
Figure 1: Mouse models of the GH/IGF axis.
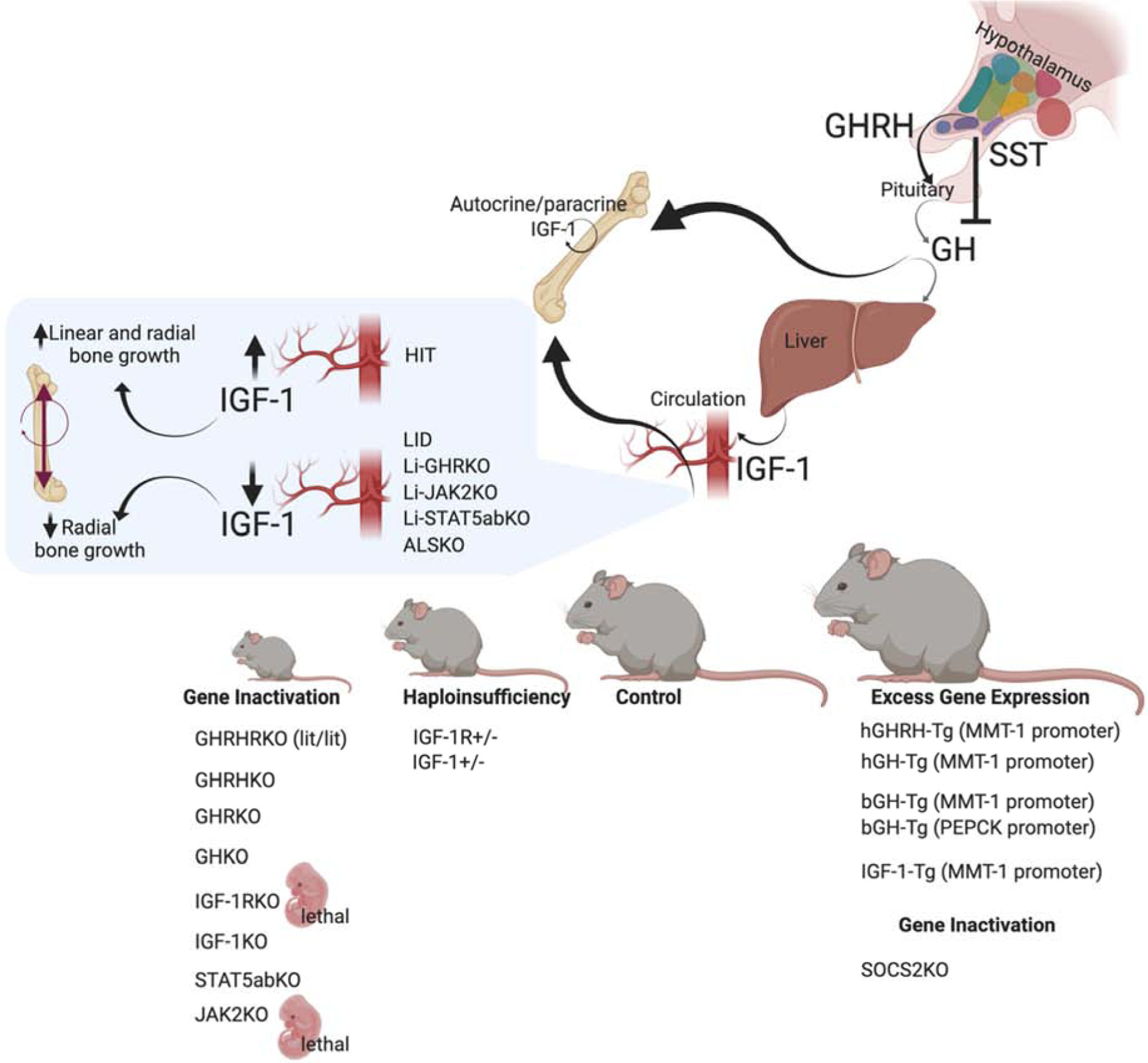
Several naturally occurring mutations in the GH/IGF-1 axis have been described over the years. Among them is the lit/lit mouse line, with a mutation in the GHRH-receptor which causes pituitary hypoplasia, reduced GH secretion, and dwarfism [220]. Similarly, targeted ablation of the GHRH gene causes GH deficiency, pituitary hypoplasia, and reductions in somatic growth [221]. GHRKO and the recently described GHKO mice [222] also show decreased linear bone growth and reduced whole-body BMD. Gene ablation of the IGF-1R was lethal [57], and caused severely retarded growth, with delayed ossification in all bones. Likewise, IGF-1KO mice showed severe dwarfism, associated with reduced BMD and decreased linear and radial bone growth [223]. In mice, inactivation of the downstream mediator of GH signaling, STAT5ab, also affected linear growth [224]. However, the details of skeletal morphology were not reported. Finally, in mice, knocking out JAK2, the GH mediator, was lethal [225], likely due to its central roles in cytokine signaling. Haploinsufficiency of IGF-1 or the IGR-1R associate with intermediate growth retardation and reduced linear and radial bone growth. Mouse models of excess GHRH [35, 226], hGH [36–42], and bGH [38, 43–49] show increased somatic growth with robust bones. Mice with excess IGF-1 resulted in selective organomegaly and as reported at the time showed minimal effects on skeletal growth [52, 227]. Lastly, SOCS2KO mice, which exhibit a “physiological” excess of GH action (as oppose to transgene driven GH expression), show increased linear and radial bone growth with increased trabecular bone volume [228].