
Extended Data Figure 9: KSR as a co-receptor for binding to trametinib.
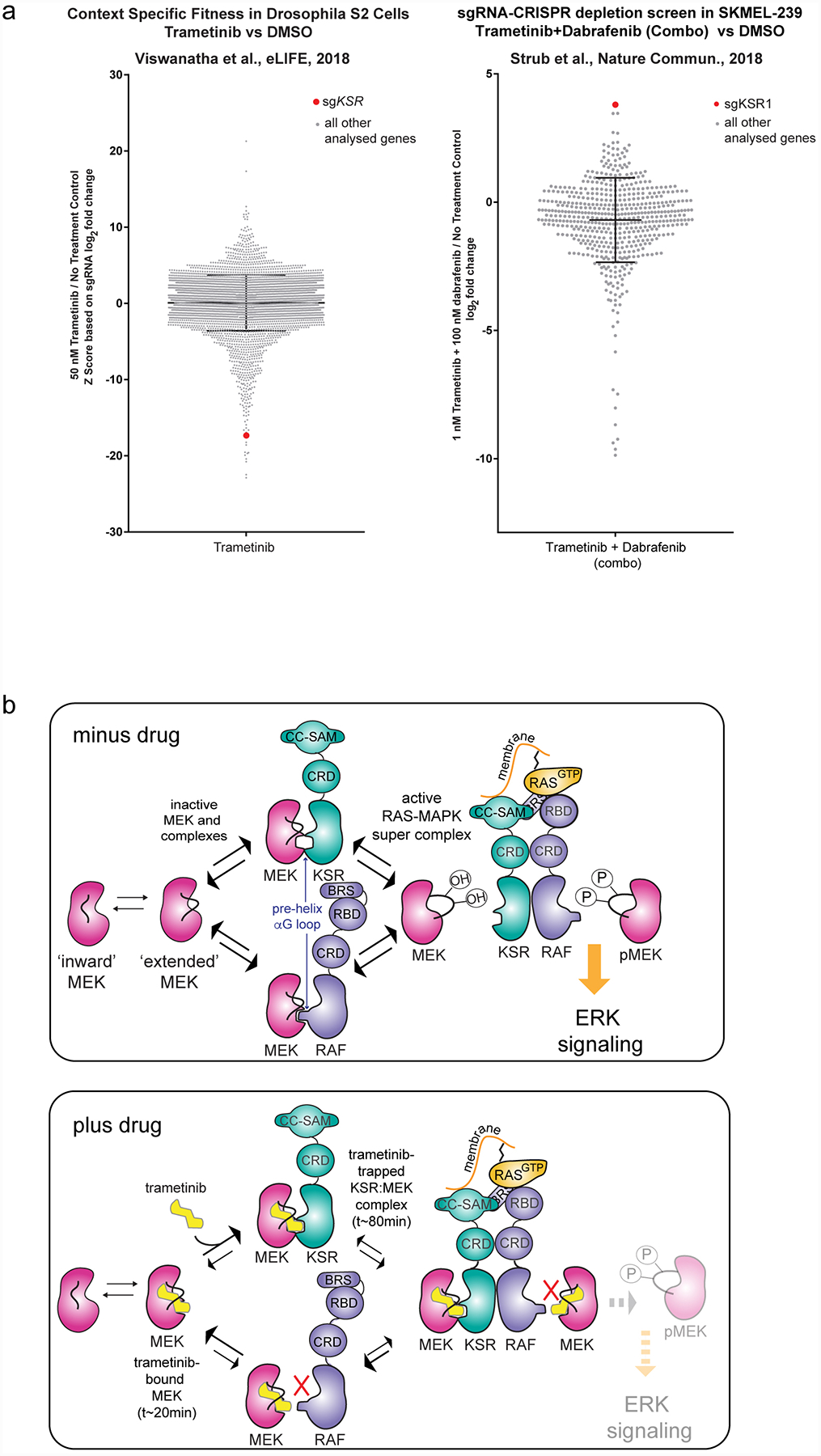
A. Literature data on CRISPR depletion screens highlight strong functional interactions between trametinib and KSR. For example, in a Drosophila cellular fitness model (left43) and a human BRAF V600E mutant cell line (right44), sgRNAs towards KSR generated relative outlier sensitivity or resistance to trametinib or a trametinib+dabrafenib combination, respectively. Raw data from Viswanatha et al. was plotted based on the authors determination of a Z-score for log2-fold change in sgRNA reads for S2 cells treated with trametinib versus a no treatment control (left). Raw data from Strub et al. was plotted based on the authors determination of log2-fold change in sgRNA reads for SKMEL-239 cells treated with a trametinib+dabrafenib combination relative to a no treatment control (right). sgRNAs towards KSR are highlighted as a red dot; all other sgRNAs analysed in the respective studies are shown as grey dots. KSR emerged as a strong outlier beyond the mean plus standard deviation (black cross hairs) of all genes analysed in each respective study. These screens could be re-investigated based on the model that KSR functions as a direct co-receptor for binding to trametinib and MEK.
B. Model for the action of trametinib on KSR:MEK and RAF:MEK complexes. In the absence of drug, MEK activation depends on heterodimerization of both RAF and KSR, with phosphorylation on the sites S218/S222 occurring through active RAF kinases. This model is adapted from structural and biochemical studies in28,29,45,46. Trametinib could down-regulate ERK signaling by impeding direct binding of MEK towards RAF in favor of KSR. In the KSR-bound state of MEK, trametinib would be expected to reside on target for extended periods of time.