phosphates on bone tissue. Furthermore, even the diffusion of cationic AMPs to the mucosal tissue will permit similar electrostatic interactions with the anionic mucins, thus retaining membrane adhesion properties. Taken together, the presence of cationic AMPs and acidic pH may not significantly affect the dual hard and soft tissue adhesion behavior of our membranes. However, we will investigate this in a future study.
5. CONCLUSION
We fabricated chitosan-based nanofiber membranes with dual adhesion to soft and hard tissue surfaces and pH-controlled delivery of antimicrobial agents, antibiotics, and peptides. Mucoadhesion was enhanced by coating the membranes with oxidized sugars/polysaccharide (pectin), whereas pristine chitosan membranes demonstrated good adhesion to hard/ enamel tissue. Absorption of AMPs (D-GL13K or IDR-1018) inside the nanofibrillar structures conferred simultaneous antimicrobial potency against relevant oral streptococci and cytocompatibility with both soft tissue and hard tissue associated cells, i.e., keratinocytes and osteoblasts. Our membranes can be potentially used for temporary preventive and/or therapeutic delivery in the oral cavity in a pH-controlled manner with a “supply on demand” release behavior. In addition, the oral dual tissue adhesive membranes might be further developed for oral biosensing of disease biomarkers, oral theranostic platforms, GBR membranes with dual hard–soft tissue functions, and synthetic muco-grafts for treating soft tissue lesions and gingival recession in chronic periodontitis.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. Wei Shen, Department of Biomedical Engineering, and Dr. Changquan Calvin Sun, School of Pharmacy, University of Minnesota, for sharing their electrospinning unit and texture analyzer, respectively. The authors also thank Dr. Alex Fok and Mr. Antonio Olivares for helping with the design of the fixtures and access to adhesion tests. Parts of this work were carried out in the Characterization Facility, University of Minnesota, which receives partial support from NSF through the MRSEC program. Portions of this work were also conducted in the Minnesota Nano Center, which is supported by the National Science Foundation through the National Nano Coordinated Infrastructure Network, Award Number NNCI - 1542202. Research reported in this publication was supported by the National Institute of Dental & Craniofacial Research of the National Institutes of Health (award number R01DE026117 to C.A. and T90DE0227232 to N.G.F.) and the National Center for Advancing Translational Sciences (Translational Research Development Program-TRDP award to Z.Y. from award number UL1TR002494). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biomac.0c01163.
Nanofiber diameter distribution plots and nanofiber membrane thicknesses (SEM images) in Figure S1; reaction scheme and FT-IR characterization for periodate oxidation of pectin in Figure S2; reaction scheme and titration of aldehyde content in oxidized pectin in Figure S3; XPS survey scan spectra in Figure S4; deconvolution of high-resolution N 1s spectra in Figure S5; tracing of the force–time graph during adhesion tests in Figure S6; degradation of chitosan vs pH and peptide release from co-electrospun membranes in Figure S7; bacterial zones of inhibition in Figure S8; adhesion of MC3T3-E1 pre-osteoblasts (fluorescence images) in Figure S9; and binding energies of deconvoluted N 1s peaks in Table S1 (PDF)
The authors declare no competing financial interest.
The raw/processed data required to reproduce these findings will be made available on request.
Contributor Information
Sunil Kumar Boda, Minnesota Dental Research Center for Biomaterials and Biomechanics, University of Minnesota, Minneapolis, Minnesota 55455, United States.
Nicholas G. Fischer, Minnesota Dental Research Center for Biomaterials and Biomechanics, University of Minnesota, Minneapolis, Minnesota 55455, United States.
Zhou Ye, Minnesota Dental Research Center for Biomaterials and Biomechanics, University of Minnesota, Minneapolis, Minnesota 55455, United States.
Conrado Aparicio, Minnesota Dental Research Center for Biomaterials and Biomechanics, University of Minnesota, Minneapolis, Minnesota 55455, United States.
REFERENCES
- (1).Zhu W; Chuah YJ; Wang D-A Bioadhesives for internal medical applications: A review. Acta Biomater. 2018, 74, 1–16. [DOI] [PubMed] [Google Scholar]
- (2).Ma S; Wu Y; Zhou F Bioinspired synthetic wet adhesives: from permanent bonding to reversible regulation. Curr. Opin. Colloid Interface Sci 2020, 47, 84–98. [Google Scholar]
- (3).Xu R; Ma S; Wu Y; Lee H; Zhou F; Liu W Adaptive control in lubrication, adhesion, and hemostasis by Chitosan–Catechol–pNIPAM. Biomater. Sci 2019, 7 (9), 3599–3608. [DOI] [PubMed] [Google Scholar]
- (4).Bhagat V; Becker ML Degradable Adhesives for Surgery and Tissue Engineering. Biomacromolecules 2017, 18 (10), 3009–3039. [DOI] [PubMed] [Google Scholar]
- (5).Kord Forooshani P; Lee BP Recent approaches in designing bioadhesive materials inspired by mussel adhesive protein. J. Polym. Sci., Part A: Polym. Chem 2017, 55 (1), 9–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Zhao Y; Wu Y; Wang L; Zhang M; Chen X; Liu M; Fan J; Liu J; Zhou F; Wang Z Bio-inspired reversible underwater adhesive. Nat. Commun 2017, 8 (1), 2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Luo J; Yang J; Zheng X; Ke X; Chen Y; Tan H; Li J A Highly Stretchable, Real-Time Self-Healable Hydrogel Adhesive Matrix for Tissue Patches and Flexible Electronics. Adv. Healthcare Mater 2020, 9 (4), 1901423. [DOI] [PubMed] [Google Scholar]
- (8).Cook MT; Khutoryanskiy VV Mucoadhesion and mucosamimetic materials—A mini-review. Int. J. Pharm 2015, 495 (2), 991–998. [DOI] [PubMed] [Google Scholar]
- (9).Buckley MJ; Beckman EJ Adhesive Use in Oral and Maxillofacial Surgery. Oral Maxil. Surg. Clin. North Am 2010, 22 (1), 195–199. [DOI] [PubMed] [Google Scholar]
- (10).Murphy MP; Kim S; Sitti M Enhanced Adhesion by Gecko-Inspired Hierarchical Fibrillar Adhesives. ACS Appl. Mater. Interfaces 2009, 1 (4), 849–855. [DOI] [PubMed] [Google Scholar]
- (11).Ballarin FM; Blackledge TA; Capitos Davis NL; Frontini PM; Abraham GA; Wong S-C Effect of topology on the adhesive forces between electrospun polymer fibers using a T-peel test. Polym. Eng. Sci 2013, 53 (10), 2219–2227. [Google Scholar]
- (12).Colley HE; Said Z; Santocildes-Romero ME; Baker SR; D’Apice K; Hansen J; Madsen LS; Thornhill MH; Hatton PV; Murdoch C Pre-clinical evaluation of novel mucoadhesive bilayer patches for local delivery of clobetasol-17-propionate to the oral mucosa. Biomaterials 2018, 178, 134–146. [DOI] [PubMed] [Google Scholar]
- (13).Kalhapure RS; Jadhav M; Rambharose S; Mocktar C; Singh S; Renukuntla J; Govender T pH-responsive chitosan nanoparticles from a novel twin-chain anionic amphiphile for controlled and targeted delivery of vancomycin. Colloids Surf., B 2017, 158, 650–657. [DOI] [PubMed] [Google Scholar]
- (14).Hagesaether E; Hiorth M; Sande SA Mucoadhesion and drug permeability of free mixed films of pectin and chitosan: An in vitro and ex vivo study. Eur. J. Pharm. Biopharm 2009, 71 (2), 325–331. [DOI] [PubMed] [Google Scholar]
- (15).Mahdavi A; Ferreira L; Sundback C; Nichol JW; Chan EP; Carter DJD; Bettinger CJ; Patanavanich S; Chignozha L; Ben-Joseph E; Galakatos A; Pryor H; Pomerantseva I; Masiakos PT; Faquin W; Zumbuehl A; Hong S; Borenstein J; Vacanti J; Langer R; Karp JM A biodegradable and biocompatible gecko-inspired tissue adhesive. Proc. Natl. Acad. Sci. U. S. A 2008, 105 (7), 2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Sriamornsak P Application of pectin in oral drug delivery. Expert Opin. Drug Delivery 2011, 8 (8), 1009–1023. [DOI] [PubMed] [Google Scholar]
- (17).Liu Y; Naha PC; Hwang G; Kim D; Huang Y; Simon-Soro A; Jung H-I; Ren Z; Li Y; Gubara S; Alawi F; Zero D; Hara AT; Cormode DP; Koo H Topical ferumoxytol nanoparticles disrupt biofilms and prevent tooth decay in vivo via intrinsic catalytic activity. Nat. Commun 2018, 9 (1), 2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Stephan RM Intra-Oral Hydrogen-Ion Concentrations Associated With Dental Caries Activity. J. Dent. Res 1944, 23 (4), 257–266. [Google Scholar]
- (19).Stephan RM Changes in Hydrogen-Ion Concentration on Tooth Surfaces and in Carious Lesions. J. Am. Dent. Assoc., JADA 1940, 27 (5), 718–723. [Google Scholar]
- (20).Jiao Y; Tay FR; Niu L-N; Chen J-H Advancing antimicrobial strategies for managing oral biofilm infections. Int. J. Oral Sci 2019, 11 (3), 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Sangsanoh P; Supaphol P Stability Improvement of Electrospun Chitosan Nanofibrous Membranes in Neutral or Weak Basic Aqueous Solutions. Biomacromolecules 2006, 7 (10), 2710–2714. [DOI] [PubMed] [Google Scholar]
- (22).Gupta B; Tummalapalli M; Deopura BL; Alam MS Functionalization of pectin by periodate oxidation. Carbohydr. Polym 2013, 98 (1), 1160–1165. [DOI] [PubMed] [Google Scholar]
- (23).Hotaling NA; Bharti K; Kiel H; Simon CG DiameterJ: A validated open source nanofiber diameter measurement tool. Biomaterials 2015, 61, 327–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Jaroslav B; Maurice P Colorimetric and Fluorimetric Determination of Aldehydes and Ketones. Pure Appl. Chem 1979, 51 (8), 1803–1814. [Google Scholar]
- (25).Wojdyr M Fityk: a general-purpose peak fitting program. J. Appl. Crystallogr 2010, 43 (5), 1126–1128. [Google Scholar]
- (26).Brako F; Thorogate R; Mahalingam S; Raimi-Abraham B; Craig DQM; Edirisinghe M Mucoadhesion of Progesterone-Loaded Drug Delivery Nanofiber Constructs. ACS Appl. Mater. Interfaces 2018, 10 (16), 13381–13389. [DOI] [PubMed] [Google Scholar]
- (27).Abdolhosseini M; Nandula SR; Song J; Hirt H; Gorr S-U Lysine substitutions convert a bacterial-agglutinating peptide into a bactericidal peptide that retains anti-lipopolysaccharide activity and low hemolytic activity. Peptides 2012, 35 (2), 231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Mansour SC; de la Fuente-Núñez C; Hancock REW Peptide IDR-1018: modulating the immune system and targeting bacterial biofilms to treat antibiotic-resistant bacterial infections. J. Pept. Sci 2015, 21 (5), 323–329. [DOI] [PubMed] [Google Scholar]
- (29).Dickson MA; Hahn WC; Ino Y; Ronfard V; Wu JY; Weinberg RA ; Louis DN; Li FP; Rheinwald JG Human Keratinocytes That Express hTERT and Also Bypass a p16INK4a-Enforced Mechanism That Limits Life Span Become Immortal yet Retain Normal Growth and Differentiation Characteristics. Mol. Cell. Biol 2000, 20 (4), 1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Nasajpour A; Ansari S; Rinoldi C; Rad AS; Aghaloo T; Shin SR; Mishra YK; Adelung R; Swieszkowski W; Annabi N; Khademhosseini A; Moshaverinia A; Tamayol A A Multifunctional Polymeric Periodontal Membrane with Osteogenic and Antibacterial Characteristics. Adv. Funct. Mater 2018, 28 (3), 1703437. [Google Scholar]
- (31).da Costa MPM; de Mello Ferreira IL; de Macedo Cruz MT New polyelectrolyte complex from pectin/chitosan and montmorillonite clay. Carbohydr. Polym 2016, 146, 123–130. [DOI] [PubMed] [Google Scholar]
- (32).Smart JD Theories of Mucoadhesion. In Mucoadhesive Materials and Drug Delivery Systems 2014, 159–174. [Google Scholar]
- (33).Beppu MM; Vieira RS ; Aimoli CG; Santana CC Crosslinking of chitosan membranes using glutaraldehyde: Effect on ion permeability and water absorption. J. Membr. Sci 2007, 301 (1), 126–130. [Google Scholar]
- (34).Diaz del Consuelo I; Pizzolato G-P; Falson F; Guy RH; Jacques Y Evaluation of pig esophageal mucosa as a permeability barrier model for buccal tissue. J. Pharm. Sci 2005, 94 (12), 2777–2788. [DOI] [PubMed] [Google Scholar]
- (35).Moussa DG; Fok A; Aparicio C Hydrophobic and antimicrobial dentin: A peptide-based 2-tier protective system for dental resin composite restorations. Acta Biomater. 2019, 88, 251–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Feng Z; Xu B Inspiration from the mirror: D-amino acid containing peptides in biomedical approaches. Biomol. Concepts 2016, 7 (3), 179–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Welin-Neilands J; Svensäter G Acid Tolerance of Biofilm Cells of Streptococcus mutans. Appl. Environ. Microbiol 2007, 73 (17), 5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Holmberg KV; Abdolhosseini M; Li Y; Chen X; Gorr S-U; Aparicio C Bio-inspired stable antimicrobial peptide coatings for dental applications. Acta Biomater. 2013, 9 (9), 8224–8231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Freire JM; Gaspar D; de la Torre BG; Veiga AS; Andreu D; Castanho MARB Monitoring antibacterial permeabilization in real time using time-resolved flow cytometry. Biochim. Biophys. Acta, Biomembr 2015, 1848 (2), 554–560. [DOI] [PubMed] [Google Scholar]
- (40).Hidalgo E; Dominguez C Mechanisms underlying chlorhexidine-induced cytotoxicity. Toxicol. In Vitro 2001, 15 (4), 271–276. [DOI] [PubMed] [Google Scholar]
- (41).Alam H; Sehgal L; Kundu ST; Dalal SN; Vaidya MM Novel function of keratins 5 and 14 in proliferation and differentiation of stratified epithelial cells. Mol. Biol. Cell 2011, 22 (21), 4068–4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Baliga S; Muglikar S; Kale R Salivary pH: A diagnostic biomarker. J. Indian Soc. Periodontol 2013, 17 (4), 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Featherstone JDB Dental caries: a dynamic disease process. Aust. Dent. J 2008, 53 (3), 286–291. [DOI] [PubMed] [Google Scholar]
- (44).Lewies A; Du Plessis LH; Wentzel JF Antimicrobial Peptides: the Achilles’ Heel of Antibiotic Resistance? Probiotics Antimicrob. Proteins 2019, 11 (2), 370–381. [DOI] [PubMed] [Google Scholar]
- (45).Hasani-Sadrabadi MM; Sarrion P; Nakatsuka N; Young TD; Taghdiri N; Ansari S; Aghaloo T; Li S; Khademhosseini A; Weiss PS; Moshaverinia A Hierarchically Patterned Polydopamine-Containing Membranes for Periodontal Tissue Engineering. ACS Nano 2019, 13 (4), 3830–3838. [DOI] [PubMed] [Google Scholar]
- (46).Shirakata Y; Sculean A; Shinohara Y; Sena K; Takeuchi N; Bosshardt DD; Noguchi K Healing of localized gingival recessions treated with a coronally advanced flap alone or combined with an enamel matrix derivative and a porcine acellular dermal matrix: a preclinical study. Clin. Oral Investig 2016, 20 (7), 1791–1800. [DOI] [PubMed] [Google Scholar]
- (47).Brouwers JEIG; Buis S; Haumann R; de Groot PPG; de Laat B; Remijn JA Successful soft and hard tissue augmentation with platelet-rich fibrin in combination with bovine bone space maintainer in a delayed implant placement protocol in the esthetic zone: A case report. Clin. Case Rep 2019, 7 (6), 1185–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Hersh EV; Ciancio SG; Kuperstein AS; Stoopler ET; Moore PA; Boynes SG; Levine SC; Casamassimo P; Leyva R; Mathew T; Shibly O; Creighton P; Jeffers GE; Corby PMA; Turetzky SN; Papas A; Wallen J; Idzik-Starr C; Gordon SM An evaluation of 10% and 20% benzocaine gels in patients with acute toothaches: Efficacy, tolerability and compliance with label dose administration directions. J. Am. Dent. Assoc., JADA 2013, 144 (5), 517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Wang Z Slanted Functional Gradient Micropillars for Optimal Bioinspired Dry Adhesion. ACS Nano 2018, 12 (2), 1273–1284. [DOI] [PubMed] [Google Scholar]
- (50).Shirzaei Sani E; Portillo Lara R; Aldawood Z; Bassir SH; Nguyen D; Kantarci A; Intini G; Annabi N An Antimicrobial Dental Light Curable Bioadhesive Hydrogel for Treatment of Peri-Implant Diseases. Matter 2019, 1 (4), 926–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Blacklow SO; Li J; Freedman BR; Zeidi M; Chen C; Mooney DJ Bioinspired mechanically active adhesive dressings to accelerate wound closure. Sci. Adv 2019, 5 (7), No. eaaw3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Serra L; Domenech J; Peppas NA Engineering design and molecular dynamics of mucoadhesive drug delivery systems as targeting agents. Eur. J. Pharm. Biopharm 2009, 71 (3), 519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Patel MM; Smart JD; Nevell TG; Ewen RJ; Eaton PJ; Tsibouklis J Mucin/Poly(acrylic acid) Interactions: A Spectroscopic Investigation of Mucoadhesion. Biomacromolecules 2003, 4 (5), 1184–1190. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.