

Abstract
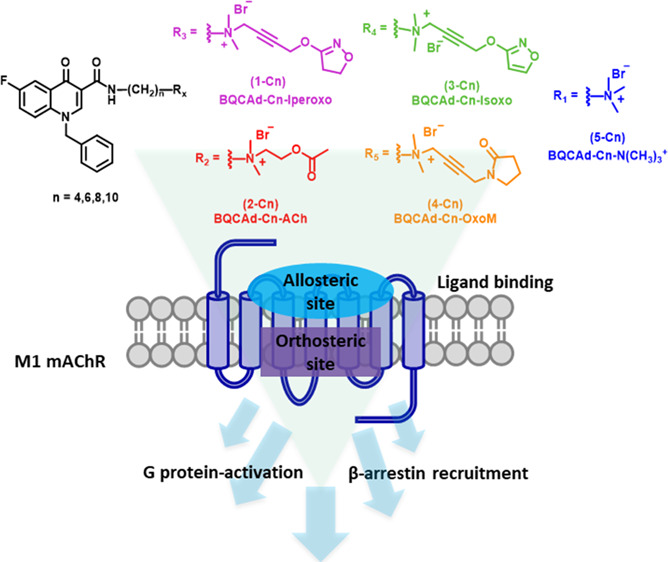
The muscarinic M1 acetylcholine receptor is an important drug target for the treatment of various neurological disorders. Designing M1 receptor-selective drugs has proven challenging, mainly due to the high conservation of the acetylcholine binding site among muscarinic receptor subtypes. Therefore, less conserved and topographically distinct allosteric binding sites have been explored to increase M1 receptor selectivity. In this line, bitopic ligands, which target orthosteric and allosteric binding sites simultaneously, may provide a promising strategy. Here, we explore the allosteric, M1-selective BQCAd scaffold derived from BQCA as a starting point for the design, synthesis, and pharmacological evaluation of a series of novel bitopic ligands in which the orthosteric moieties and linker lengths are systematically varied. Since β-arrestin recruitment seems to be favorable to therapeutic implication, all the compounds were investigated by G protein and β-arrestin assays. Some bitopic ligands are partial to full agonists for G protein activation, some activate β-arrestin recruitment, and the degree of β-arrestin recruitment varies according to the respective modification. The allosteric BQCAd scaffold controls the positioning of the orthosteric ammonium group of all ligands, suggesting that this interaction is essential for stimulating G protein activation. However, β-arrestin recruitment is not affected. The novel set of bitopic ligands may constitute a toolbox to study the requirements of β-arrestin recruitment during ligand design for therapeutic usage.
Introduction
Muscarinic acetylcholine receptors belong to the superfamily of G protein-coupled receptors. Five muscarinic receptor subtypes (M1–M5) are expressed in humans and mediate a variety of physiological functions.1,2 The binding site for the endogenous neurotransmitter acetylcholine, classical synthetic orthosteric agonists (e.g., carbachol, oxotremorine, oxotremorine-M, and iperoxo), and antagonists (e.g., atropine, N-methylscopolamine, and ipratropium) is located deeply inside the seven-fold transmembrane helical bundle.3,4 All muscarinic receptor subtypes share a high sequence similarity in their orthosteric binding pockets, which has entailed severe difficulties in the discovery of subtype-selective ligands for this receptor family.4−6 Moreover, all muscarinic receptors possess at least one allosteric binding site, which is located on top of the orthosteric binding site comprising the upper parts of transmembrane helices and extracellular loops.7−10
M1 muscarinic receptors are predominantly expressed in the central nervous system (CNS), especially in the amygdala, hippocampus, cerebral cortex, and striatum11,12 where they contribute to essential cognitive functions such as memory and learning. The M1 muscarinic receptor has been identified as an important drug target, which may be addressed for the treatment of Alzheimer’s disease (AD) and schizophrenia.13,14 Although activation of M1 receptors with synthetic orthosteric agonists has been shown to be effective in a variety of CNS diseases,15−17 all ligands exhibited severe adverse effects, which have eventually led to withdrawal from drug discovery programs, likely due to missing subtype selectivity of these drug candidates.18,19
New strategies for obtaining more selective ligands have been explored by either using positive allosteric modulators (PAMs)20−26 or by structural hybrids of orthosteric and allosteric moieties. These latter so-called bitopic ligands have been originally developed based upon the hypothesis of combining the activation properties of orthosteric ligands with the better selectivity profile of allosteric ligands.24,27−29 A number of studies have lately shown that through the design of bitopic ligand series, a differential engagement of all possible GPCR signaling pathways can be studied.30,31 We have previously synthesized bitopic agonists for the M1 receptor (Scheme 1, 1-Cn), which consist of the orthosteric agonists iperoxo or acetylcholine and the M1R-selective positive allosteric modulators (PAM) BQCAd.32−36 Benzyl quinolone carboxylic acid (BQCA) and its derivatives were reported being positive allosteric modulators (PAMs). Compared to their orthosteric counterparts, they confer a high selectivity in terms of binding and function at M1 receptors.33,37 Underlying structure–activity relationship studies suggested that the potency and efficacy of bitopic agonists basically depend on three substantial factors, i.e., linker length, geometry of the position of orthosteric and allosteric moieties, as well as the substitution pattern of the allosteric moiety.37−39
Scheme 1. Synthetic Pathway of the Bitopic M1 Receptor Ligands Composed of the PAM Benzyl Quinolone Carboxylic Acid Derivative (BQCAd 6) Covalently Linked to Non-selective Muscarinic Orthosteric Agonist Fragments (Iperoxo 1, Acetylcholine 2, Isoxo 3, Oxotremorine-M 4, and TMA (Tetramethylammonium) Head 5).
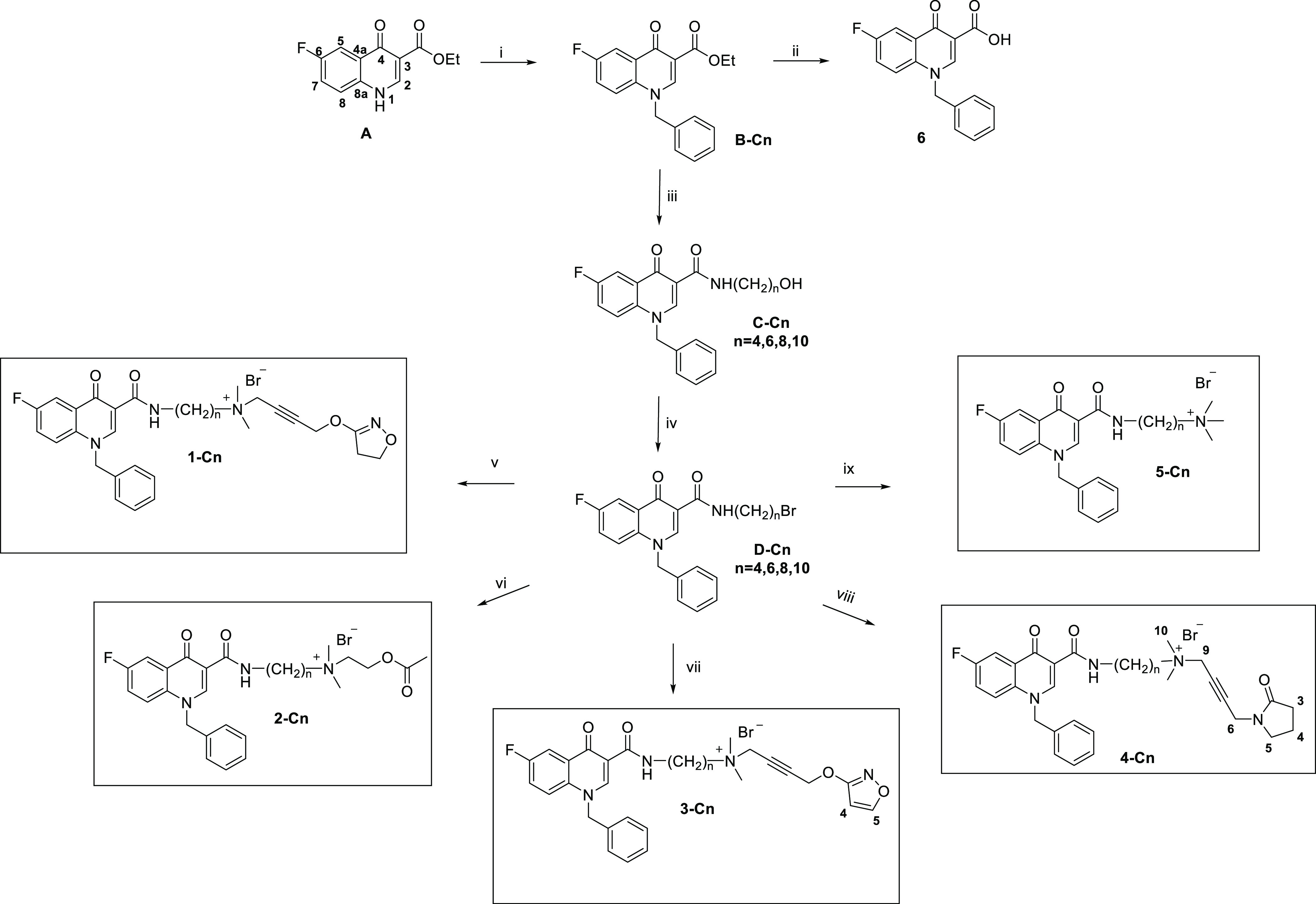
Reagents and conditions: (i) benzyl chloride, K2CO3, DMF, and 80 °C; (ii) 6 N HCl, MeOH, and reflux; (iii) H2N(CH2)nOH and 150 °C; (iv) HBr (48%), H2SO4 and reflux; (v) iperoxo base, KI/K2CO3, CH3CN, and 80 °C MW; (vi) acetylcholine, KI/K2CO3, CH3CN, and 80 °C MW; (vii) isoxo base, KI/K2CO3, CH3CN, and 80 °C MW; (viii) oxotremorine-M base, KI/K2CO3, CH3CN, and 80 °C MW; (ix) trimethylamine, CH3CN, and 40 °C.
Here, we introduce a systematic set of novel bitopic ligands for the M1 receptor, which were designed by connecting the allosteric moiety BQCAd to orthosteric muscarinic agonists using methylene linkers of varying length (Scheme 1). The allosteric interaction of BQCAd is shown in the Supporting Information (Table S4 and Figure S1 (panel F)). Classical orthosteric agonists were chosen as building blocks of the bitopic ligands aiming at a comparison of a myriad of study results from the literature and obtaining a deepened understanding of previously published works.37,38 In order to derive structure–activity relationships, the set of compounds was tested for the ability to activate G protein signaling and β-arrestin recruitment. Our data show that efficacy and potency of bitopic ligands strongly depend on the structure of the orthosteric moiety, which is supported by respective docking studies. The smallest known muscarinic agonist, tetramethylammonium (TMA),40,41 has a sufficient efficacy to activate G proteins but is not suitable for recruiting β-arrestin. Interestingly, the iperoxo hybrids 1-C6 and 1-C8 as well as the isoxo ligand 3-C8 were able to recruit β-arrestin2. Of note, our data demonstrates how subtle differences in ligand structure can impact receptor activation and simultaneously suggests that different regions of the orthosteric binding site can be explored in order to investigate M1 receptor signaling.
Results and Discussion
Chemistry
Compounds 1-Cn, 2-Cn, and 5-Cn were prepared according to previously reported procedures.37,38 Compound series 3-Cn and 4-Cn and their overall synthetic pathways are displayed in Scheme 1 (for further details, see Supporting Information Data S1). The 4-oxo-quinoline skeleton was built up using the Gould–Jacobs synthetic procedure using diethyl 2-(ethoxymethylene)-malonate for condensation with 4-fluoroaniline followed by cyclization in diphenyl ether.42−44 Both synthetic procedures were carried out under microwave assistance to promote conversion and to achieve better yields. The quinolone nitrogen was benzylated followed by amidation of the ester function utilizing an aminoalkyl alcohol of the respective spacer length. Finally, the hydroxyl function was replaced by a bromine atom using hydrobromic acid and sulfuric acid.38
All orthosteric fragments were synthesized according to the literature45−48 to obtain the tertiary base, which was subsequently used within the final synthesis step yielding the desired bitopic compounds. In brief, the synthesis of the isoxazole moiety45 starts with treating methyl propiolate with commercial hydroxylamine hydrochloride followed by subsequent reaction with 1,4-dichloro-2-butyne. The tertiary base was finally obtained by adding a two-fold excess of a solution of dimethylamine in DMF at room temperature requiring a three-step synthesis. Oxotremorine-M was obtained by starting from commercially available 2-pyrrolidinone and propargyl bromide followed by a CuCl-catalyzed Mannich reaction using dimethylamine and paraformaldehyde to obtain the corresponding tertiary bases.45,47
The final compounds were obtained by reaction of the allosteric moiety equipped with the brominated spacer and the corresponding tertiary orthosteric bases as shown in Scheme 1. All reactions were conducted using microwave assistance to shorten reaction times up to 8–48 h. The final compounds were purified by recrystallization or, in cases where crystallization was not feasible, by column chromatography using basic aluminum oxide.
Pharmacology
Recent findings correlate pronounced cholinergic adverse responses with G protein activation coupling and phosphorylation-dependent signaling with maximal clinical efficacy across various AD symptoms.49,50 In order to evaluate the impact of the orthosteric moieties on overall ligand activity and to compare the activity of the novel bitopic compounds with previously studied ones, all ligands were tested in both G protein signaling and β-arrestin recruitment assays. As a readout for G protein signaling, we applied a luminescence-based complementation assay: in intact cells, the interaction of Gαq with phospholipase Cβ3 being fused to parts of a luciferase51 can be observed. Upon stimulation of Gq-coupled receptors, spatial proximity between Gαq and PLC-β3 allows complementation of the split luciferase; thus, luminescence derived from a luciferase substrate is detected. β-Arrestin recruitment was measured by BRET between a NanoLuc-tagged M1 receptor and β-arrestin2, N-terminally fused to a Halo tag.37 Moreover, the ability of the novel ligands to interact with both the orthosteric and allosteric binding sites was tested in equilibrium and by applying dissociation binding experiments, respectively (Supporting Information Data S3).
Iperoxo-Derived Bitopic Ligands
Iperoxo is the most potent and efficacious muscarinic agonist, which has served as an orthosteric moiety of other bitopic ligands.37,38,52 We characterized iperoxo-based bitopic ligands with aliphatic linkers spanning 6 to 10 methylene groups (Scheme 1). Gq/PLC-β3 complementation assays (Figure 1A) reveal that all compounds are agonists. 1-C6 and 1-C10 are partial agonists, whereas 1-C8 behaves as a full agonist for Gq activation. Increasing the linker length from 6 to 8 methylene groups increases both potency and efficacy, whereas a further increase in linker length to 10 methylene groups decreases both potency and efficacy. The bitopic ligands were tested for their ability to stimulate β-arrestin recruitment. 1-C6 and 1-C8 are partial agonists with 1-C6 displaying a slightly higher potency than 1-C8. 1-C10 fails to stimulate β-arrestin recruitment. Interestingly, we find that 1-C8 is much more potent and efficacious than 1-C6 regarding G protein activation (Figure 1A); however, 1-C6 is more potent in recruiting β-arrestin than 1-C8 (Figure 2A).
Figure 1.

Concentration-response curves in Gα/PLC-β3 split-luciferase interaction assays in HEK293T cells expressing the human muscarinic M1 receptors (hM1), which were stimulated by utilizing varying concentrations of indicated compounds. Data is expressed as the means ± S.E.M. of 3–6 independent experiments performed in triplicate. The data were normalized to iperoxo (panel A), ACh (panel B), Isoxo (panel C), Oxo-M (panel D), and carbachol (CCh) (panel E) as a reference (maximum stimulation set to 100%).
Figure 2.

Concentration-response curves obtained with a BRET-based assay reporting the recruitment of β-arrestin to M1 receptors stimulated with various concentrations of indicated compounds (iperoxo-derived bitopic ligands (panel A), ACh-compounds (panel B), isoxazole-bitopic ligands (panel C), oxotremorine-M based ligands (panel D), and TMA-derived bitopic compounds (panel E)). β-Arrestin recruitment upon stimulation with saturating concentrations of carbachol (CCh) was set to 100%. Data are normalized to CCh and expressed as the means ± S.E.M. of 3–6 independent experiments performed in triplicate.
ACh-Derived Bitopic Ligands
In analogy to the iperoxo-based compounds, we characterized a set of ACh-derived bitopic ligands with varying linker lengths ranging from 6 to 10 methylene groups (Scheme 1). All bitopic ligands (2-C6, 2-C8, and 2-C10) are partial agonists for G protein activation (Figure 1B) and hardly induce any β-arrestin recruitment (Figure 2B). Only at very high concentrations, i.e., 100 μM, β-arrestin recruitment can be observed with all three compounds. In line with iperoxo-derived bitopic ligands (Figure 1A), potency and efficacy of ACh-based ligands are dependent on linker length. Increasing the linker length from 6 to 8 (and 10) methylene groups significantly increases the potency of 1-C8 (and 1-C10), whereas 1-C10 displays the weakest efficacy of this series of compounds. The overall efficacy of ACh-derived ligands appears to be reduced compared to the iperoxo-derived ligands; however, the structure–activity relationships seem to be rather similar.
Isoxazole-Derived Bitopic Ligands
Next, we used an orthosteric agonist, isoxo,46 which is structurally very similar to iperoxo. The molecule differs by a double bond, which was introduced into the iperoxo ring system, resulting in an aromatic isoxazole ring whose influence on the ligand–receptor interaction was studied. We used isoxo as an orthosteric fragment to synthesize the bitopic ligands 3-C6, 3-C8, and 3-C10 (Scheme 1). In line with the results obtained with iperoxo- and ACh-derived bitopic ligands, all isoxo-based bitopic ligands stimulated G protein activation (Figure 1C). However, they hardly induced any β-arrestin recruitment (Figure 2C), which is in line with ACh-based ligands. 3-C8 (a full agonist) displayed a higher efficacy than 3-C6 (a partial agonist), whereas 3-C10, in line with aforementioned observations (Figure 1A,B), demonstrated the weakest potency and efficacy of the entire series. Full concentration-response curves for β-arrestin recruitment could not be obtained. Nevertheless, the data suggest that the activation profile of the ligands follows that for G protein activation with 3-C8 and 3-C10 being the most and least efficacious ligands, respectively (Figure 2C).
Oxotremorine-M-Derived Bitopic Ligands
In the next set of compounds, another classical non-selective muscarinic agonist, oxotremorine-M, was used as an orthosteric moiety giving rise to the series of bitopic ligands 4-C6, 4-C8, and 4-C10 (Scheme 1). As seen with previous series of bitopic ligands, increasing the linker length from six to eight methylene groups seems to yield better agonists. 4-C8 displays a higher potency and a higher efficacy than 4-C6, whereas a further increase of the linker length decreases the potency and efficacy of 4-C10 (Figure 1D). All compounds only weakly stimulate β-arrestin recruitment at 100 μM and appear to behave similarly (Figure 2D).
TMA-Derived Bitopic Ligands
Lastly, we used the smallest muscarinic agonist described in the literature, i.e., TMA, as a building block for bitopic ligands. We synthesized the minimal-size bitopic ligands 5-C6, 5-C8, and 5-C10 (Scheme 1). In line with all previous data, TMA-based ligands stimulated G protein activation (Figure 1E) in a linker-length dependent manner. Again, the octamethylene linker (5-C8) seems to be more favorable for G protein activation than the hexamethylene linker (5-C6). In contrast to the other series of bitopic ligands described above, the 5-C10 compound shows a higher potency than 5-C6 (Figure 1E). Interestingly, all TMA-based ligands behave as full agonists. The ligand-induced recruitment of β-arrestin 2, determined by a BRET-based assay,51 was very weak for the entire series of ligands and could only be detected for the highest ligand concentration tested throughout the experiments (100 μM; Figure 2E). Overall, based on the experimental data, three general observations can be made: First, the majority of bitopic ligands of the series 1-Cn to 4-Cn are weaker agonists than their parent orthosteric agonists; second, all bitopic ligands show negligible to little efficacy in recruiting β-arrestin; and third, a linker length of eight methylene groups appears to be optimal for G protein activation by all bitopic ligands. This is in line with the recent finding regarding the bitopic agonist iper-8-phth capable of binding to and activating the M1 receptor as a full agonist.53 Noteworthy, shortening of the linker to four methylene groups did not lead to improved G protein activation for the TMA bitopic ligands neither in terms of maximum response nor in potency (Supporting Information Figure S4).
Binding Modes of BQCAd-Based Bitopic Ligands
In order to gain structural insights into binding poses and to link them to the observed activities, we performed computational analysis of selected bitopic ligands with focus on the n-C8 series. Docking to the active M1 receptor resulted in plausible poses for all bitopic ligands. Due to the spatial requirements and the rigidity of the BQCAd building block, the orientation of its bitopic derivatives is highly similar (Figure 3A). In the case of the M2 receptor, bitopic ligands exhibit a fixed position of the orthosteric moiety, which directs the positioning of the allosteric building blocks. Interestingly, our data strongly suggests the opposite for the M1 receptor (Supporting Information Figure S5).54 Since BQCAd is very specific for the M1 allosteric vestibule, it serves as an anchoring point, which controls the positioning of the linker and thereby the position of the orthosteric moiety in the orthosteric binding site.
Figure 3.

Proposed binding modes for BQCAd-derived bitopic ligands at the M1 receptor indicate a distinct position of the allosteric BQCAd moiety, whereas the positioning of the orthosteric building block is more diverse. (A) Superimposition of 1-C8 (green), 2-C8 (salmon), 3-C8 (blue), 4-C8 (dark gray), and 5-C8 (light grey) bound to the M1 receptor. (B) Superimposition of 5-C8 (light gray) and 5-C4 (yellow) bound to the M1 receptor. (C) Dynamic interaction pattern of 5-C8, the most potent compound from the series with the minimal orthosteric building block, derived from a 250 ns MD simulation. Yellow point clouds indicate non-polar contacts, the blue point cloud shows positive ionizable centers, the purple point cloud shows aromatic interactions, and the red point clouds indicate hydrogen bond acceptors. Next to the point clouds, the occurrence frequency and the involved residues are indicated.
With regard to the orthosteric binding pocket, the essential interaction responsible for receptor activation is the charge interaction with D1053.32.55 The positive charge is additionally surrounded by aromatic residues of the tyrosine lid (Y7.39 and Y7.43), forming cation−π interactions. Since the tyrosine lid is flexible to a certain extent and the charge interaction with D1053.32 is not restricted to a distinct geometric position, the position of the positive charge of the ammonium group shows some variance (Figure 3B). Due to the flexibility of the alkyl linker, this essential charge interaction can be observed for all studied bitopic ligands. Both the alkyl linker length and the type of the orthosteric building block control the location of the ammonium group. This might also explain why there is no direct pharmacological correlation between bitopic ligands and their purely orthosteric building blocks (Figures 1 and 2). Since the binding mode is mainly determined by the BQCAd moiety and the linker length, the orthosteric building block cannot necessarily build the same interactions as the purely orthosteric ligands themselves. Interestingly, the compound series with the smallest known orthostere (TMA) shows robust G protein activation, which suggests that only the charge interaction is crucial for G protein activation. Given that 5-C8 shows the highest potency with regard to G protein activation, we analyzed its receptor–ligand interaction by using dynamic pharmacophores.56 The initial binding mode of 5-C8 and the respective interaction pattern were conserved over 250 ns of MD simulation (Figure 3C). The largest movement was observed for the benzyl ring, which shows a more frayed distribution of its non-polar contacts, but this movement occurred at the very beginning of the trajectory. The dynamic interaction pattern indicates a key role of Y179 in the second extracellular loop (ECL2) and W4007.35 for hosting the BQCAd moiety in the allosteric vestibule of the M1 receptor. This result is in accordance with previous mutational studies that highlight key residues for allosteric modulation at the M1 receptor.57−59 Interestingly, the same epitopes have also been shown to control shape and ligand binding properties of the extracellular binding site of the M2 receptor.56,60 Since we observed a surprisingly high potency (e.g., 4-C4) or high efficacy (e.g., 1-C4) for some compounds of the C4 series, we hypothesize that these bitopic ligands might show multiple binding modes as previously reported for some bitopic muscarinic agonists.61
In conclusion, we have presented a novel set of BQCAd-based bitopic M1 receptor agonists comprising five different orthosteric agonists and alkyl linkers of varying lengths. All bitopic ligands are agonists with regard to G protein activation with efficacies ranging from partial to full agonism. On the other hand, only three ligands, i.e., 1-C6, 1-C8, and 3-C8, were able to efficiently recruit β-arrestin in the concentration range sufficient for G protein activation. Molecular modeling studies demonstrate that the overall binding mode of the bitopic ligands is mainly determined by the position of the allosteric BQCAd moiety. Hence, the ligands are “hung up” in the allosteric vestibule of the receptor, and the orthosteric moieties “dangle” into the orthosteric binding site. This may result in a situation where the orthosteric moieties of the bitopic ligands adopt different binding poses as their parent orthosteres alone. This would be in line with our experimental data showing that several bitopic ligands are less potent and efficacious than their respective orthosteric agonists. Moreover, shrinking the orthosteric moiety down to the essential ammonium ion (as in TMA, 5-Cn series) is sufficient to induce a potent and efficacious G protein response; however, these bitopic ligands fail to stimulate β-arrestin recruitment. It seems that a quite voluminous molecular portion having a higher molecular weight than that originating from the quaternary ammonium head at the orthosteric site is one of the prerequisite for β-arrestin 2 recruitment. The finding that the ligands’ binding poses are determined by their allosteric building block may point toward a higher receptor subtype selectivity than that of other bitopic ligands.62 Therefore, the whole set of bitopic ligands represents a useful toolbox of compounds to outline the chemical requirements for modulating the degree of β-arrestin recruitment and G protein activation and thus, facilitating the development of efficacious and selective drug candidates for the treatment of diseases such as Alzheimer and schizophrenia.
Materials and Methods
Materials
A Chinese hamster ovary (CHO) cell line stably expressing hM1 was obtained from Wyeth Research (Princeton, NJ). 96-Well round-bottom and white 96-well plates were purchased from ThermoFisher and from Greiner Bio One, Germany. Dulbecco’s modified Eagle’s medium (DMEM) and phosphate-buffered saline (PBS) were from Sigma (Schnelldorf, Germany). Leibovitz’s L-15 medium (L-15) and Hank’s balanced salt solution (HBSS) were from ThermoFisher (Dreieich, Germany). Fetal calf serum (FCS), trypsin, and geneticin (G418) were from Merck Biochrom (Berlin, Germany). d-Luciferin was purchased as a potassium salt from Pierce (ThermoFisher, Dreieich, Germany) and was dissolved in HBSS at a concentration of 400 mM. Puromycin was obtained from Invivogen (Toulouse, France). [3H]N-Methylscopolamine, 250 μCi (9.25 MBq), was purchased from PerkinElmer (Rodgau, Germany) and polyethylenimine solution (1%, PEI) from Sigma (Schnelldorf, Germany). For chromatographic applications (HPLC, LC–MS), deionized water produced by means of a Milli-Q system (Merck, Darmstadt, Germany) was used. HPLC grade and LC–MS grade solvents were from Sigma-Aldrich (Munich, Germany). Unless stated otherwise, all chemicals were purchased from Sigma-Aldrich (Schnelldorf, Germany), VWR (Darmstadt, Germany), and TCI (Eschborn, Germany) and were used without prior purification.
General Medicinal Chemistry Methods
1H (400.132 MHz) and 13C (100.613 MHz) NMR spectra were recorded using a Bruker AV 400 NMR spectrometer (Bruker Biospin, Ettlingen, Germany). As an internal standard, the signals of the deuterated solvents were used (DMSO-d6: 1H 2.5 ppm, 13C 39.52 ppm; CDCl3: 1H 7.26 ppm, 13C 77.16 ppm). Abbreviations for data quoted are s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; b, broad; dd, doublet of doublets; dt, doublet of triplets; tt, triplet of triplets; and tq, triplet of quartets. Coupling constants (J) are given in Hz. The NMR signals were assigned polarization transfer experiments (DEPT) and two-dimensional experiments, such as 1H-1H correlation (COSY) and 1H-13C-proton-carbon heteronuclear correlation (HMQC, HMBC). TLC analyses were performed on silica gel 60 F254, C18 silica-coated aluminum panels ALUGRAM RP-18W/UV254 and on precoated TLC plates Alox-25/UV254 (Macherey-Nagel, Düren, Germany); the detection was made using UV light at 254 nm, intrinsic fluorescence at 366 nm or with ethanolic KMnO4, Dragendorff reagent, or phosphomolybdic acid ethanolic solution. For classical purification, column chromatography was performed using silica gel with a grain size of 63–200 μm (Merck, Darmstadt, Germany). Flash chromatography on a puriFlash430 system (Interchim, Montluçon, France) was performed using prepacked columns (Interchim, Montluçon, France) with silica gel filling (particle size, 30 or 50 μm) for the normal phase or with C18-silica gel filling (particle size, 15 μm) for the reverse phase. The detection was carried out by means of a UV detector and an evaporative light scattering detector (ELSD). Microwave-assisted reactions were carried out using an MLS-rota PREP or synthWAVE instruments (Milestone). The LC–MS analyses of all the test compounds were performed using a Shimadzu LC-MS-2020 mass spectrometer (Shimadzu Deutschland GmbH, Duisburg, Germany) containing a DGU-20A3R degassing unit, an LC20AB liquid chromatograph, and SPD-20A UV/Vis detector, and an LC/MSD ion trap (Agilent Technologies, Waldbronn, Germany) connected to an Agilent 1100 modular system. A Synergi Fusion-RP (150 mm × 4.6 mm, i.d., 4 μm; Phenomenex Ltd., Aschaffenburg, Germany) column and a gradient consisting of solvent A, water with 0.1% formic acid, and solvent B, MeOH with 0.1% formic acid, were used. Solvent B was increased from 0 to 90% in 13 min then decreased to 5% in 1 min and 5% for 4 min. The method was run with a flow rate of 1.0 mL/min and UV detection at 254 nm. All compounds were found to have a purity of ≥95%. Mass spectra were recorded in ESI-positive mode, and the data are reported as the mass-to-charge ratio (m/z) of the corresponding positively charged molecular ions. Detailed syntheses and spectral data of intermediate and target compounds are reported in the Supporting Information (Data S1).
Cell Culture
Chinese hamster ovary cells (CHO) stably expressing the hM1 receptor (CHO-hM1 cells) were cultured in Ham’s nutrient mixture F-12 (HAM- F12) supplemented with 10% (v/v) FCS (FCS), 100 U/mL penicillin, 100 μg/mL streptomycin, 0.2 mg/mL G418, and 2 mM l-glutamine at 37 °C in a 5% CO2-humidified atmosphere. HEK293T cells stably cotransfected with the human M1 receptor and the Gαq-PLC-β3 sensor51 were kindly provided by Timo Littmann (University of Regensburg). Cells were cultured in DMEM containing 10% FCS (full medium) at 37 °C in a water-saturated atmosphere containing 5% CO2 as reported previously.51 HEK293T cells for BRET assays were cultured in a DMEM high-glucose medium containing 10% FCS, 2 mM glutamine, 500 units × mL–1 penicillin, and 0.5 mg × mL–1 streptomycin.
Split-Luciferase Bioluminescence Assay
HEK293T cells stably cotransfected with the human M1 receptor and the Gαq-PLC-β3 sensor51 were detached from a 75 cm2 flask by trypsinization and centrifuged (700g for 5 min). The pellet was resuspended in an assay medium consisting of L-15 with 5% FCS, and the density of the suspension was adjusted to 1.25 × 106 cells/mL. Then, 80 μL of this suspension was seeded into each well of a 96-well plate, and the plate was subsequently incubated at 37 °C in a humidified atmosphere (without additional CO2) overnight. On the next day, 10 μL of 10 mM d-luciferin (Pierce) was added to the cells, and the plate was transferred into a prewarmed microplate luminescence reader (Mithras LB 940 Multimode Microplate Reader, Berthold Technologies, Bad Wildbad, Germany). The cells were allowed to equilibrate inside the reader for 10 min before basal luminescence was determined by recording luminescence for the entire plate ten times with an integration time of 0.5 s per well. In the meantime, serial dilutions of agonists were prepared. The resulting solutions were also prewarmed to 37 °C and subsequently added to the cells. Thereafter, luminescence was recorded for 15 plate repeats amounting to a time period of 20 min. Negative controls (solvent) and positive controls (reference full agonist, carbachol (hM1R)) eliciting a maximal response (100%) were included for subsequent normalization of the data. After acquisition of the data, the peak luminescence intensities obtained after stimulation were used for quantitative analysis using GraphPad Software (San Diego, CA, USA).
β-Arrestin 2 Recruitment Assay
β-Arrestin 2 recruitment was determined by measuring BRET by using the NanoBRET system.63 BRET was measured between a full length human M1 receptor N-terminally carrying a FLAG-tag and C-terminally carrying a nanoluciferase. The β-arrestin 2 was N-terminally modified with a HALO-tag and labeled with a HALO-618 fluorescent ligand (Promega, Mannheim, Germany). Therefore, 1 × 106 HEK293T cells were seeded in a 6 cm dish and after 20 h transiently transfected with 1 μg of the receptor, 2 μg of β-arrestin 2, and 1 μg of human GRK2 with the Effectene transfection reagent in accordance to the user manual. Twenty hours after transfection, cells were transferred from 6-well plates to 96-well plates. Cells were counted, and 20,000 cells per well were seeded into white 96-well plates (Brand GmbH & Co. KG, Wertheim, Germany). The next day, BRET was measured using the Synergy Neo2 Hybrid Multi-Mode Reader (BioTek Instruments GmbH, Bad Friedrichshall, Germany), and the BRET ratio was corrected against buffer conditions. The highest ligand concentration tested was 100 μM due to limited solubility in the buffer used for BRET assays.
Computational Analysis Methods
All receptor-ligand docking experiments were carried out with CCDC’s software GOLD version 5.1.64 A previously reported active homology model of the M1 receptor, which is in accordance with the available active M1 receptor cryo-EM structure, was used for docking.60 All residues of the receptor core region and the extracellular loop regions were defined as potential binding sites. Default settings were applied for receptor–ligand docking using GoldScore as a primary scoring function. All docking poses and receptor–ligand interactions were analyzed with LigandScout 4.2,65 using a 3D pharmacophore approach. The molecular dynamics simulations were carried out on GPUs (Nvidia RTX 2080 Ti) at the Freie Universität Berlin with Desmond 2018-3 following the previously published procedure.56 Subsequently, the MD trajectories were analyzed with software VMD66 and a dynamic pharmacophore approach (dynophores) implemented in the LigandScout framework.56,65,67
Data Treatment
The binding data from individual experiments were analyzed by computer-aided nonlinear regression analysis using Prism 5 (GraphPad Software, San Diego, CA, USA). All sigmoidal concentration-response curves were obtained by fitting three-parameter (Hill slope constrained to 1) nonlinear regression curves (GraphPad Prism, San Diego, CA, USA). The BRET raw data were analyzed and corrected in Excel (Microsoft Corporation, WA, USA) and plotted in GraphPad Prism 7 (GraphPad Software, San Diego, CA, USA).
Acknowledgments
Special thanks goes to Dr. Timo Littmann for the kind concession of the cells expressing the Split-Luc construct.
Glossary
ABBREVIATIONS
- ACh
acetylcholine
- AD
Alzheimer disease
- BQCAd
benzyl quinolone carboxylic acid derivative
- CCh
carbachol
- CHO
Chinese hamster ovary
- DMEM
Dulbecco’s modified Eagle’s medium
- FBS
fetal bovine serum
- GPCR
G protein-coupled receptor
- [3H]NMS
N-[3H]methylscopolamine
- mAChR
muscarinic acetylcholine receptor
- PAM
positive allosteric modulator
- PLC
phospholipase C
- RLB
radioligand binding
- SARs
structure–activity relationships
- 7TMR
seven transmembrane receptor
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c04220.
Synthesis and experimental data of target compounds, maximum agonist effect (Emax) and potency (pEC50) of bitopic compounds in Gα/PLC-β3 split-luciferase complementation assays, radioligand binding experiments, additional G protein activation experiments for n-C4 compounds, and a comparison of binding modes of bitopic ligands at the M1 and the M2 receptor (PDF)
Author Contributions
D.V., M.K., A.B., M.B., G.W., C.H., and U.H. participated in research design. D.V., M.K., R.M., and M.B. conducted experiments. D.V. and M.K. contributed to new reagents or analytic tools and performed data analysis. The manuscript was written by D.V., M.B., A.B., and U.H. All authors have given approval to the final version of the manuscript.
D.V. and M.K. were supported by the international doctoral college “Receptor Dynamics: Emerging Paradigms for Novel Drugs” funded within the framework of the Elite Network of Bavaria (grant number K-BM-2013-247). M.B. was supported by the Deutsche Forschungsgemeinschaft (German Research Foundation – DFG 407626949). A.B. acknowledges funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) through SFB1423, project number 421152132, subproject C05. This publication was supported by the Open Access Publication Fund of the University of Wuerzburg.
The authors declare no competing financial interest.
Supplementary Material
References
- Bonner T. I.; Buckley N. J.; Young A. C.; Brann M. R. Identification of a family of muscarinic acetylcholine receptor genes. Science 1987, 237, 527–532. 10.1126/science.3037705. [DOI] [PubMed] [Google Scholar]
- Bonner T. I. The molecular basis of muscarinic receptor diversity. Trends Neurosci. 1989, 12, 148–151. 10.1016/0166-2236(89)90054-4. [DOI] [PubMed] [Google Scholar]
- Wess J. Molecular biology of muscarinic acetylcholine receptors. Crit. Rev. Neurobiol. 1996, 10, 69–99. 10.1615/critrevneurobiol.v10.i1.40. [DOI] [PubMed] [Google Scholar]
- Thal D. M.; Sun B.; Feng D.; Nawaratne V.; Leach K.; Felder C. C.; Bures M. G.; Evans D. A.; Weis W. I.; Bachhawat P.; et al. Crystal structures of the M1 and M4 muscarinic acetylcholine receptors. Nature 2016, 531, 335–340. 10.1038/nature17188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubík J.; Zimčík P.; Randáková A.; Fuksová K.; El-Fakahany E. E.; Doležal V. Molecular mechanisms of methoctramine binding and selectivity at muscarinic acetylcholine receptors. Mol. Pharmacol. 2014, 86, 180–192. 10.1124/mol.114.093310. [DOI] [PubMed] [Google Scholar]
- Bradley S. R.; Lameh J.; Ohrmund L.; Son T.; Bajpai A.; Nguyen D.; Friberg M.; Burstein E. S.; Spalding T. A.; Ott T. R.; et al. AC-260584, an orally bioavailable M(1) muscarinic receptor allosteric agonist, improves cognitive performance in an animal model. Neuropharmacology 2010, 58, 365–373. 10.1016/j.neuropharm.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Dror R. O.; Green H. F.; Valant C.; Borhani D. W.; Valcourt J. R.; Pan A. C.; Arlow D. H.; Canals M.; Lane J. R.; Rahmani R.; et al. Structural basis for modulation of a G-protein-coupled receptor by allosteric drugs. Nature 2013, 503, 295–299. 10.1038/nature12595. [DOI] [PubMed] [Google Scholar]
- Gnagey A. L.; Seidenberg M.; Ellis J. Site-directed mutagenesis reveals two epitopes involved in the subtype selectivity of the allosteric interactions of gallamine at muscarinic acetylcholine receptors. Mol. Pharmacol. 1999, 56, 1245–1253. 10.1124/mol.56.6.1245. [DOI] [PubMed] [Google Scholar]
- Venkatakrishnan A. J.; Deupi X.; Lebon G.; Tate C. G.; Schertler G. F.; Babu M. M. Molecular signatures of G-protein-coupled receptors. Nature 2013, 494, 185–194. 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- Spalding T. A.; Trotter C.; Skjærbæk N.; Messier T. L.; Currier E. A.; Burstein E. S.; Li D.; Hacksell U.; Brann M. R. Discovery of an Ectopic Activation Site on the M1Muscarinic Receptor. Mol. Pharmacol. 2002, 61, 1297–1302. 10.1124/mol.61.6.1297. [DOI] [PubMed] [Google Scholar]
- Buckley N. J.; Bonner T. I.; Brann M. R. Localization of a family of muscarinic receptor mRNAs in rat brain. J. Neurosci. 1988, 8, 4646–4652. 10.1523/JNEUROSCI.08-12-04646.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levey A. I.; Edmunds S. M.; Koliatsos V.; Wiley R. G.; Heilman C. J. Expression of m1-m4 muscarinic acetylcholine receptor proteins in rat hippocampus and regulation by cholinergic innervation. J. Neurosci. 1995, 15, 4077–4092. 10.1523/JNEUROSCI.15-05-04077.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anagnostaras S. G.; Murphy G. G.; Hamilton S. E.; Mitchell S. L.; Rahnama N. P.; Nathanson N. M.; Silva A. J. Selective cognitive dysfunction in acetylcholine M1 muscarinic receptor mutant mice. Nat. Neurosci. 2003, 6, 51–58. 10.1038/nn992. [DOI] [PubMed] [Google Scholar]
- Kruse A. C.; Kobilka B. K.; Gautam D.; Sexton P. M.; Christopoulos A.; Wess J. Muscarinic acetylcholine receptors: novel opportunities for drug development. Nat. Rev. Drug Discovery 2014, 13, 549–560. 10.1038/nrd4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodick N. C.; Offen W. W.; Shannon H. E.; Satterwhite J.; Lucas R.; van Lier R.; Paul S. M. The selective muscarinic agonist xanomeline improves both the cognitive deficits and behavioral symptoms of Alzheimer disease. Alzheimer Dis. Assoc. Disord. 1997, 11 Suppl 4, S16–S22. [PubMed] [Google Scholar]
- Bartus R. T. On neurodegenerative diseases, models, and treatment strategies: Lessons learned and lessons forgotten a generation following the cholinergic hypothesis. Exp. Neurol. 2000, 163, 495–529. 10.1006/exnr.2000.7397. [DOI] [PubMed] [Google Scholar]
- Fisher A. M1 muscarinic agonists target major hallmarks of Alzheimer’s disease--the pivotal role of brain M1 receptors. Neurodegener. Dis. 2008, 5, 237–240. 10.1159/000113712. [DOI] [PubMed] [Google Scholar]
- Langmead C. J.; Watson J.; Reavill C. Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol. Ther. 2008, 117, 232–243. 10.1016/j.pharmthera.2007.09.009. [DOI] [PubMed] [Google Scholar]
- Bender A. M.; Jones C. K.; Lindsley C. W. Classics in Chemical Neuroscience: Xanomeline. ACS Chem. Neurosci. 2017, 8, 435–443. 10.1021/acschemneuro.7b00001. [DOI] [PubMed] [Google Scholar]
- Lange H. S.; Cannon C. E.; Drott J. T.; Kuduk S. D.; Uslaner J. M. The M1 Muscarinic Positive Allosteric Modulator PQCA Improves Performance on Translatable Tests of Memory and Attention in Rhesus Monkeys. J. Pharmacol. Exp. Ther. 2015, 355, 442–450. 10.1124/jpet.115.226712. [DOI] [PubMed] [Google Scholar]
- Puri V.; Wang X.; Vardigan J. D.; Kuduk S. D.; Uslaner J. M. The selective positive allosteric M1 muscarinic receptor modulator PQCA attenuates learning and memory deficits in the Tg2576 Alzheimer’s disease mouse model. Behav. Brain Res. 2015, 287, 96–99. 10.1016/j.bbr.2015.03.029. [DOI] [PubMed] [Google Scholar]
- Bradley S. J.; Bourgognon J.-M.; Sanger H. E.; Verity N.; Mogg A. J.; White D. J.; Butcher A. J.; Moreno J. A.; Molloy C.; Macedo-Hatch T.; et al. M1 muscarinic allosteric modulators slow prion neurodegeneration and restore memory loss. J. Clin. Invest. 2017, 127, 487–499. 10.1172/JCI87526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardigan J. D.; Cannon C. E.; Puri V.; Dancho M.; Koser A.; Wittmann M.; Kuduk S. D.; Renger J. J.; Uslaner J. M. Improved cognition without adverse effects: Novel M1 muscarinic potentiator compares favorably to donepezil and xanomeline in rhesus monkey. Psychopharmacology 2015, 232, 1859–1866. 10.1007/s00213-014-3813-x. [DOI] [PubMed] [Google Scholar]
- Bock A.; Schrage R.; Mohr K. Allosteric modulators targeting CNS muscarinic receptors. Neuropharmacology 2018, 427. 10.1016/j.neuropharm.2017.09.024. [DOI] [PubMed] [Google Scholar]
- Sako Y.; Kurimoto E.; Mandai T.; Suzuki A.; Tanaka M.; Suzuki M.; Shimizu Y.; Yamada M.; Kimura H. TAK-071, a novel M1 positive allosteric modulator with low cooperativity, improves cognitive function in rodents with few cholinergic side effects. Neuropsychopharmacology 2019, 44, 950–960. 10.1038/s41386-018-0168-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurimoto E.; Matsuda S.; Shimizu Y.; Sako Y.; Mandai T.; Sugimoto T.; Sakamoto H.; Kimura H. An Approach to Discovering Novel Muscarinic M1 Receptor Positive Allosteric Modulators with Potent Cognitive Improvement and Minimized Gastrointestinal Dysfunction. J. Pharmacol Exp. Ther. 2018, 364, 28–37. 10.1124/jpet.117.243774. [DOI] [PubMed] [Google Scholar]
- Bock A.; Mohr K. Dualsteric GPCR targeting and functional selectivity: The paradigmatic M2 muscarinic acetylcholine receptor. Drug Discovery Today: Technol. 2013, 10, e245–e252. 10.1016/j.ddtec.2012.12.003. [DOI] [PubMed] [Google Scholar]
- Lane J. R.; Sexton P. M.; Christopoulos A. Bridging the gap: bitopic ligands of G-protein-coupled receptors. Trends Pharmacol. Sci. 2013, 34, 59–66. 10.1016/j.tips.2012.10.003. [DOI] [PubMed] [Google Scholar]
- de Amici M.; Dallanoce C.; Holzgrabe U.; Tränkle C.; Mohr K. Allosteric ligands for G protein-coupled receptors: a novel strategy with attractive therapeutic opportunities. Med. Res. Rev. 2010, 30, 463–549. 10.1002/med.20166. [DOI] [PubMed] [Google Scholar]
- Smith J. S.; Lefkowitz R. J.; Rajagopal S. Biased signalling: From simple switches to allosteric microprocessors. Nat. Rev. Drug Discovery 2018, 243. 10.1038/nrd.2017.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootten D.; Christopoulos A.; Marti-Solano M.; Babu M. M.; Sexton P. M. Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 2018, 19, 638–653. 10.1038/s41580-018-0049-3. [DOI] [PubMed] [Google Scholar]
- Kuduk S. D.; Di Marco C. N.; Saffold J. R.; Ray W. J.; Ma L.; Wittmann M.; Koeplinger K. A.; Thompson C. D.; Hartman G. D.; Bilodeau M. T.; et al. Identification of a methoxynaphthalene scaffold as a core replacement in quinolizidinone amide M1 positive allosteric modulators. Bioorg. Med. Chem. Lett. 2014, 24, 1417–1420. 10.1016/j.bmcl.2014.01.012. [DOI] [PubMed] [Google Scholar]
- Kuduk S. D.; Chang R. K.; Di Marco C. N.; Ray W. J.; Ma L.; Wittmann M.; Seager M. A.; Koeplinger K. A.; Thompson C. D.; Hartman G. D.; et al. Quinolizidinone carboxylic acids as CNS penetrant, selective m1 allosteric muscarinic receptor modulators. ACS Med. Chem. Lett. 2010, 1, 263–267. 10.1021/ml100095k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L.; Seager M. A.; Wittmann M.; Jacobson M.; Bickel D.; Burno M.; Jones K.; Graufelds V. K.; Xu G.; et al. Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 15950–15955. 10.1073/pnas.0900903106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrage R.; Seemann W. K.; Klöckner J.; Dallanoce C.; Racké K.; Kostenis E.; de Amici M.; Holzgrabe U.; Mohr K. Agonists with supraphysiological efficacy at the muscarinic M2 ACh receptor. Br. J. Pharmacol. 2013, 169, 357–370. 10.1111/bph.12003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallanoce C.; de Amici M.; Barocelli E.; Bertoni S.; Roth B. L.; Ernsberger P.; de Micheli C. Novel oxotremorine-related heterocyclic derivatives: Synthesis and in vitro pharmacology at the muscarinic receptor subtypes. Bioorg. Med. Chem. 2007, 15, 7626–7637. 10.1016/j.bmc.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Messerer R.; Kauk M.; Volpato D.; Alonso Canizal M. C.; Klöckner J.; Zabel U.; Nuber S.; Hoffmann C.; Holzgrabe U. FRET Studies of Quinolone-Based Bitopic Ligands and their Structural Analogues at the Muscarinic M1 Receptor. ACS Chem. Biol. 2017, 833. 10.1021/acschembio.6b00828. [DOI] [PubMed] [Google Scholar]
- Chen X.; Klöckner J.; Holze J.; Zimmermann C.; Seemann W. K.; Schrage R.; Bock A.; Mohr K.; Tränkle C.; Holzgrabe U.; et al. Rational design of partial agonists for the muscarinic m1 acetylcholine receptor. J. Med. Chem. 2015, 58, 560–576. 10.1021/jm500860w. [DOI] [PubMed] [Google Scholar]
- Agnetta L.; Kauk M.; Canizal M. C. A.; Messerer R.; Holzgrabe U.; Hoffmann C.; Decker M. A Photoswitchable Dualsteric Ligand Controlling Receptor Efficacy. Angew. Chem., Int. Ed. 2017, 56, 7282–7287. 10.1002/anie.201701524. [DOI] [PubMed] [Google Scholar]
- Kennedy R. H.; Wyeth R. P.; Gerner P.; Liu S.; Fontenot H. J.; Seifen E. Tetramethylammonium is a muscarinic agonist in rat heart. Am. J. Physiol. 1995, 268, C1414–C1417. 10.1152/ajpcell.1995.268.6.C1414. [DOI] [PubMed] [Google Scholar]
- Zakharov S. I.; Overholt J. L.; Wagner R. A.; Harvey R. D. Tetramethylammonium activation of muscarinic receptors in cardiac ventricular myocytes. Am. J. Physiol. 1993, 264, C1625–C1630. 10.1152/ajpcell.1993.264.6.C1625. [DOI] [PubMed] [Google Scholar]
- Gould R. G.; Jacobs W. A. The Synthesis of Certain Substituted Quinolines and 5,6-Benzoquinolines. J. Am. Chem. Soc. 1939, 61, 2890–2895. 10.1021/ja01265a088. [DOI] [Google Scholar]
- Niedermeier S.; Singethan K.; Rohrer S. G.; Matz M.; Kossner M.; Diederich S.; Maisner A.; Schmitz J.; Hiltensperger G.; Baumann K.; et al. A small-molecule inhibitor of Nipah virus envelope protein-mediated membrane fusion. J. Med. Chem. 2009, 52, 4257–4265. 10.1021/jm900411s. [DOI] [PubMed] [Google Scholar]
- Microwave assisted Gould-Jacob reaction: Sinthesis of 4-quinolones under solvent free reactions.
- Disingrini T.; Muth M.; Dallanoce C.; Barocelli E.; Bertoni S.; Kellershohn K.; Mohr K.; de Amici M.; Holzgrabe U. Design, synthesis, and action of oxotremorine-related hybrid-type allosteric modulators of muscarinic acetylcholine receptors. J. Med. Chem. 2006, 49, 366–372. 10.1021/jm050769s. [DOI] [PubMed] [Google Scholar]
- Dallanoce C.; Conti P.; de Amici M.; de Micheli C.; Barocelli E.; Chiavarini M.; Ballabeni V.; Bertoni S.; Impicciatore M. Synthesis and functional characterization of novel derivatives related to oxotremorine and oxotremorine-M. Bioorg. Med. Chem. 1999, 7, 1539–1547. 10.1016/s0968-0896(99)00107-8. [DOI] [PubMed] [Google Scholar]
- Bebbington A.; Shakeshaft D. An Improved Synthesis of Oxotremorine. J. Med. Chem. 1965, 8, 274–275. 10.1021/jm00326a037. [DOI] [PubMed] [Google Scholar]
- Kloeckner J.; Schmitz J.; Holzgrabe U. Convergent, short synthesis of the muscarinic superagonist iperoxo. Tetrahedron Lett. 2010, 51, 3470–3472. 10.1016/j.tetlet.2010.04.130. [DOI] [Google Scholar]
- Kliewer A.; Gillis A.; Hill R.; Schmiedel F.; Bailey C.; Kelly E.; Henderson G.; Christie M. J.; Schulz S. Morphine-induced respiratory depression is independent of β-arrestin2 signalling. Br. J. Pharmacol. 2020, 177, 2923–2931. 10.1111/bph.15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley S. J.; Molloy C.; Valuskova P.; Dwomoh L.; Scarpa M.; Rossi M.; Finlayson L.; Svensson K. A.; Chernet E.; Barth V. N.; et al. Biased M1-muscarinic-receptor-mutant mice inform the design of next-generation drugs. Nat. Chem. Biol. 2020, 16, 240–249. 10.1038/s41589-019-0453-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littmann T.; Ozawa T.; Hoffmann C.; Buschauer A.; Bernhardt G. A split luciferase-based probe for quantitative proximal determination of Gαq signalling in live cells. Sci. Rep. 2018, 8, 17179. 10.1038/s41598-018-35615-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock A.; Merten N.; Schrage R.; Dallanoce C.; Bätz J.; Klöckner J.; Schmitz J.; Matera C.; Simon K.; Kebig A.; et al. The allosteric vestibule of a seven transmembrane helical receptor controls G-protein coupling. Nat. Commun. 2012, 3, 1044. 10.1038/ncomms2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holze J.; Bermudez M.; Pfeil E. M.; Kauk M.; Bödefeld T.; Irmen M.; Matera C.; Dallanoce C.; de Amici M.; Holzgrabe U.; et al. Ligand-Specific Allosteric Coupling Controls G-Protein-Coupled Receptor Signaling. ACS Pharmacol. Transl. Sci. 2020, 859. 10.1021/acsptsci.0c00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermudez M.; Bock A.; Krebs F.; Holzgrabe U.; Mohr K.; Lohse M. J.; Wolber G. Ligand-Specific Restriction of Extracellular Conformational Dynamics Constrains Signaling of the M2 Muscarinic Receptor. ACS Chem. Biol. 2017, 12, 1743–1748. 10.1021/acschembio.7b00275. [DOI] [PubMed] [Google Scholar]
- Vass M.; Podlewska S.; de Esch I. J. P.; Bojarski A. J.; Leurs R.; Kooistra A. J.; de Graaf C. Aminergic GPCR-Ligand Interactions: A Chemical and Structural Map of Receptor Mutation Data. J. Med. Chem. 2019, 62, 3784–3839. 10.1021/acs.jmedchem.8b00836. [DOI] [PubMed] [Google Scholar]
- Bock A.; Bermudez M.; Krebs F.; Matera C.; Chirinda B.; Sydow D.; Dallanoce C.; Holzgrabe U.; de Amici M.; Lohse M. J.; et al. Ligand Binding Ensembles Determine Graded Agonist Efficacies at a G Protein-coupled Receptor. J. Biol. Chem. 2016, 291, 16375–16389. 10.1074/jbc.M116.735431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui H.; Lazareno S.; Birdsall N. J. Probing of the location of the allosteric site on m1 muscarinic receptors by site-directed mutagenesis. Mol. Pharmacol. 1995, 47, 88–98. [PubMed] [Google Scholar]
- Keov P.; López L.; Devine S. M.; Valant C.; Lane J. R.; Scammells P. J.; Sexton P. M.; Christopoulos A. Molecular mechanisms of bitopic ligand engagement with the M1 muscarinic acetylcholine receptor. J. Biol C.hem. 2014, 289, 23817–23837. 10.1074/jbc.M114.582874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdul-Ridha A.; López L.; Keov P.; Thal D. M.; Mistry S. N.; Sexton P. M.; Lane J. R.; Canals M.; Christopoulos A. Molecular determinants of allosteric modulation at the M1 muscarinic acetylcholine receptor. J. Biol. C.hem. 2014, 289, 6067–6079. 10.1074/jbc.M113.539080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermudez M.; Rakers C.; Wolber G. Structural Characteristics of the Allosteric Binding Site Represent a Key to Subtype Selective Modulators of Muscarinic Acetylcholine Receptors. Mol. Inf. 2015, 34, 526–530. 10.1002/minf.201500025. [DOI] [PubMed] [Google Scholar]
- Design of Hybrid Molecules for Drug Development; Elsevier, 2017. [Google Scholar]
- Bradley S. J.; Molloy C.; Bundgaard C.; Mogg A. J.; Thompson K. J.; Dwomoh L.; Sanger H. E.; Crabtree M. D.; Brooke S. M.; Sexton P. M.; et al. Bitopic Binding Mode of an M1 Muscarinic Acetylcholine Receptor Agonist Associated with Adverse Clinical Trial Outcomes. Mol. Pharmacol. 2018, 93, 645–656. 10.1124/mol.118.111872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machleidt T.; Woodroofe C. C.; Schwinn M. K.; Méndez J.; Robers M. B.; Zimmerman K.; Otto P.; Daniels D. L.; Kirkland T. A.; Wood K. V. NanoBRET--A Novel BRET Platform for the Analysis of Protein-Protein Interactions. ACS Chem. Biol. 2015, 10, 1797–1804. 10.1021/acschembio.5b00143. [DOI] [PubMed] [Google Scholar]
- Jones G.; Willett P.; Glen R. C.; Leach A. R.; Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Wolber G.; Seidel T.; Bendix F.; Langer T. Molecule-pharmacophore superpositioning and pattern matching in computational drug design. Drug Discovery Today 2008, 13, 23–29. 10.1016/j.drudis.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Schaller D.; Šribar D.; Noonan T.; Deng L.; Nguyen T. N.; Pach S.; Machalz D.; Bermudez M.; Wolber G. Next generation 3D pharmacophore modeling. WIREs Comput. Mol. Sci. 2020, 10, e1468 10.1002/wcms.1468. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.