

Abstract
The novel 2′-deoxyguanosine analog Entecavir (ETV) is a potent inhibitor of hepatitis B virus (HBV) replication and is recommended for treatment in human immunodeficiency virus type 1 (HIV-1) and HBV-co-infected patients because it had been reported that ETV is HBV-specific. Recent clinical observations, however, have suggested that ETV may indeed demonstrate anti-HIV-1 activity. To investigate this question at a molecular level, kinetic studies were used to examine the interaction of 5′-triphosphate form of ETV with wild type (WT) HIV-1 reverse transcriptase (RT) and the nucleoside reverse transcriptase inhibitor-resistant mutation M184V. Using single turnover kinetic assays, we found that HIV-1 WT RT and M184V RT could use the activated ETV triphosphate metabolite as a substrate for incorporation. The mutant displayed a slower incorporation rate, a lower binding affinity, and a lower incorporation efficiency with the 5′-triphosphate form of ETV compared with WT RT, suggesting a kinetic basis for resistance. Our results are supported by cell-based assays in primary human lymphocytes that show inhibition of WT HIV-1 replication by ETV and decreased susceptibility of the HIV-1 containing the M184V mutation. This study has important therapeutic implications as it establishes ETV as an inhibitor for HIV-1 RT and illustrates the mechanism of resistance by the M184V mutant.
Entecavir (ETV)2 is a guanosine nucleoside analogue approved for the treatment of chronic hepatitis B virus (HBV) infection. The 5′-triphosphate form of ETV (ETVTP) inhibits HBV polymerase. Similar to other nucleoside analogs, upon entry into the cell, metabolic activation of ETV requires phosphorylation by cellular kinases to generate the active triphosphate form of the drug. ETV and the other three other approved nucleoside analogs lamivudine (3TC), adefovir dipivoxil (ADV), and telbivudine also target the HBV polymerase (Pol), which is essential to viral replication. Pol is responsible for converting the plus-strand pre-genomic RNA into a partially double-stranded circular DNA genome during a multi-step process (1–3). ETV-5′ monophosphate is incorporated by HBV Pol resulting in inhibition of the protein-linked priming activity, the reverse transcription of the pre-genomic mRNA, and the DNA-directed DNA synthesis activities of HBV Pol (4). ETV binds to the HBV Pol with high affinity. Its increased affinity over the other nucleoside reverse transcriptase inhibitors (NRTIs) may in part be ascribed to its novel structure (see Fig. 1A) (4). Like the other approved drugs, it is potent in cell culture systems and in humans (5–7). This potency may be attributed to multiple modes of action on several targets essential for viral replication (4).
FIGURE 1. Incorporation of ETVTP by HIV-1 RT.
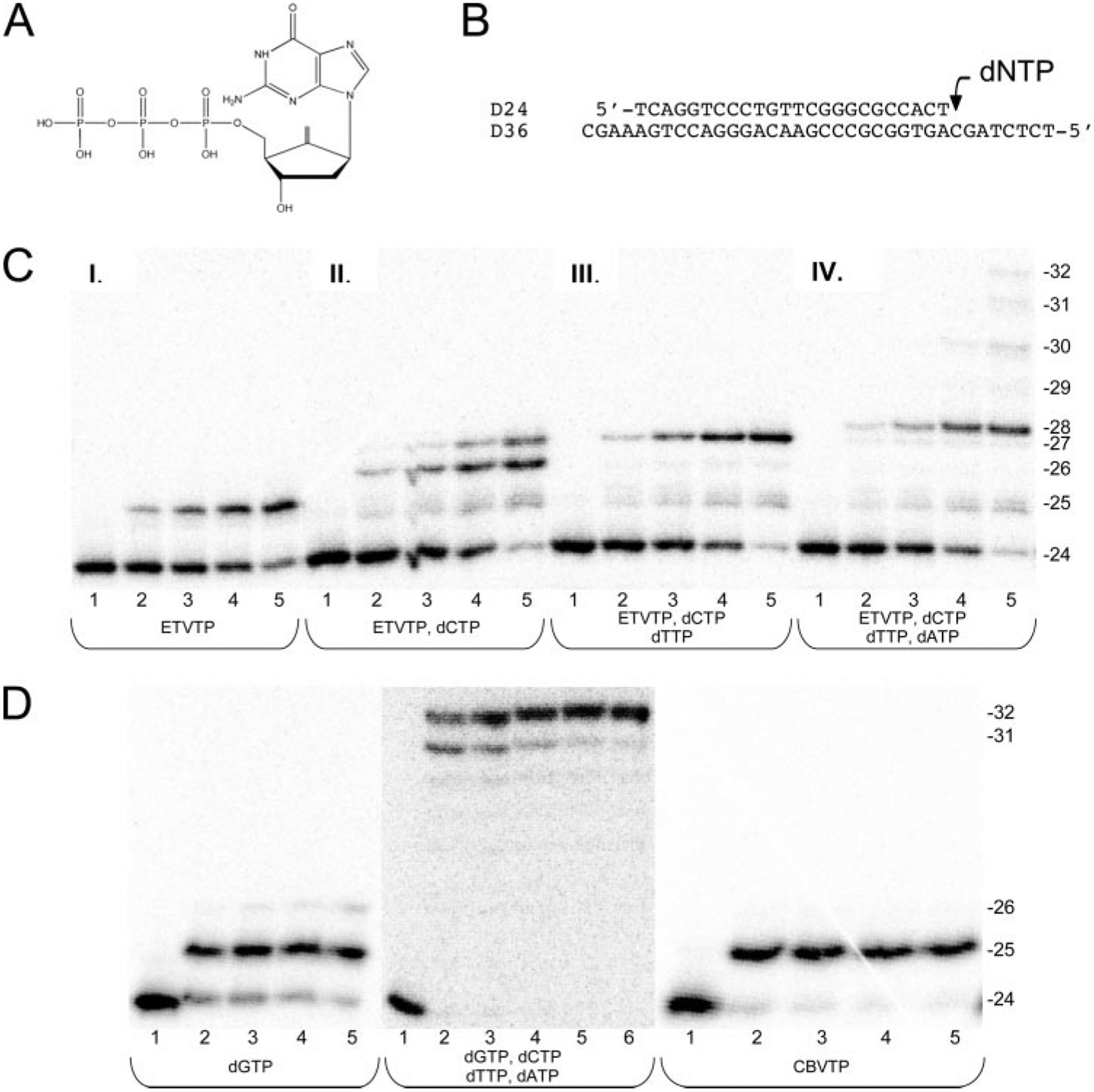
A, the structure of ETVTP. B, the 24-nucleotide (nt) DNA primer annealed to a 36-nt DNA template that was used for all experiments. The arrow denotes the position of the next dNTP to be incorporated. C, products from single-turnover incorporation of ETVTP alone (100 μm) or with the natural dNTPs (100 μm of each) into the D24/D36 primer/template (50 nm) by WT RT (250 nm). Lanes represent the increasing reaction time; Lane 1, 0 min; lane 2, 1 min; lane 3, 2 min; lane 4, 3 min; lane 5, 5 min; lane 6, 10 min. The sizes of the DNA primer (24-mer) and the extended products are indicated. D, products from single-turnover incorporation of dGTP alone (100 μm), all four natural dNTPs (100 μm each) and CBVTP alone (200 μm) into the D24/D36 primer/template (50 nm) by WT RT (250 nm). Lanes represent the increasing reaction times as described above. The sizes of the DNA primer (24-mer) and the extended products are indicated.
Unlike other nucleoside analogs approved for HBV therapy that are obligate chain terminators of DNA elongation because they lack a 3′-hydroxyl required for nucleotide addition, ETV is a nonobligate chain terminator that has been suggested to stall the HBV Pol at sites two or three residues downstream from dG incorporation sites (4) (see Fig. 1A). Chain termination occurs through the increasing steric constraints that affect the efficiency of subsequent nucleotide additions (8).
Because of its high potency and selectivity, ETV is recommended as a first line treatment in human immunodeficiency virus type 1 (HIV-1) and HBV co-infected patients who do not require anti-HIV therapy (9, 10). Earlier studies suggested that ETV displays no significant activity against HIV in cell culture (5), although recent clinical data have called this observation into question (11). Also, it remains unclear whether ETV could inhibit the HIV-1 reverse transcriptase (RT) similar to other approved HIV-1 NRTIs or fixed conformation nucleoside analogs (12).
Antiviral therapy against HIV-1 includes the nonnucleoside reverse transcriptase inhibitors and the NRTIs. The NRTIs target the HIV-1 RT and act as chain terminators similar to the HBV-approved drugs. They require phosphorylation to become metabolically active and act as substrates for the viral polymerase. They act as obligate chain terminators because of the lack of a 3′-hydroxyl group. 3TC is also approved for use against HIV, whereas adefovir dipivoxil and telbivudine are only approved for HBV infections but can be used for co-infected individuals when treatment of only HBV is needed.
A recent report revealed that during ETV therapy of HIV- and HBV-infected persons, a decline in HIV-1 RNA was observed along with appearance in one subject of the NRTI-resistant mutation M184V in HIV-1 RT (11). In addition, ETV was shown to be a potent, but partial, inhibitor of HIV-1 infectivity in a sensitive in vitro assay and to prevent the accumulation of viral cDNA in recently infected cells (11). This suggests that ETV could potentially inhibit HIV replication and target HIV-1 RT. It is unclear whether HIV-1 RT could incorporate the activated ETV triphosphate metabolite, ETVTP. To date, no studies examining the interaction of ETVTP with HIV-1 RT have been reported. Using a kinetic approach that has been previously employed in our laboratory to study nucleotide analog incorporation, the current study was performed to examine the interaction of ETVTP and HIV-1 RT. We also looked at the ability of the NRTI resistant mutation M184V to affect incorporation of ETVTP. In parallel, the ability of ETV to inhibit WT HIV-1 and HIV-1 harboring the M184V mutation in human peripheral blood mononuclear (PBM) cells was re-examined.
Our results show that HIV-1 RT can use ETVTP as a substrate for incorporation and that ETVTP has an affinity similar to other HIV-1 NRTIs. HIV-1 RT with the M184V mutation can also incorporate ETVTP but with a reduced efficiency due to a slower incorporation rate and a weaker affinity. The data suggest a unique mechanism of inhibition of RT that is examined by molecular modeling studies. This work provides a molecular and kinetic basis for the HIV-1 inhibition seen in patients. The cell-based results showing inhibition of HIV-1 by ETV and decreased susceptibility to the HIV-1 containing the M184V mutation further supports the cell-free enzymatic results.
EXPERIMENTAL PROCEDURES
Expression and Purification of HIV-1 RT—
The WT RT and M184V RT clones were generously provided by Stephen Hughes, Paul Boyer, and Andrea Ferris (Frederick Cancer Research and Development Center, Frederick, MD). N-terminal histidine-tagged heterodimeric p66/p51 reverse transcriptase was purified as previously described (13).
Materials—
ETVTP was synthesized as previously described (14). dGTP was purchased from Amersham Biosciences. The (−)-CBVTP was generously provided by Dr. William B. Parker (Southern Research Institute, Birmingham, AL) and further purified as previously described (14).
Labeling and Annealing of Oligonucleotides—
Primers and templates used for incorporation studies are shown in Fig. 1, and all of the DNA oligonucleotides were synthesized on an Applied Biosystems 380A DNA Synthesizer (Keck DNA synthesis facility, Yale University) and purified using 20% polyacrylamide denaturing gel electrophoresis. Primer/templates were labeled and annealed as previously described (15).
Single-turnover and Pre-steady-state Burst Experiments—
Rapid chemical quench experiments were performed as previously described with a KinTek Instruments model RQF-3 rapid quench-flow apparatus (13, 16). The reactions were carried out by rapid mixing of a solution containing the preincubated complex of 250 nm HIV-1 RT (active site concentration) and 50 nm 5′-labeled DNA/DNA duplex with a solution of 10 mm MgCl2 and varying concentrations of either ETVTP alone or along with dNTPs in the presence of 50 mm Tris-Cl, 50 mm NaCl at pH 7.8 and 37 °C (all concentrations represent the final concentrations after mixing). The reactions were stopped by quenching at various time points with 0.3 m (final) EDTA. Pre-steady-state burst experiments were done under the same conditions as those described for a single-turnover experiment, except the amount of primer template (300 nm final) was in 3-fold excess of enzyme (100 nm active sites final). Products were separated on a 20% polyacrylamide gel and quantitated on a Bio-Rad Molecular Imager FX.
Determination of Kd,DNA for Primer/Template—
The equilibrium dissociation constant (Kd,DNA) for primer/template was determined as described previously (16). Briefly, 200 nm WT HIV-1 RT (active sites) was incubated with varying concentrations of radiolabeled primer/template for 1 min or longer on ice. Reactions were initiated by mixing with a solution containing 10 mm MgCl2 and either 300 μm dGTP or 50 μm ETVTP. All concentrations represent final concentrations after mixing. Reactions were stopped by the addition of 0.3 m EDTA (final) after 0.5 s for dGTP and 1 min for ETVTP incorporation. Burst amplitudes were determined as described under “Single-turnover and Pre-steady-state Burst Experiments” under “Experimental Procedures” and plotted as a function of primer/template concentration.
HIV-1 Antiviral Assay in Primary Human Lymphocytes—
Data Analysis—
Data were fit by nonlinear regression using the program Kaleidagraph (Synergy Software, Reading, PA). Results from single-turnover incorporation experiments were fit to a single exponential equation, [product] = A(1 − exp(−kobsdt)), where A represents the amplitude, and kobsd (k observed) is the first-order rate constant for dNTP or analogue incorporation. Data from pre-steady-state burst experiments were fit to a burst equation, [product] = A(1 − exp(−kobsdt) + ksst), where kss is the observed steady-state rate constant. The dissociation constant (Kd,dNTP) of dNTP or analogue binding to the complex of RT and primer/template during dNMP incorporation was calculated by fitting the observed rate constants at different concentrations of dNTP or analogue to one of the following hyperbolic equation, kobsd = kpol[dNTP]/Kd,dNTP + [dNTP] or kobsd = A[dNTP]/Kd,dNTP + [dNTP], where kpol is the maximum first-order rate constant for dNMP incorporation, and Kd,dNTP is the equilibrium dissociation constant for the interaction of dNTP or analogue with the E•DNA complex. The data from DNA binding experiments were fitted to the quadratic equation [E•DNA] = [(Kd,DNA + E + D) − ((Kd,DNA + E + D)2 − 4ED)1/2], where E is the active enzyme concentration, and D is the total DNA concentration. Errors reported represent the deviation of points from the curve fit generated by Kaleidagraph or were calculated by standard statistical analysis (19).
Modeling—
The modeling efforts were started from the crystal structure of the RT template-primer complex from Huang et al. (20), PDB entry 1RTD. A single molecule of the complex was kept, including the four Mg2+ associated with it. The crystal structure was initially read into the UCSF Chimera program (21), and the incoming dTTP was replaced for dGTP. Also, the corresponding dAMP in template position 5 was replaced for dC while maintaining the hydrogen bonding within them. Then the dCMPs in primer positions +3, +4, and +6 (17, 19, 20) were swapped for dGMP, and dGMPs were swapped for dCMPs in the corresponding template positions (8, 9, and 11) to maintain CG pairing. This completed the first initial model (GA) that features dGMPs in all primer positions from +1 to +7 with minimal amount of disruption from the crystal structure.
Separately, the initial structure of ETV was built and optimized at the HF/6–31g(d) level using GaussView and Gaussian 03 (22). This ETV model was over-laid on the dGTP of the GA model to create E0. The same procedure was used separately on each of the primer dGMPs from +1 to +7 to create models E1 through E7. The creation of the initial models was completed by reading the PDB files created by Chimera in Schrödinger’s Maestro 7.5 (23) and adding the hydrogen atoms needed at protonation states appropriate to pH 7. All the remaining calculations were done using the Impact program (24) through the Maestro interface. For each of the models created in this fashion, the following set of energy minimizations was sequentially run using the truncated Newton-Raphson algorithm with the OPLS_2001 force field, a distance-dependent dielectric ϵ = 4r, and a 12-Å cutoff for non-bonded interactions.
First, the position of all the hydrogen atoms was optimized. Then the newly created ETV-C pair was fully minimized while keeping the rest of the atoms fixed. The next minimization optimized the ETV-C pair plus the pair above it (the preceding nucleotide in the primer chain and the next in the template). In the final optimization all nucleotides in primer positions +1 to +7 and their hydrogen-bonding partners in the template dCMP were allowed to move while keeping the chain atoms (P, O3′, C3′, C4′, C5′, and O5′) fixed. The dNTP and the two closer Mg2+ ions were fully optimized as well during this run.
RESULTS
In the current study we used a pre-steady-state kinetic approach to determine the kinetic parameters of HIV-1 RT incorporation of the active form of the guanosine analog ETVTP (shown in Fig. 1A). The kinetic constants determined were the maximum rate of polymerization (kpol) and the equilibrium dissociation constant (Kd,dNTP). The incorporation efficiency (kpol/Kd,dNTP) was calculated from these values and used to compare WT and M184V mutant forms of the HIV-1 RT enzymes. This analysis was used to assess the ability of HIV-1 RT to use ETVTP as a substrate for incorporation.
A single turnover experiment in which HIV-1 RT is in excess of primer/template was performed to determine whether RT could incorporate ETVTP onto the 24/36-mer DNA/DNA primer-template based on the HIV-1 genomic sequence as shown in Fig. 1B. It was found that HIV-1 RT does indeed incorporate ETVMP similar to other nucleotide substrates as illustrated by the elongation of the DNA 24-mer to a 25-mer (Fig. 1C, panel I). It is incorporated similar to the natural substrate dGTP (Fig. 1D, first panel, left) and the NRTI guanosine analog, carbovir triphosphate (CBVTP), the active triphosphate metabolite of abacavir (Fig. 1D, third panel, right). These data directly demonstrate that even though ETV is used to target HBV Pol, it can also be used as substrate for HIV-1 RT similar to the natural dGTP substrate and CBVTP. This has important consequences for the treatment of individuals co-infected by HBV and HIV.
Unlike other NRTI inhibitors, ETV has a 3′-hydroxyl group that can be used for subsequent nucleotide addition. To further investigate the molecular mechanism of inhibition, the ability of HIV-1 RT to incorporate additional nucleotides after ETV incorporation was assessed (Fig. 1C, panels II–IV). For this experiment, the next nucleotide, dCTP (100 μm), was added to the reaction mixture containing excess HIV-1 RT (250 nm), DNA/DNA duplex (50 nm), ETVTP (100 μm), and MgCl2 (10 mm). We observed that after ETVMP is incorporated, dCMP could also be incorporated using the ETV 3′-hydroxyl group for addition to form a 26-mer opposite the template dG and at longer times (>2 min) 27-mer likely due to a pyrimidine-purine mismatch from misincorporation opposite the template dA (see gel in Fig. 1C, panel II). The inclusion of the natural nucleotide, dTTP, in addition to dCTP in the reaction resulted in the further elongation to form a 27-mer (see gel in Fig. 1C, panel III). The addition of dCTP, dTTP, and dATP with ETVTP showed that multiple nucleotides are incorporated after ETV to form a 28-mer before chain termination occurred (see gel, Fig. 1C, panel IV). Extension to the end of the template would normally occur when all four natural nucleotides are in the reaction to form a 32-mer (Fig. 1D, second panel, middle). These results establish that ETVMP may be incorporated onto the growing DNA strand by RT during reverse transcription. Moreover, when additional nucleotides are added with ETVTP, multiple nucleotides are incorporated after ETV addition before chain termination occurs. This would suggest a new unique mechanism for inhibition of HIV RT and reverse transcription in which there is a delayed chain termination effect that is somewhat similar to that suggested for HBV Pol inhibition in which the HIV-1 RT stalls after multiple incorporations before another ETV is incorporated (4, 8).
To determine the maximum rate of polymerization (kpol), the binding affinity (Kd,ETVTP), and efficiency of ETVTP incorporation (kpol/Kd,ETVTP), the concentration dependence of ETVTP incorporation was examined with single turnover experiments with HIV-1 RT using increasing concentrations of ETVTP (500 nm to 50 μm). The reaction kinetics for single nucleotide incorporation exhibited complex behavior for incorporation of ETVMP by HIV-1 RT. No change in rate was observed with increasing concentrations of ETVTP. Upon removal of contaminating pyrophosphate, there was an increase in observed rate as a function of ETVTP concentration (Fig. 2A). A hyperbolic fit of the observed rate versus ETVTP concentration (Fig. 2B) suggests that WT HIV-1 RT appears to bind ETVTP with a Kd,ETVTP of 2.23 ± 0.67 μm, kpol of 0.107 ± 0.007 s−1, and corresponding efficiency of 0.048 μm−1 s−1 (see Table 1). The natural nucleotide dGTP is incorporated at a faster rate, kpol of 18.3 ± 1.30 s−1, and binds with a greater affinity, Kd,dGTP of 1.76 ± 0.51 μm, than ETVTP, resulting in a greater efficiency of incorporation of 10.4 ± 3.11 μm−1 s−1. The guanosine analog CBVTP, an effective inhibitor of HIV-1 RT, is also not as efficiently incorporated as dGTP. In comparison to ETVTP, the binding affinity for CBVTP is weaker (Kd,CBVTP of 27.8 ± 7.68 μm). However, the rate of incorporation is much faster (kpol of 2.26 ± 0.24 s−1, Table 1), resulting in a 2-fold higher efficiency.
FIGURE 2. Concentration dependence of the observed rate on ETVTP concentration for WT HIV-1 RT.
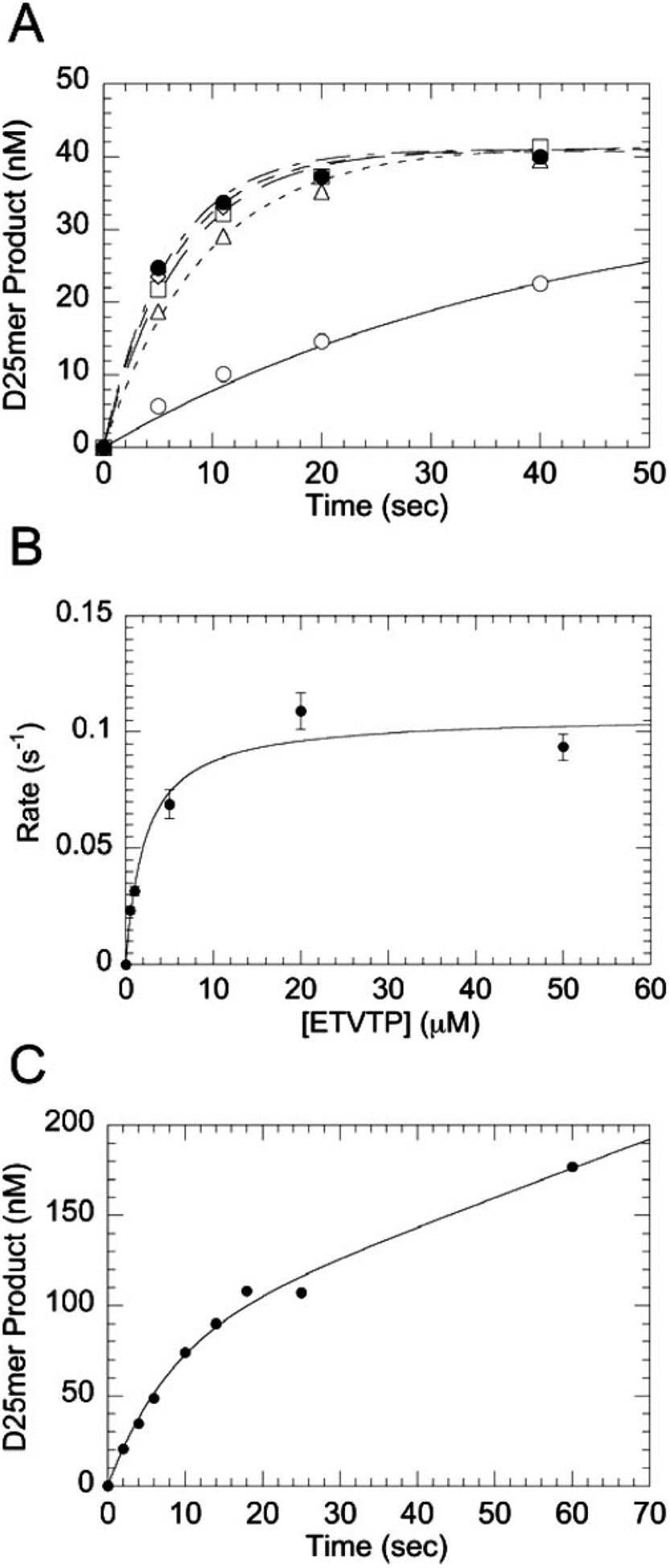
A, single-turnover incorporation was measured by mixing a preincubated solution of RT (250 nm) and DNA/DNA primer/template (50 nm) with various concentrations of ETVTP (curves displayed represent 0.5 μm (○), 1 μm (□), 5 μm (◇), 20 μm (●) and 50 μm (△)) and MgCl2 (10 mm) at 37 °C (all concentrations are final after mixing). B, Kd,ETVTP curve was generated from all ETVTP concentrations tested (500 nm to 50 μm) where the observed rate (kobsd) was plotted against ETVTP concentration. The fit to the data gives an equilibrium binding constant (Kd,ETVTP) of 2.23 ± 0.67 μm and a maximum rate of incorporation (kpol) of 0.107 ± 0.007 s−1. C, pre-steady-state burst kinetics of incorporation of ETVMP into a DNA/DNA primer/template by WT RT were measured by mixing a preincubated solution of RT (100 nm) and primer/template (300 nm) with ETVTP (50 μm) and Mg2+ (10 mm) under rapid quench conditions (all concentrations are final after mixing). The reaction was quenched at indicated times and analyzed by 20% sequencing gel electrophoresis. The solid line represents a fit to a burst equation with an amplitude (A) equal to 79 ± 12 nm, an observed first-order rate constant for the burst phase (kobsd) equal to 0.125 ± 0.031 s−1, and an observed rate for the linear phase (kss) equal to 0.020 ± 0.006 s−1.
TABLE 1.
Enzyme assay showing effect of ETVTP on HIV RT
| HIV RT | dNTP | kpol | Kd,dNTP | kpol/Kd,dNTP |
|---|---|---|---|---|
| s−1 | μM | μM−1s−1 | ||
| WT | dGTP | 18.3 ± 1.30 | 1.76 ± 0.51 | 10.4 ± 3.11 |
| ETVTP | 0.107 ± 0.007 | 2.23 ± 0.67 | 0.048 ± 0.015 | |
| CBVTP | 2.26 ± 0.24 | 27.8 ± 7.68 | 0.081 ± 0.024 | |
| M184V | dGTP | 34.4 ± 3.36 | 5.93 ± 2.04 | 5.8 ± 2.1 |
| ETVTP | 0.019 ± 0.002 | 27.8 ± 6.12 | 0.0007 ± 0.0002 | |
| CBVTP | 0.887 ± 0.078 | 14.1 ± 3.9 | 0.063 ± 0.018 |
A pre-steady-state burst was done to determine whether the rate-limiting step follows chemistry similar to a natural nucleotide. A pre-steady-state burst of product formation was observed for ETVMP incorporation by WT RT (Fig. 2C). The presence of a pre-steady-state burst during incorporation of an analog suggests that it is being incorporated by a comparable kinetic mechanism as natural nucleotides. Slow chemical catalysis and product release, however, were observed with the burst phase (kobsd) equal to 0.125 ± 0.031 s−1 and an observed rate for the linear phase (kss) equal to 0.020 ± 0.006 s−1. A titration of primer/template with WT HIV-1 RT was performed using either dGTP or ETVTP as the substrate for nucleotide incorporation (Fig. 3). A significant difference was noted in the dissociation constant for the primer/template when using 50 μm ETVTP as the substrate (Kd,DNA of 127.5 ± 4.9 nm) versus the natural substrate dGTP (Kd,DNA of 31.3 ± 9.3 nm) indicating that the presence of ETVMP alters the DNA affinity.
FIGURE 3. Effect of ETVTP versus dGTP incorporation on primer/template binding to WT HIV-1 RT.
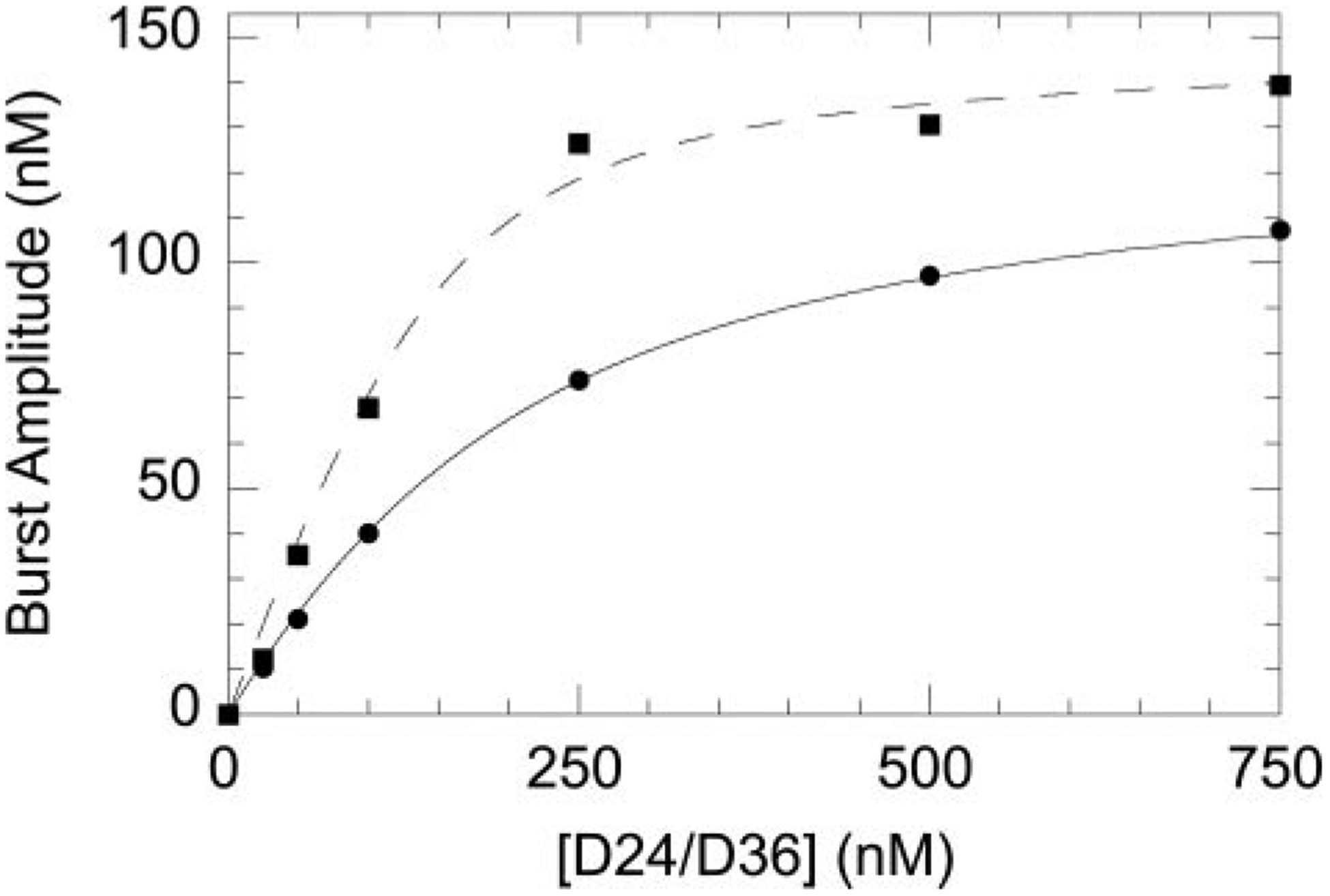
Burst amplitudes were measured at varying concentrations of D24/D36 using dGTP (■) or ETVTP (●) as the substrate for incorporation. Data were fit to the quadratic equation (see “Experimental Procedures”). The Kd,DNA for D24/D36 during dGTP incorporation was 31.3 ± 9.3 nm, whereas during ETVTP incorporation the Kd,DNA was 127.5 ± 4.9 nm.
The appearance of M184V in subjects during ETV therapy (11) suggests that viruses with the M184V mutation may be resistant to ETV. In single-round assays, infection of primary CD4+ T cells by the M184V virus is not inhibited by ETV (11). To establish a kinetic basis for M184V resistance to ETV, we determined the kinetic constants for ETVTP incorporation by M184V mutant HIV-1 RT (Table 1). As illustrated in Table 1, M184V has a Kd,ETVTP of 27.8 ± 6.12 μm and a kpol of 0.019 ± 0.002 s−1. This indicates that compared with WT RT, the RT of M184V binds to ETV with a lower affinity and at a slower rate, resulting in an 68-fold lower efficiency of incorporation. The incorporation of dGTP with M184V RT is only 2-fold less efficient than WT RT, and the efficiency for CBVTP is similar (Table 1). A similar reduction in efficiency was noted for HIV-RT containing the M184I mutation.3 This mutation is also selected by l-oxathiolane nucleosides such as 3TC. The effect of ETV against WT and cloned M184V HIV-1 in human PBM cells was also re-examined. It was observed that ETV was effective in inhibiting WT with an EC50 of 1.8 μm on day 6 after infection. This is much lower than previously published data testing ETV against HIV in cell culture (5) but higher than the EC50 observed in single round infectivity assays (11). M184V exhibited a much higher EC50 of 34.2 μm compared with WT. Our enzymatic data agree with the cell culture data used to test the effect of ETV on HIV-1 viral replication (Table 2 and Ref.11) and support the observed appearance of ETV resistance by M184V seen in HIV/HBV coinfected persons on ETV monotherapy (11). Resistance to ETV was not observed for the NRTI resistance mutations K65R or the AZT-resistant complex (Table 2), suggesting that M184V is specific for ETV resistance. Because ETV acts as an NRTI, it is not surprising that a known NRTI mutation would cause resistance. The M184V mutation in HIV-RT causes resistance to 3TCTP by reducing the ability of 3TCTP to bind RT (Table 2). It is likely that M184V also provides ETV resistance by a similar mechanism.
TABLE 2.
PBM cell assay showing the effects of ETV in HIV infected cells
| HIV-cloned virus | Nucleoside analog | EC50 | EC90 | FI50 | FI90 |
|---|---|---|---|---|---|
| μM | μM | ||||
| WT (xxBRU) | AZT | 0.007 | 0.03 | ||
| 3TC | 0.020 | 0.19 | |||
| ETV | 1.8 | 8.4 | |||
| M184V | AZT | 0.005 | 0.03 | 0.71 | 1.0 |
| 3TC | >100 | >100 | >100 | >100 | |
| ETV | 34.2 | 73.2 | 19.0 | 8.7 | |
| K65R | AZT | 0.003 | 0.011 | 0.43 | 0.37 |
| 3TC | 0.09 | 1.0 | 4.5 | 5.3 | |
| ETV | 0.21 | 1.8 | 0.12 | 0.21 | |
| 4xAZT | AZT | 0.017 | 2.0 | 2.4 | 66.7 |
| 3TC | 0.10 | 0.7 | 5.0 | 3.7 | |
| ETV | 0.63 | 5.7 | 0.35 | 0.68 |
Molecular modeling studies were used to understand the interaction of ETV with RT at a molecular level. The computational procedure followed here was purposely designed to provide realistic yet attainable models to assess the effects of ETV substitution in the growing DNA chain and the strain introduced at different positions to better understand the experimental results. If instead, one wanted to create models to measure the actual geometrical distortion in the chain, a large number of very long molecular dynamics simulations in explicit solvent would need to be undertaken. Qualitatively, the results of our calculations are shown in Table 3. The energy of the model when ETV is in the +5 position is considerably higher than all other positions, and the distances to the closest carbon atoms (C2′ and C1′, highlighted in bold type in Table 3) of the neighboring deoxyribose are shorter than all other substitutions. This is also shown in Fig. 4, where the overlap of the molecular surface of the ETV methylene with the closest deoxyribose is larger in area and more distorted at position 5 than in all the other models. This suggestion is in accord with our experimental data showing (Fig. 1C, panel IV) that when ETVTP and three additional dNTPs are incorporated such that ETVMP would be the +4 position, a buildup of 28-mer product is observed and rather than the incorporation of an additional dNTP that would place ETV in the unfavorable +5 position. The different steric demand at this position stems from the geometry imposed by the protein surface on the DNA. As mentioned in the paper by Huang et al. (20) describing the crystal structure, the initial part of the DNA chain is formed in an A-like conformation but becomes gradually more B-like starting at this position. The initial portion of the sequence studied here is entirely composed of GC pairs; it is likely that a more flexible chain, i.e. with a higher percent of AT pairs, may be more flexible and overcome this steric requirement.
TABLE 3.
Results of the energy minimizations of models E0 through E7
| ETVa | E | rc C6′n-C2′n +1 | rc C6′n-C1′n − 1 |
|---|---|---|---|
| kcal/molb | Åc | Åc | |
| 0 | −47.627 | 3.28 | 4.09 |
| +1 | −47.194 | 3.28 | 4.02 |
| +2 | −62.252 | 3.20 | 3.80 |
| +3 | −50.038 | 3.11 | 3.79 |
| +4 | −42.632 | 3.15 | 3.96 |
| +5d | −36.721 | 2.95 | 3.29 |
| +6 | −70.262 | 3.81 | 3.69 |
| +7 | −88.219 | 3.16 | 3.54 |
Position of the ETV in the primer chain.
Total energy of the last minimization.
Distance between the exocyclic methylene carbon in ETV and the closest carbons in the deoxyribose in the next nucleotide in the primer chain.
The energy and distances at position +5 are indicated in boldface to highlight the higher distortion.
FIGURE 4. Overlap between ETV and the next 2′-deoxyribose in the primer chain.
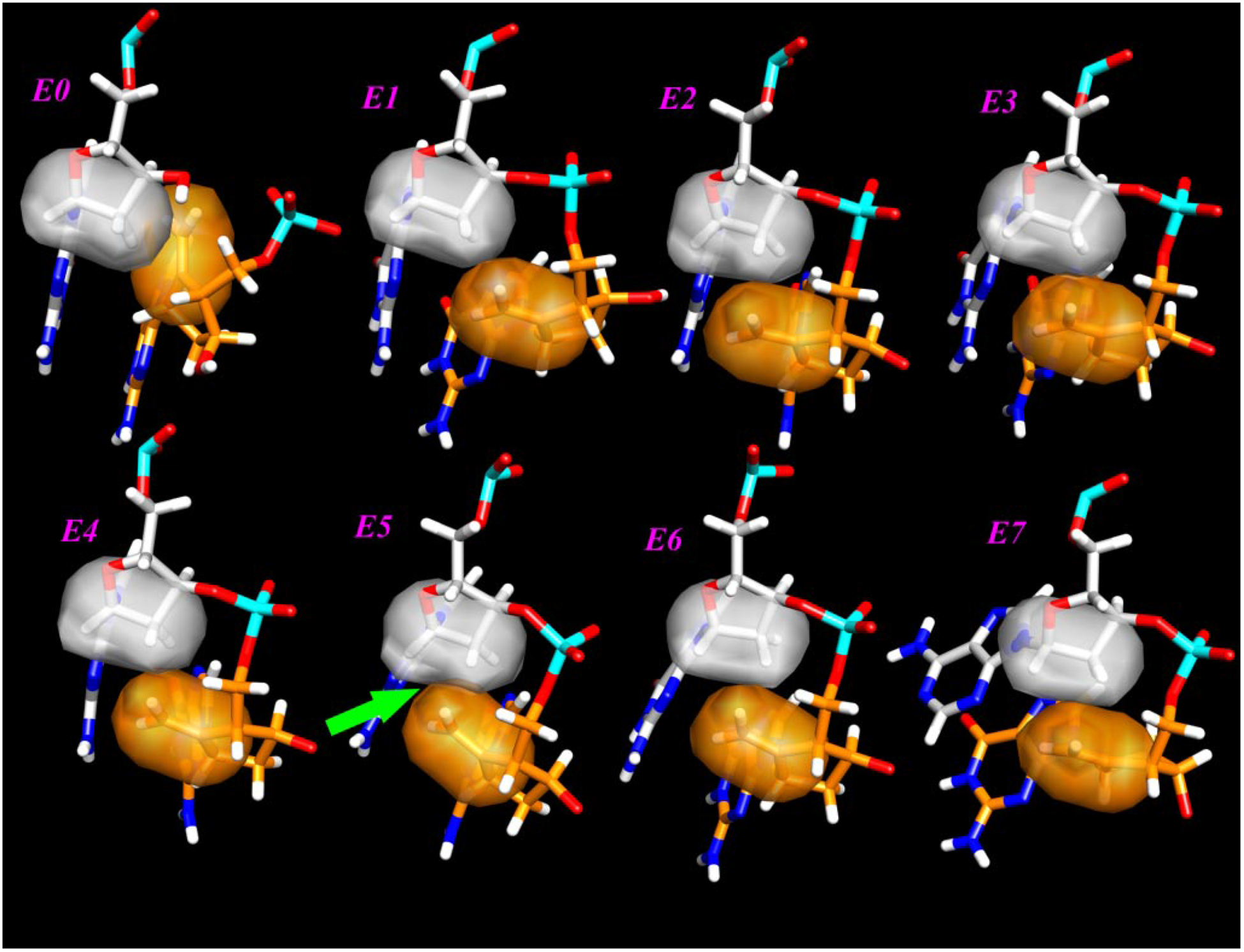
The surface of the methylene moiety is colored orange, and the closest atoms in the next 2′-deoxyribose are in gray. The green arrow near the lower left corner shows the largest overlap found when ETV is in position +5 in the primer.
DISCUSSION
Our results show that HIV-1 RT can use ETVTP as a substrate and that ETV is capable of inhibiting reverse transcription despite the fact that ETV has been previously reported to have no effect on HIV-1 replication in cell culture (5). There have been no previous reports examining if the active metabolite of ETV is a substrate for HIV-1 RT. Because ETV is a guanosine analog, it is not surprising that RT is able to incorporate ETV as readily as it does the natural nucleotide, dGTP, or another guanosine analog, CBVTP (Fig. 1). ETV is hypothesized to inhibit HBV Pol by increasing steric constraints after incorporation by causing a decrease in the efficiency of subsequent dNTP additions (8). We observed that the incorporation of a single ETV is not enough to inhibit polymerization (Fig. 2); polymerization is inhibited after subsequent nucleotides are added after ETVTP incorporation. Our results suggest that inhibition of HIV-1 RT replication by ETV may be similar to the mechanism previously reported for ETV inhibition of HBV Pol (4, 8). The concept of delayed chain termination is a unique mechanism for HIV RT inhibition. Although the details of the reaction kinetics have not been fully explored, previous studies with 4′-azidothymidine suggested similar chain extension/termination behavior (25, 26). Very recent studies with 4′-ethynyl-2-fluoro-2′-deoxyadenosine have proposed an analogous mechanism (27).
We determined the kinetic constants for ETVTP incorporation by HIV RT. Earlier studies suggested that ETVTP displayed a complex kinetic behavior for incorporation; however, additional studies with another batch of ETVTP revealed this was due to the presence of pyrophosphate. As such, the increase in rate was used to determine the binding affinity of ETVTP to HIV-1 RT. The delayed chain termination behavior has not been described previously for the other NRTIs currently approved to treat HIV-1 infections. ETV had a slower maximum rate of incorporation and incorporation efficiency compared with dGTP or CBVTP. In cell culture, the EC50 for ETV is1.8 μm and much higher than the NRTIs approved for use against HIV, suggesting that it is not a potent inhibitor and yet still can lead to resistance.
The factors contributing to the complex kinetic behavior for incorporation of ETVTP are still under investigation, however, in part due to slow product dissociation and effects on DNA binding. A pre-steady-state burst experiment (Fig. 2C) showed a biphasic formation of product similar to other NRTIs, indicating that the overall rate-limiting step is still product release; however, this rate was slow compared with all other NRTIs examined. Interestingly, our data from primer/template binding experiments using dGTP or ETVTP as the substrate for incorporation suggest that the presence or incorporation of ETVTP may have an effect on primer/template binding (Fig. 3). Further studies are in progress to understand the unique kinetic behavior for ETVTP that may contribute to the unusual dose response behavior in cell culture that was recently observed in single round infectivity assays (11).
The selection of M184V in patients receiving ETV monotherapy suggested that M184V might be resistant to ETV (11). We found that M184V RT was less efficient at incorporating ETVTP compared with WT mostly due to a decrease in the binding affinity for ETVTP (Table 2). This indicates that M184V is less susceptible to inhibition by ETV and that the M184V mutation affects binding. Our kinetic data are supported by a 19-fold increase in the EC50 of M184V for ETV in primary lymphocytes, whereas the change is not shared by other NRTI-resistant mutations such as K65R and AZT resistance complex mutants. This suggests that ETV treatment selects for the M184V mutation as a mechanism of resistance. This has been corroborated by the Schinazi laboratory by selecting M184I/V HIV in the presence of ETV in primary lymphocytes.3
ETV utilizes a unique mechanism for inhibition of HIV-1 RT for a nucleoside analog as it does not act as an obligate chain terminator. Our kinetic and modeling data suggest that the incorporation of ETV causes a distortion in the growing strand when it reaches the +5 position, having similarities to its mechanism against HBV Pol (8). This increase could cause RT to fall off the template and stop polymerization. The steric inhibition would be enhanced by the incorporation of more than one ETV during polymerization.
In summary, the current study provides an answer to the unexplained clinical findings with ETV by establishing that ETVTP serves as a substrate for WT HIV-1 reverse transcriptase and that the efficiency of its incorporation is decreased by the presence of the M184V mutation in RT. These results offer insight into HIV-1/HBV-coinfected patients taking ETV monotherapy. Our results heighten concern that ETV in treatment may cause an increase in the selection of drug resistant HIV. The selection of known NRTI resistance mutations like M184V against ETV could lead to the limiting of treatment options for those infected patients, particularly treatment with 3TC or emtricitabine.
Acknowledgments
This work was supported in part by National Institutes of Health Grants GM-49551 (to K. S. A.), and AI-44616 (to W. Jorgensen in support of J. T.-R.), AI-51178, a grant from AI-43222, Howard Hughes Medical Institute (to R. S.), Emory’s Center for AIDS Research National Institutes of Health Grants 5P30-AI-50409 and 5R37-AI-041980, and a grant from the Department of Veterans Affairs (to R. F. S.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: HIV, human immunodeficiency virus; ETV, Entecavir; ETVTP, 5′-triphosphate ETV; HBV, hepatitis B virus; Pol, polymerase; NRTI, nucleoside reverse transcriptase inhibitor; RT, reverse transcriptase; PBM, peripheral blood mononuclear; WT, wild type; CBVTP, carbovir triphosphate; AZT, azidothymidine.
R. F. Schinazi, unpublished information.
Note Added in Proof—The print version differs from the earlier Papers in Press version due to our finding that our Entecavir triphosphate (ETVTP) sample was contaminated with pyrophosphate. The presence of pyrophosphate affected the rate, affinity, and incorporation efficiency of ETVTP. The Kd and kpol values for ETVTP incorporation by WT and M184V are modified in Table 1, Fig. 2, and text to reflect the values determined after removal of contaminating pyrophosphate.
REFERENCES
- 1.Ganem D, Pollack JR, and Tavis J (1994) Infect. Agents Dis 3, 85–93 [PubMed] [Google Scholar]
- 2.Locarnini SA, Civitico GM, and Newbold JE (1996) Antiviral Chem. Chemother 7, 53–64 [Google Scholar]
- 3.Seeger C, Summers J, and Mason WS (1991) Curr. Top. Microbiol. Immunol 168, 41–60 [DOI] [PubMed] [Google Scholar]
- 4.Seifer M, Hamatake RK, Colonno RJ, and Standring DN (1998) Antimicrob. Agents Chemother 42, 3200–3208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Innaimo SF, Seifer M, Bisacchi GS, Standring DN, Zahler R, and Colonno RJ (1997) Antimicrob. Agents Chemother 41, 1444–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ono SK, Kato N, Shiratori Y, Kato J, Goto T, Schinazi RF, Carrilho FJ, and Omata M (2001) J. Clin. Investig 107, 449–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rivkin A (2007) Drugs Today (Barc) 43, 201–220 [DOI] [PubMed] [Google Scholar]
- 8.Langley DR, Walsh AW, Baldick CJ, Eggers BJ, Rose RE, Levine SM, Kapur AJ, Colonno RJ, and Tenney DJ (2007) J. Virol 81, 3992–4001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Benson CA, Kaplan JE, Masur H, Pau A, and Holmes KK (2004) MMWR Recomm. Rep 53, 1–112 [PubMed] [Google Scholar]
- 10.Soriano V, Miro JM, Garcia-Samaniego J, Torre-Cisneros J, Nunez M, del Romero J, Martin-Carbonero L, Castilla J, Iribarren JA, Quereda C, Santin M, Gonzalez J, Arribas JR, Santos I, Hernandez-Quero J, Ortega E, Asensi V, del Pozo MA, Berenguer J, Tural C, Clotet B, Leal M, Mallolas J, Sanchez-Tapias JM, Moreno S, Gatell JM, Tellez MJ, Rubio R, Ledesma E, Domingo P, Barreiro P, Pedreira J, Romero M, Gonzalez-Lahoz J, and Lissen E (2004) J. Viral. Hepat 11, 2–17 [DOI] [PubMed] [Google Scholar]
- 11.McMahon MA, Jilek BL, Brennan TP, Shen L, Zhou Y, Wind-Rotolo M, Xing S, Bhat S, Hale B, Hegarty R, Chong CR, Liu JO, Siliciano RF, and Thio CL (2007) N. Engl. J. Med 356, 2614–2621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boyer PL, Julias JG, Marquez VE, and Hughes SH (2005) J. Mol. Biol 345, 441–450 [DOI] [PubMed] [Google Scholar]
- 13.Kerr SG, and Anderson KS (1997) Biochemistry 36, 14064–14070 [DOI] [PubMed] [Google Scholar]
- 14.Ray AS, Yang Z, Shi J, Hobbs A, Schinazi RF, Chu CK, and Anderson KS (2002) Biochemistry 41, 5150–5162 [DOI] [PubMed] [Google Scholar]
- 15.Murakami E, Basavapathruni A, Bradley WD, and Anderson KS (2005) Antiviral Res. 67, 10–17 [DOI] [PubMed] [Google Scholar]
- 16.Kati WM, Johnson KA, Jerva LF, and Anderson KS (1992) J. Biol. Chem 267, 25988–25997 [PubMed] [Google Scholar]
- 17.Schinazi RF, McMillan A, Cannon D, Mathis R, Lloyd RM, Peck A, Sommadossi J-P, St. Clair M, Wilson J, Furman PA, Painter G, Choi W-B, and Liotta DC. (1992) Antimicrob. Agents Chemother 36, 2423–2431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schinazi RF, Sommadossi JP, Saalmann V, Cannon DL, Xie MY, Hart GC, Smith GA, and Hahn EF (1990) Antimicrob. Agents Chemother 34, 1061–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skoog DA, and Leary JJ (1992) Principles of Instrumental Analysis, 4th Ed., Saunders Collge Publishing, New York [Google Scholar]
- 20.Huang H, Verdine GL, Chopra R, and Harrison SC (1998) Science 282, 1669–1675 [DOI] [PubMed] [Google Scholar]
- 21.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004) J. Comput. Chem 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 22.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JJA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, and Pople JA (2003) Gaussian 03, Revision B.01, Gaussian, Inc., Pittsburgh, PA [Google Scholar]
- 23.Maestro (2006) Version 7.5, Schrödinger, LLC, New York, NY [Google Scholar]
- 24.Impact (2006) Version 4.0, Schrödinger, LLC, New York, NY [Google Scholar]
- 25.Chen MS, Suttmann RT, Papp E, Cannon PD, McRoberts MJ, Bach C, Copeland WC, and Wang TS (1993) Biochemistry 32, 6002–6010 [DOI] [PubMed] [Google Scholar]
- 26.Chen MS, Suttmann RT, Wu JC, and Prisbe EJ (1992) J. Biol. Chem 267, 257–260 [PubMed] [Google Scholar]
- 27.Nakata H, Amano M, Koh Y, Kodama EI, Yang G, Kohgo S, Hayakawa H, Matsuoka M, Anderson KS, Cheng YC, and Mitsuya H (2007) Antimicrob. Agents Chemother 51, 2701–2708 [DOI] [PMC free article] [PubMed] [Google Scholar]