

Abstract
Of the more than 100 studies that have examined relationships between excessive ethanol consumption and the brain transcriptome, few rodent studies have examined chronic consumption. Heterogeneous stock collaborative cross mice freely consumed ethanol vs. water for 3 months. Transcriptional differences were examined for the central nucleus of the amygdala, a brain region known to impact ethanol preference. Early preference was modestly predictive of final preference and there was significant escalation of preference in females only. Genes significantly correlated with female preference were enriched in annotations for the primary cilium and extracellular matrix. A single module in the gene co-expression network was enriched in genes with an astrocyte annotation. The key hub node was the master regulator, orthodenticle homeobox 2 (Otx2). These data support an important role for the extracellular matrix, primary cilium and astrocytes in ethanol preference and consumption differences among individual female mice of a genetically diverse population.
Keywords: Alcohol, Addiction, Drinking, Ethanol preference, RNA-Seq, Sex differences
1. Introduction
Using either microarrays or RNA-Seq, there are now more than 100 studies across multiple species that have examined some aspect of the relationships between excessive alcohol (ethanol) consumption and the brain transcriptome. The first meta-study was in mice [1] and illustrated the breadth of the transcriptional features associated with genetic predisposition for high ethanol intake or preference. Subsequent studies, in mice [2–7], rats [8–10], non-human primates [11,12] and humans [13–15], have all confirmed the breadth of the transcriptional effects associated with genetic risk for ethanol use and consequent to its use. Further, all aspects of the addiction circuitry [16,17] appear to be affected (e.g. [5,18]). Some common transcriptional features across species have emerged; genes associated with synaptic plasticity and especially glutamate plasticity, genes associated with cell adhesion and genes associated with neuro-immune function have all been repeatedly detected [see [13] and references therein]. These studies have been important for identifying risk and consequence mechanisms that could lead to new risk markers and therapeutic developments.
Our research has emphasized the use of selective breeding in mice to detect genes and gene ensembles associated with risk for high ethanol preference and consumption [3–6,19]. The founding populations for all selectively bred lines are genetically heterogeneous to provide genetic diversity critical to successful selective breeding. However, the genetic diversity among the mouse selective breeding projects that have been performed for ethanol intake or preference has varied. For selective breeding, in addition to genetic variation, individual differences for the selection phenotype within the founding population are critical. During the course of our studies, we observed that 10 to 15% of Heterogeneous Stock-Collaborative Cross (HS-CC) [20] mice have a preference for consuming more of their fluid from the ethanol- than water-containing tube. In contrast, less than 5% of our heterogeneous stock-Northport (HS/NPT) mice [21] have a similar ethanol preference (unpublished observations). The HS-CC were formed by crossing 8 inbred mouse strains, including three wild-derived strains (CAST, WSB and PWK). The HS/NPT were formed by crossing 8 standard laboratory mouse strains. The HS-CC encompasses 89% of the genetic diversity available in Mus musculus, whereas the HS/NPT encompasses 36% of the available genetic diversity [22]. The Diversity Outbred population maintained by The Jackson Laboratory (Bar Harbor, ME) has a related genetic pedigree to the HS-CC and a similar percentage of mice exhibit ethanol preference (unpublished observations).
Colville et al. [4,5] used short-term selective breeding from HS-CC founders to rapidly produce High and Low preference selected lines. Across the prelimbic cortex, the nucleus accumbens (NAc) shell and the central nucleus of the amygdala (CeA), transcriptome analyses established significant differences between the selected lines in the expression of cell adhesion genes (particularly cadherins and protocadherins) and genes associated with synaptic plasticity. Regarding the latter, Dlg2, which encodes for PSD-93, was found to be a key selection-relevant hub node. The current study uses a random population of HS-CC mice in a different context, one of chronic (3 month) ethanol consumption (24 h/7 d per week two bottle choice). Here, the focus is on 3 different but likely related parameters, addressed by the following questions: 1. Given that a moderate proportion of the mice would begin with an ethanol preference ≥ 0.5, would this higher preference be present at the end of the 3 month trial? 2. What proportion of mice that begin the trial with a preference ratio < 0.5 would escalate to a preference ratio ≥ 0.5? 3. At the end of the three month trial, is it possible to detect a transcriptional signature associated with high preference and is this signature different from the risk signature previously detected [5], when comparing the HS-CC High and Low selected lines? The transcriptional analyses focused on the central nucleus of the amygdala (CeA) which is known to have a key role in the regulation of ethanol preference (e.g., [23]).
2. Materials and methods
2.1 . Animals and husbandry
The HS-CC mice used in the current study were obtained from the breeding colony at the VA Portland Health Care System, an AAALAC accredited facility. The HS-CC colony is maintained as 48 families using a rotational breeding design. All procedures were in accordance with the VA Institutional Animal Care and Use Committee and were performed according to NIH Guidelines for the Care and Use of Laboratory Animals. Mice within the breeding colony were maintained at 21±1°C on a 12-h light:dark schedule, with lights on at 0600 h. They were housed in filtered acrylic plastic cages (19×31×13 cm) on Eco-Fresh bedding (Absorption Corp., Ferndale, WA) with tap water and Purina 5001 chow (PMI Nutrition International, Brentwood, MO, USA) freely available. Pups were weaned and housed with same-sex littermates at postnatal day 21±2 d. One male and one female were randomly chosen from each of the 48 families for testing. An additional 12 males and 12 females were randomly chosen such that there were no more than 3 mice from any family. Thus, a total of 120 mice entered the phenotyping protocol; 114 finished (there was some loss due mostly to drinking tubes leaking). Median age for both males and females was 15 weeks at the initiation of ethanol preference testing.
2.2. Three-month 2-bottle choice trial
Prior to beginning the trial, mice were individually housed for 1 week in cages of the same type as during rearing, under the same lighting and temperature conditions. At the end of the 1 week period, filter tops were removed from the cages to allow placement of two water-filled glass graduated cylinders fitted with rubber stoppers and stainless steel sipper tubes, introduced for a 4-day period. On day 5, one of the tubes was replaced with a 5% (v/v) ethanol solution, diluted with tap water from 100% ethanol (Fisher BioReagents, Waltham, MA). The side containing the ethanol solution was switched every 2 days across a 4-day period. On day 9, the ethanol concentration was increased to 10% for 4 days of acclimation to the 10% concentration. The 3-month ethanol trial then began, during which mice had access to water and 10% ethanol. The amounts of ethanol and water consumed were measured daily, and mice were weighed weekly. At the end of the 3 month (week 13) trial, all mice were euthanized between 1000 and 1400 h; ethanol was available until the time of euthanasia. In preliminary studies using HS-CC mice that had access to ethanol in the 2-bottle choice procedure, we found that between 1000 and 1400 h, blood ethanol concentrations are essentially 0, even for mice with a high preference. Four hours into the dark phase (2200 h), mice with a preference ≥ 0.5 have blood ethanol concentrations in the 50 to 90 mg/dl range. The mice were euthanized in four passes; thus, the ethanol trial lasted for 90–93 days. The key behavioral outcome measure was ethanol preference calculated as ml consumed from the ethanol tube divided by total ml consumed from both tubes. The weekly preference averages for each individual animal are presented in Supplementary Material Table S1. Repeated measures ANOVA (Statistica, TIBCO software, Inc., Palo Alto, CA, version 13.3) was used to examine the data for sex and time (initial week 1 vs. final week 13) effects on ethanol preference and consumption. Pearson correlations (Statistica) were used to follow individual animal week to week relationships and the initial (week 1) vs. final (week 13) week relationship. To determine if the correlations were significantly different, we followed Steiger [24] as implemented in Lee and Preacher [25].
2.3. Dissection of tissue and extraction of RNA
Brains were removed from euthanized mice and immediately frozen on dry ice. Ninety-six samples balanced for sex, family and preference quartile were chosen for dissection. Our previous studies [4,5] empirically established that in order to construct co-expression networks of high quality (see below), sample sizes of ~ 30 to 40 are required; given our previous observation of sex-specific transcriptional effects associated with excessive ethanol consumption [6] and the long-established observation that female mice have a higher preference than male mice, sufficient animals were funneled through dissection, RNA extraction and RNA-Seq to analyze the male and female data separately.
Frozen brains were sliced in 25 micron coronal sections on a cryostat (Leica Biosystems CM1800, Buffalo Grove, IL) at −13°C; slices containing the amygdala were mounted on PEN slides (Thermo Fisher Scientific, Waltham, MA). Mounted slices were lightly thionin stained under RNAse free conditions and dehydrated in increasing concentrations of ethanol diluted in RNAse free water (50%, 70%, 95%, & 100%) for 30 seconds each and then air-dried. The CeA was dissected bilaterally on a Leica LMD-6000 using known anatomical landmarks [26]. Dissected tissue was processed with the Arcturus Picopure kit (Thermo Fisher Scientific) yielding on average 200ng of total RNA. RNA quality was assessed using the 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) and RNA Quality Scores. Only samples with RNA Quality Scores of > 7 and > 100 ng of total RNA were used for library formation.
2.4. RNA-Seq
Library formation (polyA+, stranded) and sequencing were all performed according to Illumina’s specifications at the OHSU Massively Parallel Sequencing Shared Resource. Briefly, libraries with strand orientation were prepared using the TruSeq RNA Sample Preparation Kit (Illumina, San Diego CA, USA). Libraries were sequenced on a HiSeq 2500 (Illumina). Eighty-six samples were sequenced. Libraries were multiplexed 6 per lane, balanced for sex, family and preference quartile, yielding approximately 25 to 30 million total reads per sample. In some lanes, there were only 5 samples. Data were aligned to the mouse genome (mm10, build 38). FastQC was used for quality checks on the raw sequence data. Two samples failed the Q/C and were not carried forward. Sequence data were aligned using STAR (Spliced Transcripts Alignment to a Reference [27]), allowing for a maximum of three mismatches per 100bp read. For all samples, > 85% of the reads uniquely aligned. Using the featureCounts suite [28], reads were aligned to known genomic features to generate counts at both exon and gene levels. Gene expression data were imported into the R application environment; upper-quartile normalization was performed using the DESeq2 Bioconductor package [29]. The gene read density threshold for inclusion in all analyses was an average of ≥ 1 CPM across samples. These data have been deposited into NCBI’s Gene Expression Omnibus (RNA180116RH).
2.5. Correlations between gene expression and ethanol preference
Pearson correlations (uncorrected p < 0.05) were used to detect relationships between ethanol preference and individual gene expression. Data were analyzed by pooling data for males and females and also by examining each sex separately, based on our a priori interest in sex-specific outcomes; 14,447 genes met the threshold (see above) for inclusion in the analysis across both sexes. Principal components analysis was used to detect data clustering.
2.6. Co-Expression network construction
Gene co-expression networks were constructed using the Weighted Gene Co-expression Network Analysis (WGCNA) algorithm [30] as described in detail elsewhere [4,5,31]. Briefly, all genes meeting the threshold for inclusion (see above) were entered into the WGCNA. The number of modules is kept at 25 to 30, with no module having < 100 genes. Empirically, we have found that these conditions and keeping sample sizes at ~40 results in modules of high quality. Next, genes contributing the bottom 10% of network connectivity were culled from the networks [5]; these culled genes are generally denoted as “leaf” nodes within the networks. The culling step reduces the number of genes by 30 to 40% and reduces the subsequent computational burden. The next steps vary depending on the experimental design, but the focus is similar – which network module(s) contain the genes correlated with the phenotype under investigation. In the current study, this step was simplified by the observation that essentially all of those genes were found in a single module (brown). Once the module or modules are identified, the hub nodes are interrogated.
2.7. Gene ontology (GO) annotation
The GOrilla algorithm [32] was used to provide a representation of GO annotation enrichment against a background set of genes. Given that the correlations for the genes associated with preference or consumption were uncorrected, 1000 permutations of the data were used to set the minimum threshold for significance. No annotations with a FDR < 0.001 were detected. Thus, the threshold for a significant annotation was set at a FDR < 0.001. Cell-type enrichment analysis used the procedure outlined in Cahoy et al. [33].
3. Results
3.1. Three-month 2-bottle choice trial
One hundred and fourteen HS-CC mice (58 females and 56 males) completed the trial. Weekly preference averages are found in Supplementary Material Table S1. Figures 1A and 1B illustrate the week 1 compared to week 13 ethanol preference relationships for females and males, respectively. The correlation for the entire group of 114 mice was 0.47 (p < 0.0001). The correlation for each of the sexes is shown in Figure 1 and was statistically significant for both sexes (ps < 0.001). However, week 1 preference explained somewhat more of the variance in week 13 for female mice (26%), compared to male mice (19%). When the preference data for weeks 1 and 13 (Figure 2) were analyzed by repeated measures ANOVA, a significant sex x week interaction was found (F[1,112] = 3.8, p = 0.05). In females, there was a significant increase in preference from week 1 to week 13 (0.20week1 vs 0.31week13, F[1,112] = 17.8; p < 0.0001), whereas there was no significant change in the males (0.19week1 vs 0.23 week13, p = 0.16). Among the females, 7 mice began the trial with a preference ratio of ≥ 0.5 and 6 of these 7 females (86%) had an average preference ratio of ≥ 0.5 in week 13 (Figure 3A). Among the males, 5 mice began the trial with a preference ratio in week 1 ≥ 0.5 and 3 of these males (60%) had an average preference ratio of ≥ 0.5 in week 13 (Figure 3B). Note: preference values of > 0.45 have been rounded up to 0.5.
Figure 1. Ethanol preference during the first week of a chronic 3-month drinking trial modestly predicts ethanol preference in the final week.

Correlations are shown for (A) females and (B) males for the first (Week 1) and last (Week 13) weeks of the ethanol drinking trial. Pearsons correlations were significant (ps < 0.001). The solid line is the regression with the dotted lines representing the 95% confidence intervals. N=58 for females; N=56 for males
Figure 2. In females, there was a significant increase in preference from week 1 to week 13, whereas there was no significant change in males.

Shown are means ± SEM for ethanol preference ratio of all 58 females and 56 males during the first (week1) and final (week 13) weeks of the 3-month ethanol drinking trial. N=58 for females; N=56 for males. ***p < 0.0001
Figure 3. Initial ethanol preference greater than or equal to 0.5 is partially predictive of higher preference in final week of drinking.

Shown are individual preference ratios for (A) females and (B) males that had preference ratios ≥ 0.5 during the first week of drinking. Six out of 7 females had a preference ratio at this level in week 13, compared to 3 of 5 males. Assigned subject numbers are given in the figure legends.
With regard to more profound increases in preference over time, among the females, 9 mice began the trial with a preference ratio < 0.5 in week 1 and had a preference ratio of ≥ 0.5 in week 13 (Figure 4A). For 4 of these animals, the total 13 week average preference ratio was ≥ 0.5, as the result of escalation. Overall, 15/58 females (26%) ended the trial with a preference ratio in Week 13 of ≥ 0.5. Among the males, 8 mice began the trial with a preference ratio < 0.5 in week 1 and had a preference ratio of ≥ 0.5 in week 13 (Figure 4B). For only 1 of these animals was the escalation sufficient such that the average preference ratio across the 13-week trial was ≥ 0.5. Overall, 11/56 males (20%) ended the trial with a preference ratio in Week 13 of ≥ 0.5.
Figure 4. For some individual mice, ethanol preference escalated across the weeks of the 3-month trial.

Shown are individual preference ratios for (A) females and (B) males that had initial preference ratios < 0.5 during the first week of drinking that increased to ratios ≥ 0.5 in the final week. Assigned subject numbers are given in the figure legends.
The data on consumption are found in Supplementary Material Table S2. The overall correlations between preference and consumption for weeks 1, 13 and across all weeks were 0.90, 0.90 and 0.93 (N =114; ps < 0.00001). For females, the correlations for these time periods were 0.91, 0.93 and 0.95 (N = 58; ps < 0.00001). For males the correlations were 0.92, 0.88 and 0.94 (N = 56; ps < 0.00001). Frequency distributions illustrating total amount of ethanol consumed are presented in Figure 5 to provide information about cumulative intake. Mean ± SEM total consumption was 427 ± 29 g/kg for the sexes combined for the 13-week period. ANOVA identified a significant effect of sex (F[1,112] = 16.9, p < 0.0001), with females consuming an average of 537 ± 45 g/kg (range = 63 – 1508 g/kg) and males consuming 313 ± 30 g/kg (range = 41 – 1104 g/kg). Six females consumed ≥ 9 g/kg in week 1 and this level of consumption was found in week 13 for 4 mice. Twelve females escalated consumption from < 9 g/kg in week 1 to ≥ 9 g/kg at the end of the trial. Four males consumed ≥ 9 g/kg in week 1 and 2 of these mice had this level of consumption at the end of the trial. Six males escalated consumption from < 9 g/kg in week 1 to ≥ 9 g/kg over the course of the trial.
Figure 5. There was considerable individual animal variation in total amount of ethanol consumed during the 3-month drinking period, with females consuming more than males.

Frequency distributions for females and males representing the number of mice with levels of ethanol intake summed across 3 months. The range of values was 41 – 1508 g/kg (537 ± 45 g/kg for females; 313 ± 30 g/kg for males). N=58 for females; N=56 for males
3.2. RNA-Seq analysis - central nucleus of the amygdala (CeA)
Ninety-six samples were chosen for RNA-Seq analysis, balanced for sex and quartile of ethanol preference. Data are provided in Supplementary Material Table S3 for 84 samples. Samples were lost for several reasons: failed dissection, low RNA yield, poor RNA quality, failed library preparation and poor sequencing. The final group included 12 high preference (≥ 0.5) females and 8 high preference males; the remaining 64 were distributed across the preference range from < 0.5 to 0. Results for principal components analysis of the data are illustrated in Figure 6. The data clustered into two components. Component 1 (the larger) was formed from 33 female and 31 male samples. Component 2 (the smaller) was formed from 10 female and 10 male samples. The differences between clusters 1 and 2 were largely driven by the higher expression in cluster 2 of a group of pseudogenes that included Gm13341 (p< 6 × 10−35), Gm11407 (p< 6 × 10−27), Gm13339 (p< 7 × 10−23), Gm29216 (p< 1 × 10−19), Gm13340 (p< 5 × 1015), Gm4076 (p< 1 × 10−14) and Gm28438 (p< 4 × 10−5) (see far right of Supplementary Material Table S3). These pseudogenes were of small to intermediate transcript length (345 to 1545 bp; see “Transcript Length” in Table S3). Removing these genes from the analysis had no effect on the results described below; further, if just the samples in cluster 1 are used, the results are essentially similar to those obtained by using all samples.
Figure 6. Principal components analysis separates RNA-Seq data for the central nucleus of the amygdala into two components for mice with varying levels of ethanol preference.

Component 1 (PC1) accounted for 21% of the variance and was formed from 33 female and 31 male samples. Component 2 (PC2) accounted for 19% of the variance and was formed from 10 female and 10 male samples.
The analysis of the RNA-Seq data focused on preference phenotype x expression correlations, beginning with the data for all 84 mice, then moving to the 43 females and 41 males in separate analyses. Correlations were calculated for week 1 average, week 13 average and the average across all weeks. For all comparisons, it was always the week 13 data that provided the strongest annotation structure and it is these results that are discussed here and provided in Supplementary Material Tables S3–S5; however, using the data in Supplementary Material Tables S1 and S3, the annotation structure for the other intervals can be easily calculated. Annotations are provided for the positive and negative correlations; in no case was combining the positive and negative correlations more informative than examining them separately. Given the differences in the number of high preference females and males (see above), a sample matching approach was used so that the female and male samples had equal preference; this increased average male preference from 0.21 to 0.28 and decreased average female preference from 0.32 to 0.28. The mice included in the match are noted in Supplementary Material Table S3. Qualitatively, the results were no different from those described below for the entire male and female samples.
Gene-preference correlations are listed in Supplementary Material Table S4, and the GO annotations for the genes significantly correlated with preference (average week 13) are found in Supplementary Material Table S5. The positive annotations for all 84 samples included, motile cilium (FDR < 3 × 10−9) and ciliary part (FDR < 4 × 10−8). [Note: there is no GO annotation for primary cilium; the genes with an annotation for motile cilium would also be found in primary cilium. In brain, motile cilia are only found in the linings of the third and fourth ventricles [34]]. There were no significant (FDR < 0.001) GO annotations for the genes negatively correlated with preference.
For females, the ethanol preference cilia-related annotations improved in significance and included cilium movement (FDR < 2 × 10−8) and cilium organization (FDR < 1 × 10−7). Annotations associated with the extracellular matrix emerged and included extracellular region (FDR < 5 × 10−10) and collagen-containing extracellular matrix (FDR < 1 × 10−6). The latter category included 10 collagens: Col4a1, Col4a3, Col4a4, Col4a5, Col5a1, Col5a3, Col8a1, Col8a3, Col9a3 & Col18a1. There were no significant annotations in females for the negatively correlated genes. In males, there were no significant annotations for the positive or negatively correlated genes.
Annotation data were also extracted for consumption (average week 13) and can be found in Supplementary Material Table S6; gene-consumption correlations are in Supplementary Material Table S4. For females and the positive correlations with ethanol consumption, in addition to the cilium and extracellular matrix annotations seen for preference, there was an enrichment in annotations associated with biological adhesion (FDR < 9 × 10−7 and cell adhesion (FDR < 6 × 10−7). For the negative correlations, there was no significant annotation enrichment.
3.3. Network analysis – females
Given the data above, the network analysis focused on the female sample for ethanol preference. The female gene co-expression network was formed as described elsewhere [5]. The network modules were color coded. The network was culled by removing those genes that contribute the bottom 10% of network connectivity; these genes are leaf nodes that in many applications of the Weighted Gene Co-expression Network Analysis (WGCNA) are found in the grey module. Beginning with 14,447 genes, culling reduced the number of genes in the female network to 8,492 (see Supplementary Material Table S7). The correlation and network data were integrated with a focus on aligning the hub nodes with preference. After integration, 461 of the remaining genes (86%) positively correlated with female preference (i.e., were found in the culled network; Table S7) and 403 (87%) were found in a single network module (brown). The brown module represented by the network shown in Figure 7 was enriched (p < 0.0001) in genes with an astrocyte annotation; Supplementary Material Table S7 lists the probabilities that the genes in each module are expressed in neurons, astrocytes or oligodendrocytes as per Cahoy et al. [33]. Results for ethanol consumption are also presented in Supplementary Material Table S7, for which 416 of the genes positively correlated with female consumption were in the culled network, and 376 (90%) were in the brown network module. All of the genes positively correlated with consumption are also in the preference gene list.
Figure 7. Composite gene-gene interaction network for differential expression in the CeA, associated with differences in ethanol preference in female HS-CC mice.
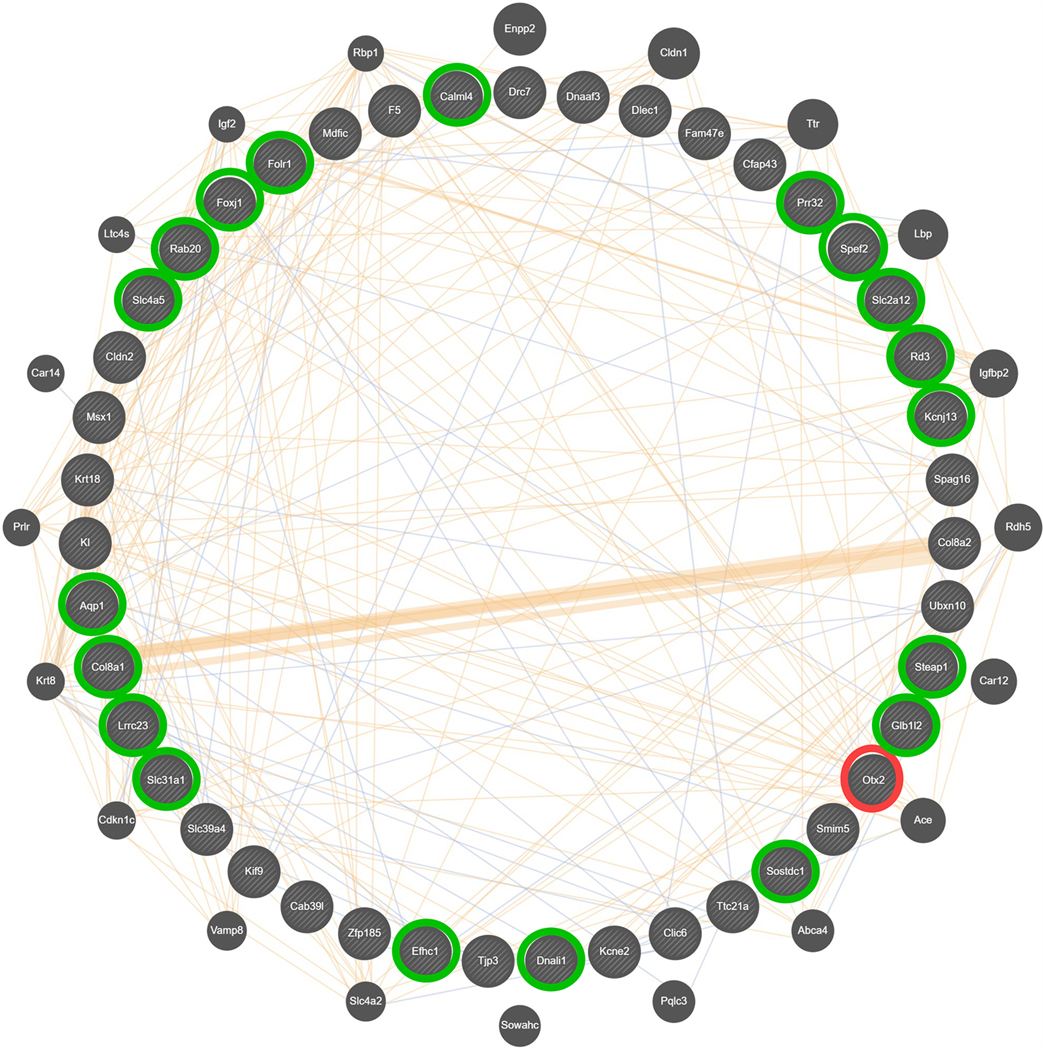
This network was created using GeneMANIA [80] and derived from 43 hub nodes (inner circle) found within the brown module, enriched in genes with an astrocyte annotation. The genes in the outer circle were identified by GeneMANIA [80] as functional associates, with symbol size, based on number of network connections. Otx2 (circled in red) was a top hub node and 19 other top hub nodes (circled in green) were down-regulated when Otx2 was absent. Blue lines represent known colocalization (5.47%) and tan lines represent predicted interactions (9.11%). The majority of network interactions are accounted for by co-expression (84.97%), which is the basis for the network. Since representation of co-expression obstructs visualization of colocalization and predicted interactions, co-expression is shown in Supplementary Information Figure S1.
Annotation of the female brown module (Supplementary Material Table S7) was consistent with the results above and included extracellular matrix (FDR < 1 × 10−8), cilium organization (FDR < 3 × 10−10), and cilium (FDR < 2 × 10−23). Among the positively correlated genes in the brown module, 43 had a relative intra-modular connectivity ≥ 0.9; i.e., they were top hub nodes. Enrichr [35,36] was used to search for key transcription factors among top hub nodes. A key finding extracted was that 19 of the top nodes (highlighted in green in Figure 7) were down-regulated in an orthodenticle homeobox 2 (Otx2) knockout mouse (GSE27630; [37]) (FDR < 5 × 10−20). Otx2 was one of the positively correlated top hub nodes. Top hub nodes down-regulated in the Otx2 knockout included Rd3, Prr32, Lrrc23, Spef2, Slc31a1, Kcnj13, Slc2a12, Foxj1, Calml4, Slc4a5, Dnali1, Glb1l2, Aqp1 Rab20, Efhc11, Sostdc1, Col8a1, Steap1 and Folr1. The entire list of genes down-regulated in the Otx2 knockout animals are found in Supplementary Material Table S2 of [37].
Finally, given the outcomes of the annotation analysis, and particularly an enrichment (females only) in genes associated with the extracellular matrix, we considered the possibility that the apparent enrichment was an artifact associated with transcript length (see [38]). The length bias was examined from 2 perspectives. One, following on the experimental design in Mandelboum et al. [38], we randomly divided the female expression data into two samples (N=22 and 21) and calculated differential expression between groups; by definition all significant calls were false positives. The range of correlations between p-values and transcript length after 100 iterations ranged from −0.03 to 0.04. Fold change vs length was similar. Two, if length bias is driving the extracellular matrix annotations, it would follow that the brown network module would be enriched in longer transcripts (87% of the genes positively correlated with preference were found in the brown module). This argument was simplified by focusing on the top hub nodes since it is these nodes that are driving the module annotations. The range of average top hub node transcript length varies > 5-fold (see “Hub Node Transcript Length” in Supplementary Material Table S7), and the brown module hub nodes are intermediate in transcript length.
4. Discussion
Rosenwasser et al. [39] appears to be the only report of a study in which HS mice (in that publication, HS/NPT as well as mice bred from HS/Ibg) were allowed to consume alcohol for a prolonged period (30 weeks). During the trial, there were periods of intermittent access which led to significant increases in ethanol consumption. However, after the termination of the intermittent access condition, the mice returned to baseline levels of consumption under continuous access. Importantly, that study found that the intermittent access-induced escalation of consumption was not genotype-dependent, but its robustness was. The Rosenwasser et al. [39] study examined only males and no data on individual responses were reported; thus, it could not be determined, from the individual perspective, if preference and consumption during the continuous access periods remained constant, increased or decreased. The current long-term continuous access study illustrates the very significant effects of sex, individual variation and the sex x individual interaction. Although we report here data only for HS-CC mice [20], we have obtained essentially identical results in Diversity Outbred (DO) mice raised locally from DO parents (unpublished observations). As noted in the Introduction, HS-CC and DO mice have a similar genetic background, but a different breeding history [40]. Some of the key behavioral observations in the current study may be summarized: (1) More female than male mice both began the trial with a preference ratio ≥ 0.5, and had a preference ratio ≥ 0.5 at the end of the 3-month trial; (2) Week 1 preference data were only moderately predictive of week 13 preference; (3) There was a subgroup of both males and females that showed a significant escalation of preference over the 13 week trial. There appear to be several different trajectories of escalation but given the relatively small numbers this may simply reflect experimental noise rather than some biological phenomenon; and (4) Preference and consumption remained tightly coupled over the course of the trial.
We observed preference escalation in 16% of the female mice and 14% of the males. Some, but not all, clinical data support a sex difference in escalation [41,42]. The escalation phenomenon in humans is often referred to as telescoping. Becker and Koob [43] have noted that the importance of sex may in part be related to the population being surveyed. Data collected from a general population may well differ from a population seeking treatment. Distant from this inconsistency has been the observation for nearly 50 years, that female rodents consume more alcohol (g/kg) than males (see references in [43]), which we also clearly observed. Finally, we make mention that there is evidence for other drugs of abuse, most notably cocaine, of greater escalation in females than males [44–46].
We observed a strong alignment of the transcriptional data with female, but not male, preference and consumption. Regarding this observation, we begin by noting the limitations of collecting data from only a single brain region, the CeA. However, previous studies (e.g., [23]) found that from the perspective of ethanol preference, the CeA is a key region. To our knowledge, the CeA is the only region in mice where lesions affect preference without affecting fluid consumption. The other limitation with the current study is that we only have transcriptional data from the end of the 3 month trial; obviously, a within-subject comparison prior to ethanol exposure and at the end of the trial would have been ideal. While such a study is not feasible in mice, the advantages of such a study in non-human primates, in which biopsy studies were obtained prior to and after ethanol drinking, has been recently demonstrated [12]. A key question to be addressed in the current study is whether the transcriptional signature detected at the end of the three-month trial is similar or different from the signature previously detected for preference risk [4,5]. The risk signature was extracted from HS-CC mice selectively bred for High and Low preference, but never exposed to ethanol. The comparison of the current study with our previous studies is not perfect --- the ideal comparison would be with mice selectively bred based on their three-month drinking profiles.
With this caveat in mind, we can clearly conclude that the transcriptional signature associated with initial preference risk differs significantly from the preference signature detected in the current study. Risk was clearly associated, across three brain regions (CeA, NAc shell and prelimbic cortex), with a cluster of genes associated with synaptic plasticity and cell adhesion. The latter group was enriched in cadherins and protocadherins. The key hub node for the risk cluster was Dlg2 which encodes for PSD-93. No sex effect was detected for the risk transcriptome, although the sample sizes were not sufficient to generate high quality female and male co-expression networks. In contrast, for the preference signature, we found that in female HS-CC mice, high preference was associated with the increased expression of a gene cluster enriched in annotations associated with the extracellular matrix and cilia. While it is known that alcohol affects the motile cilia in the brain’s ventricles and other tissues (see e.g., [47]), in the CeA and other brain regions there will only be primary cilia in neurons and astrocytes; there are no primary cilia associated with microglia. There are a number of proteins highly, if not entirely, localized in the neuronal primary cilia. These include ADCY3, SSTR3 and HT6R. There is some evidence that the manipulation of these cilia-specific molecules affects ethanol preference and consumption. For example, de Bruin et al. [48] found that a highly selective HT6R antagonist (CMP 42) attenuated both nicotine- and alcohol-seeking behaviors in Wistar rats. Further, Ht6r knockout mice are less sensitive to alcohol-induced ataxia and sedation [49], and HT6R antagonists reduce cocaine self-administration, attenuate cue-induced reinstatement, attenuate the expression of cocaine-induced conditioned place preference, and reduce the acquisition and expression of nicotine-induced sensitization (see references in [48]). However, in the CeA, Ht6r was either not expressed or was expressed but below our level of detection. Adcy3 & Sstr3 were substantially expressed but not correlated with preference. The orphan receptor, GPR88, is enriched in striatal neuronal primary cilia [50] and detectable expression is found in the CeA. The GPR88 agonist, RTI-13951–33, significantly reduces alcohol self-administration and intake in female Long-Evans rats in a dose-dependent manner, without effects on locomotion and sucrose self-administration. Consistent with these observations, we found that Gpr88 expression in females was moderately negatively correlated with preference (Supplementary Material Table S4). Of interest, Gpr88, Adcy3 & Sstr3 were not expressed in the brown module, which contained the ciliary structural constituents. Given that the brown module is enriched in astrocyte annotation genes, it could be reasonably argued that our attention should focus on astrocyte primary cilia. However as noted by Sterpka and Chen [51], “Presently, little is known about the function, signaling pathways, and structural dynamics of astrocytic primary cilia in the mature brain, although astrocytes fulfill a wide range of functions including providing trophic support, maintaining homeostasis, and protecting neurons from acute insults or brain injury [52]. Since astrocytes can proliferate under certain pathological conditions [53], astrocytic primary cilia are not static but subject to dynamic changes.”
The data presented here extend the observations from several laboratories that the extracellular matrix has a key role(s) in the regulation and consequences of ethanol consumption (e,g., [54,55]). The brain extracellular matrix has 3 main features: perineuronal nets (PNNs), basement membrane, and neural interstitial matrix. The precise effects of ethanol on the basement membrane are not clear. Lasek et al. [55] have summarized the effects of ethanol and addictive drugs on PNNs. In the cortex, PNNs are most prominently associated with parvalbumin-containing GABAergic interneurons [56]. Chen et al. [57] found that chronic binge drinking increases insular cortex PNNs, as measured by an increase in binding of Wisteria floribunda agglutinin, commonly used to label PNNs. Coleman et al. [58] observed that adolescent binge ethanol drinking increased several PNN proteins derived from astrocytes, including brevican, neurocan and tenascin-C. Zhang et al. [59] found that fetal ethanol exposure inhibited arylsulfatase B, which in turn decreased the turnover of the PNN proteoglycans with marked effects on synaptic development.
The neural interstitial matrix occupies most of the extracellular space of the brain parenchyma and is comprised of proteoglycans, tenascins, and link proteins [60]. A key component is hyaluronan (or hyaluronic acid), a long linear polysaccharide not attached to a protein core, but that interacts with and crosslinks extracellular matrix proteins [61]. Hyaluronan is synthesized by hyaluronan synthases present on the surface of neurons [61,62]; smaller amounts of laminin, fibronectin, and collagen are also present in the interstitial matrix [60]. Components of the interstitial matrix, fibronectin, laminin, collagen, tenascins, and proteoglycans are all processed by various proteases, including plasmin, tissue plasminogen activator (tPA), matrix metallopeptidases (MMPs), a disintegrin and metalloproteases (ADAMs), and ADAMTs (ADAMs with a thrombospondin motif) [63]. These proteases are directly implicated in the regulation of synapse formation, synaptic plasticity, and learning and memory (e.g., [64]). There is compelling evidence to link MMPs and other extracellular matrix proteases to ethanol exposure. Some key findings include: (1) Chronic broad spectrum MMP inhibition during ethanol dependence induction and withdrawal prevented escalated ethanol self-administration in rats; moreover, acute MMP inhibition after induction of ethanol dependence prevented expression of escalated ethanol intake [65]. (2) Go et al. [66] focused on the role(s) of the MMPs in withdrawal-induced escalation of ethanol self-administration and observed that MMP-9 activity was increased in the CeA and NAc as a function of escalation. Further, they observed that site-specific inhibition of MMP-9 blocked the withdrawal-induced escalation. (3) MMP-9 has key role(s) in the regulation of neuroimmune function [67], which has been shown to have a key functional role in the regulation of ethanol consumption [68,69]. (4) Some substituted tetracyclines reduce ethanol consumption [70,71], an effect not related to their antibiotic efficacy. While these drugs have a multitude of mechanisms, they are broad spectrum MMP inhibitors [72].
We observed that the genes positively correlated with female preference are largely localized in a single network module, brown, and a number of these genes (N=43) are top intramodular hub nodes. Queries revealed that 19 of these top hub nodes are regulated by Otx2 which is one of the 43 top hub nodes. Otx2 is often referred to a master regulator and is known to have key roles in normal brain patterning and postnatal plasticity. Through its interactions with various downstream targets, Otx2 is further required for the generation of various neuronal subpopulations, including ocular motor and midbrain dopaminergic neurons [73,74], and the development and the maintenance of the PNNs. In the adult brain, the existing evidence suggests that Otx2 expression is largely localized to the choroid plexus [75] and the OTX2 protein is captured by the perineuronal nets and accumulated in parvalbumin type GABA-ergic neurons throughout the brain, including in the cingulate cortex, the somatosensory cortex, the hippocampus, and the basolateral amygdala [76]. The data presented here suggest that there is low but detectable expression of Otx2 in the CeA. The cellular source of the mRNA is unknown. We have examined our published RNA-Seq datasets; the data found in Hitzemann et al. [77] appears typical. No Otx2 expression was detected in the prelimbic cortex, low expression was detected in the NAc and low to moderate expression was detected in the ventral midbrain. Walter et al. [12] have reported that Otx2 is not expressed in macaque cortical area 46. Thus, our data are consistent with reports that Otx2 is not expressed in the adult cortex (e.g. [76]); however, there appears to be some residual expression that persists in adult subcortical areas. This expression is affected by ethanol exposure and may have a role in the escalation of ethanol preference.
As noted above, the data presented here examine for the first time, unlimited ethanol availability for an extended period in a highly diverse mouse population. The HS-CC (and DO) mice appear to be approximately 10 times more genetically diverse than Homo sapiens [78,79]. Further, the HS-CC have no rare alleles that can complicate and/or diffuse the integration of the phenotypic and gene expression data. The data indicate that long-term ethanol exposure is associated with changes in the extracellular matrix, as well as the primary cilium. Some data suggest that Otx2 may have a role as a master regulator of these changes. Finally, we must acknowledge that during the review process, the issue was raised that our focus on extracellular matrix-related genes was associated with a gene length bias [38]. We believe that this bias will be especially notable for small sample sizes, and given the relatively large sample sizes in the present study the bias would not substantially affect our conclusions. Analytical results reported herein support this conclusion, yet we strongly encourage the reanalysis of our rich data set by others, and we contend that gene length is an issue that needs to be taken seriously. As we have shown in Supplementary Material Table S7, there is wide variation in the length of hub nodes across the network modules. To our knowledge, this is the first time this phenomenon has been reported and illustrates that the physical properties of genes and gene clusters cannot be ignored.
Supplementary Material
HIGHLIGHTS.
Chronic alcohol consumption alters the brain transcriptome
Highly diverse genetically heterogeneous mice vary in alcohol preference
Chronically drinking females but not males escalate preference
Primary cilium and extracellular matrix transcriptome impact female preference
A gene co-expression network for alcohol preference was enriched in astrocyte genes
Acknowledgments
Funding
This research was supported by the National Institutes of Health [grants NIH P60AA010760, U01AA013484, and R24AA020245]; and the VA Research Career Scientist Program.
Footnotes
Conflicts of interest
The authors declare that they have no competing financial interests. The contents of this article do not represent the views of the U.S. Department of Veterans Affairs or the United States government.
Appendix A. Supplementary Material
Supplementary data to this article can be found online.
Supplementary data
Supplementary material
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Mulligan MK, Ponomarev I, Hitzemann RJ, et al. Toward understanding the genetics of alcohol drinking through transcriptome meta-analysis. Proc Natl Acad Sci U S A. 2006;103(16):6368–6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hitzemann R, Malmanger B, Belknap J, Darakjian P, McWeeney S. Short-term selective breeding for high and low prepulse inhibition of the acoustic startle response; pharmacological characterization and QTL mapping in the selected lines. Pharmacol Biochem Behav. 2008;90(4):525–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Metten P, Iancu OD, Spence SE, et al. Dual-trait selection for ethanol consumption and withdrawal: genetic and transcriptional network effects. Alcohol Clin Exp Res. 2014;38(12):2915–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Colville AM, Iancu OD, Oberbeck DL, et al. Effects of selection for ethanol preference on gene expression in the nucleus accumbens of HS-CC mice. Genes Brain Behav. 2017;16(4):462–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Colville AM, Iancu OD, Lockwood DR, et al. Regional Differences and Similarities in the Brain Transcriptome for Mice Selected for Ethanol Preference From HS-CC Founders. Front Genet. 2018;9:300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Iancu OD, Colville AM, Wilmot B, et al. Gender-specific effects of selection for drinking in the dark on the network roles of coding and noncoding RNAs. Alcohol Clin Exp Res. 2018;42(8):1454–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Mulligan MK, Rhodes JS, Crabbe JC, Mayfield RD, Harris RA, Ponomarev I. Molecular profiles of drinking alcohol to intoxication in C57BL/6J mice. Alcohol Clin Exp Res. 2011;35(4):659–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Rodd ZA, Kimpel MW, Edenberg HJ, et al. Differential gene expression in the nucleus accumbens with ethanol self-administration in inbred alcohol-preferring rats. Pharmacol Biochem Behav. 2008;89(4):481–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Saba LM, Flink SC, Vanderlinden LA, et al. The sequenced rat brain transcriptome--its use in identifying networks predisposing alcohol consumption. FEBS J. 2015;282(18):3556–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].McBride WJ, Kimpel MW, McClintick JN, et al. Gene expression within the extended amygdala of 5 pairs of rat lines selectively bred for high or low ethanol consumption. Alcohol. 2013;47(7):517–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Iancu OD, Colville A, Walter NAR, et al. On the relationships in rhesus macaques between chronic ethanol consumption and the brain transcriptome. Addict Biol. 2018;23(1):196–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Walter NAR, Zheng CL, Searles RP, McWeeney SK, Grant KA, Hitzemann R. Chronic voluntary ethanol drinking in cynomolgus macaques elicits gene expression changes in prefrontal cortical area 46. Alcohol Clin Exp Res. 2020;44(2):470–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Farris SP, Riley BP, Williams RW, et al. Cross-species molecular dissection across alcohol behavioral domains. 2018;Alcohol;72:19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rao X, Thapa KS, Chen AB, et al. Allele-specific expression and high-throughput reporter assay reveal functional genetic variants associated with alcohol use disorders. Mol Psychiatry. 2019;Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Warden AS, Mayfield RD. Gene expression profiling in the human alcoholic brain. Neuropharmacology. 2017;122:161–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35(1):217–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Koob GF, Volkow ND. Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry. 2016;3(8):760–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Contet C Gene Expression Under the influence: Transcriptional profiling of ethanol in the brain. Curr Psychopharmacol. 2012;1(4):301–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hitzemann R, Edmunds S, Wu W, et al. Detection of reciprocal quantitative trait loci for acute ethanol withdrawal and ethanol consumption in heterogeneous stock mice. Psychopharmacology (Berl). 2009;203(4):713–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Iancu OD, Darakjian P, Walter NA, et al. Genetic diversity and striatal gene networks: focus on the heterogeneous stock-collaborative cross (HS-CC) mouse. BMC Genomics. 2010;11:585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Hitzemann B, Dains K, Kanes S, Hitzemann R. Further studies on the relationship between dopamine cell density and haloperidol-induced catalepsy. J Pharmacol Exp Ther. 1994;271:969–976. [PubMed] [Google Scholar]
- [22].Roberts A, Pardo-Manuel de Villena F, Wang W, McMillan L, Threadgill DW. The polymorphism architecture of mouse genetic resources elucidated using genome-wide resequencing data: implications for QTL discovery and systems genetics. Mamm Genome. 2007;18(6–7):473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Dhaher R, Finn D, Snelling C, Hitzemann R. Lesions of the extended amygdala in C57BL/6J mice do not block the intermittent ethanol vapor-induced increase in ethanol consumption. Alcohol Clin Exp Res. 2008;32(2):197–208. [DOI] [PubMed] [Google Scholar]
- [24].Steiger JH. Tests for comparing elements of a correlation matrix. Psychological Bulletin 1980; 87(2): 245–251. 10.1037/0033-2909.87.2.245 [DOI] [Google Scholar]
- [25].Lee IA, Preacher KJ. Calculation for the test of the difference between two dependent correlations with one variable in common [Computer software] September 2013. Available from http://quantpsy.org.
- [26].Franklin KBJ, Paxinos, G. The mouse brain in stereotaxic coordinates. Third Edition, 2007, Elsevier, Inc., Academic Press, San Diego, CA. [Google Scholar]
- [27].Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–930. [DOI] [PubMed] [Google Scholar]
- [29].Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Iancu OD, Kawane S, Bottomly D, Searles R, Hitzemann R, McWeeney S. Utilizing RNA-Seq data for de novo coexpression network inference. Bioinformatics. 2012;28(12):1592–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cahoy JD, Emery B, Kaushal A, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J Neurosci. 2008; 28(1):264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ringers C, Olstad EW, Jurisch-Yaksi N. The role of motile cilia in the development and physiology of the nervous system. Philos Trans R Soc Lond B Biol Sci. 2020;375(1792):2019.0156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chen EY, Tan CM, Kou Y, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kuleshov MV, Jones MR, Rouillard AD, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44(W1):W90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Johansson PA, Irmler M, Acampora D, et al. The transcription factor Otx2 regulates choroid plexus development and function. Development. 2013;140(5):1055–1066. [DOI] [PubMed] [Google Scholar]
- [38].Mandelboum S, Manber Z, Elroy-Stein O, Elkon R. Recurrent functional misinterpretation of RNA-seq data caused by sample-specific gene length bias. PLoS Biol. 2019; 17(11):e3000481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Rosenwasser AM, Fixaris MC, Crabbe JC, Brooks PC, Ascheid S. Escalation of intake under intermittent ethanol access in diverse mouse genotypes. Addict Biol 2013;18(3):496–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chesler EJ, Gatti DM, Morgan AP, et al. Diversity Outbred Mice at 21: Maintaining Allelic Variation in the Face of Selection. G3 (Bethesda). 2016;6(12):3893–3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Keyes KM, Martins SS, Blanco C, Hasin DS. Telescoping and gender differences in alcohol dependence: new evidence from two national surveys. Am J Psychiatry. 2010;167(8):969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sharrett-Field L, Butler TR, Reynolds AR, Berry JN, Prendergast MA. Sex differences in neuroadaptation to alcohol and withdrawal neurotoxicity. Pflugers Arch. 2013;465(5):643–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Becker JB, Koob GF. Sex Differences in Animal Models: Focus on Addiction. Pharmacol Rev. 2016;68(2):242–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Anker JJ, Carroll ME. Females are more vulnerable to drug abuse than males: evidence from preclinical studies and the role of ovarian hormones. Curr Top Behav Neurosci. 2011;8:73–96. [DOI] [PubMed] [Google Scholar]
- [45].Becker JB, Hu M. Sex differences in drug abuse. Front Neuroendocrinol. 2008;29(1):36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Becker JB, Perry AN, Westenbroek C. Sex differences in the neural mechanisms mediating addiction: a new synthesis and hypothesis. Biol Sex Differ. 2012;3(1):14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Omran AJA, Saternos HC, Althobaiti YS, et al. Alcohol consumption impairs the ependymal cilia motility in the brain ventricles. Sci Rep. 2017;7(1):13652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].de Bruin NMWJ McCreary AC, van Loevezijn A, et al. A novel highly selective 5-HT6 receptor antagonist attenuates ethanol and nicotine seeking but does not affect inhibitory response control in Wistar rats. Behav Brain Res. 2013;236(1):157–165. [DOI] [PubMed] [Google Scholar]
- [49].Bonasera SJ, Chu HM, Brennan TJ, Tecott LH. A null mutation of the serotonin 6 receptor alters acute responses to ethanol. Neuropsychopharmacology. 2006;31(8):1801–1813. [DOI] [PubMed] [Google Scholar]
- [50].Ehrlich AT, Semache M, Bailly J, et al. Mapping GPR88-Venus illuminates a novel role for GPR88 in sensory processing. Brain Struct Funct. 2018;223(3):1275–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Sterpka A, Chen X. Neuronal and astrocytic primary cilia in the mature brain. Pharmacol Res. 2018;137:114–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Molofsky AV, Krencik R, Ullian EM, et al. Astrocytes and disease: a neurodevelopmental perspective. Genes Dev. 2012;26(9):891–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119(1):7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Lasek AW. Effects of Ethanol on Brain Extracellular Matrix: Implications for Alcohol Use Disorder. Alcohol Clin Exp Res. 2016;40(10):2030–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Lasek AW, Chen H, Chen WY. Releasing Addiction Memories Trapped in Perineuronal Nets. Trends Genet. 2018;34(3):197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Fawcett JW, Oohashi T, Pizzorusso T. The roles of perineuronal nets and the perinodal extracellular matrix in neuronal function. Nat Rev Neurosci. 2019;20(8):451–465. [DOI] [PubMed] [Google Scholar]
- [57].Chen H, He D, Lasek AW. Repeated Binge Drinking Increases Perineuronal Nets in the Insular Cortex. Alcohol Clin Exp Res. 2015;39(10):1930–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Coleman LG Jr, Liu W, Oguz I, Styner M, Crews FT. Adolescent binge ethanol treatment alters adult brain regional volumes, cortical extracellular matrix protein and behavioral flexibility. Pharmacol Biochem Behav. 2014;116:142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zhang X, Bhattacharyya S, Kusumo H, Goodlett CR, Tobacman JK, Guizzetti M. Arylsulfatase B modulates neurite outgrowth via astrocyte chondroitin-4-sulfate: dysregulation by ethanol. Glia. 2014;62(2):259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lau LW, Cua R, Keough MB, Haylock-Jacobs S, Yong VW. Pathophysiology of the brain extracellular matrix: a new target for remyelination. Nat Rev Neurosci. 2013;14(10):722–729. [DOI] [PubMed] [Google Scholar]
- [61].Kwok JC, Dick G, Wang D, Fawcett JW. Extracellular matrix and perineuronal nets in CNS repair. Dev Neurobiol. 2011;71(11):1073–1089. [DOI] [PubMed] [Google Scholar]
- [62].Kwok JC, Carulli D, Fawcett JW. In vitro modeling of perineuronal nets: hyaluronan synthase and link protein are necessary for their formation and integrity. J Neurochem. 2010;114(5):1447–1459. [DOI] [PubMed] [Google Scholar]
- [63].Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. 2014;15(12):786–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Baranes D, Lederfein D, Huang YY, Chen M, Bailey CH, Kandel ER. Tissue plasminogen activator contributes to the late phase of LTP and to synaptic growth in the hippocampal mossy fiber pathway. Neuron 1998;21(4):813–825. [DOI] [PubMed] [Google Scholar]
- [65].Smith AW, Nealey KA, Wright JW, Walker BM. Plasticity associated with escalated operant ethanol self-administration during acute withdrawal in ethanol-dependent rats requires intact matrix metalloproteinase systems. Neurobiol Learn Mem. 2011;96(2):199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Go BS, Sirohi S, Walker BM. The role of matrix metalloproteinase-9 in negative reinforcement learning and plasticity in alcohol dependence. Addict Biol 2020;25(2):e12715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Vafadari B, Salamian A, Kaczmarek L. MMP-9 in translation: from molecule to brain physiology, pathology, and therapy. J Neurochem. 2016;139 Suppl 2:91–114. [DOI] [PubMed] [Google Scholar]
- [68].Ray LA, Roche DJ, Heinzerling K, Shoptaw S. Opportunities for the development of neuroimmune therapies in addiction. Int Rev Neurobiol. 2014;118:381–401. [DOI] [PubMed] [Google Scholar]
- [69].Cui C, Shurtleff D, Harris RA. Neuroimmune mechanisms of alcohol and drug addiction. Int Rev Neurobiol. 2014;118:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Bergeson SE, Nipper MA, Jensen J, Helms ML, Finn DA. Tigecycline reduces ethanol intake in dependent and nondependent male and female C57BL/6J mice. Alcohol Clin Exp Res. 2016;40(12):2491–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Syapin PJ, Martinez JM, Curtis DC, et al. Effective reduction in high ethanol drinking by semisynthetic tetracycline derivatives. Alcohol Clin Exp Res. 2016;40(12):2482–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Ryan ME, Usman A, Ramamurthy NS, Golub LM, Greenwald RA. Excessive matrix metalloproteinase activity in diabetes: inhibition by tetracycline analogues with zinc reactivity. Curr Med Chem. 2001;8(3):305–316. [DOI] [PubMed] [Google Scholar]
- [73].Tripathi PP, Di Giovannantonio LG, Sanguinetti E, et al. Increased dopaminergic innervation in the brain of conditional mutant mice overexpressing Otx2: effects on locomotor behavior and seizure susceptibility. Neuroscience. 2014;261:173–183. [DOI] [PubMed] [Google Scholar]
- [74].Sherf O, Nashelsky Zolotov L, Liser K, et al. Otx2 requires Lmx1b to control the development of mesodiencephalic dopaminergic neurons. PLoS One. 2015;10(10):e0139697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Planques A, Oliveira Moreira V, Dubreuil C, Prochiantz A, Di Nardo AA. OTX2 signals from the choroid plexus to regulate adult neurogenesis. eNeuro. 2019;6(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Spatazza J, Lee HH, Di Nardo AA, et al. Choroid-plexus-derived Otx2 homeoprotein constrains adult cortical plasticity. Cell Rep. 2013;3(6):1815–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Hitzemann R, Iancu OD, Reed C, Baba H, Lockwood DR, Phillips TJ. Regional Analysis of the Brain Transcriptome in Mice Bred for High and Low Methamphetamine Consumption. Brain Sci. 2019;9(7):E155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Phifer-Rixey M, Bonhomme F, Boursot P, et al. Adaptive evolution and effective population size in wild house mice. Mol Biol Evol. 2012;29(10):2949–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Tenesa A, Navarro P, Hayes BJ, et al. Recent human effective population size estimated from linkage disequilibrium. Genome Res. 2007;17(4):520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Warde-Farley D, Donaldson SL, Comes O, et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38:W214–W220. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.