

Summary
KPNA2/importin-alpha1 (karyopherin subunit alpha 2) is the primary nucleocytoplasmic transporter for some transcription factors to activate cellular proliferation and differentiation. Aberrant increase of KPNA2 level is identified as a prognostic marker in a variety of cancers. Yet, the turnover mechanism of KPNA2 remains unknown. Here, we demonstrate that KPNA2 is degraded via the chaperone-mediated autophagy (CMA) and that Zika virus (ZIKV) enhances the KPNA2 degradation. KPNA2 contains a CMA motif, which possesses an indispensable residue Gln109 for the CMA-mediated degradation. RNAi-mediated knockdown of LAMP2A, a vital component of the CMA pathway, led to a higher level of KPNA2. Moreover, ZIKV reduced KPNA2 via the viral NS2A protein, which contains an essential residue Thr100 for inducing the CMA-mediated KPNA2 degradation. Notably, mutant ZIKV with T100A alteration in NS2A replicates much weaker than the wild-type virus. Also, knockdown of KPNA2 led to a higher ZIKV viral yield, which indicates that KPNA2 mediates certain antiviral effects. These data provide insights into the KPNA2 turnover and the ZIKV-cell interactions.
KEYWORDS: Chaperone-mediated autophagy (CMA), importin-alpha-1, KPNA2, NS2A, Zika virus, ZIKV
Introduction
Karyopherins are a group of proteins mediating the nucleocytoplasmic trafficking of numerous proteins, among which many are transcription factors that are involved in cellular proliferation and differentiation, and host immunity [1–4]. In the classical nuclear import pathway, cytoplasmic cargoes bearing a classical nuclear localization sequence (NLS) are first recognized by an importin alpha, which then associates with importin beta [2]. The ternary complex of importin beta/alpha/NLS-cargo translocates through the nuclear pore complex (NPC) to the nucleus.
Seven isoforms of importin alpha (KPNA1-KPNA7 [karyopherin subunit alpha]) are reported and grouped into three subfamilies [2,5,6]. KPNA2 and KPNA7 belong to the same subfamily of KPNA isoforms with 53% identical amino acids [6]. All KPNA isoforms are expressed in all adult cells to some extent with the exception of KPNA5, which is restricted to the testis [7], and KPNA7, which is limited to the ovaries and early-stage embryos [8,9]. The KPNAs are composed of a flexible N-terminal importin beta binding (IBB) domain, and a highly structured domain of ten tandem armadillo (ARM) repeats. The helical ARM repeats form a groove for NLS binding. The IBB domain interacts in trans with importin beta and in cis with the NLS-binding groove. The competitive binding of the IBB domain regulates the binding of NLS-containing cargos [10]. Each isoform of KPNAs has its preference for cargos. KPNA2 transports the POU domain transcription factor POU5F1/OCT4 (POU class 5 homeobox 1) 11], transcription factor MYC/c-Myc [12], E2F1 [13], the small GTPase RAC1 [14], the deoxynucleotide-triphosphate (dNTP) hydrolase SAMHD1 (sterile alpha motif and histidine/aspartic acid domain 1) [15], RELA/NF-κB p65 [16], NBN/NBS1 [17] and so on. However, KPNA2 inhibits the nuclear translocation of POU transcription factor POU3F1/OCT6 and POU3F2/BRN2 [18].
Aberrant KPNA2 expression levels are found in a variety of cancers, including melanoma, brain, liver, lung, ovarian, and prostate cancers [19–22]. Increased KPNA2 protein level is considered as a prognostic marker for various human cancers, including colorectal cancer [23,24], bladder cancer [25], epithelial ovarian cancer [26], hepatocellular carcinoma [27], oral cancer [28], lung cancer [29], gastric adenocarcinoma [30], epithelial ovarian cancer [31], prostate cancer [20], and breast cancer [32].
KPNA2 is essential for the maintenance and differentiation of embryonic stem cells (ESCs). The knockdown of KPNA2 expression in ESCs leads to a reduction of the transcription factors that maintain pluripotency (e.g. POU5F1, NANOG, and SOX2) and induces cell differentiation into several lineages [18]. KPNA2 plays a crucial role in the maintenance of undifferentiated ESCs through the inhibition of the nuclear import of specific transcription factors like POU3F1/OCT6 that induce cell differentiation [18]. Therefore, the intracellular distribution of transcription factors is carefully controlled for the regulation of their activities. Yet, the turnover mechanism of KPNA2 is unknown.
ZIKV is a mosquito-borne RNA virus of the genus Flavivirus, the family Flaviviridae. ZIKV was first isolated from a monkey in the Zika forest of Uganda in 1947 [33,34]. The virus drew little attention until the first documented human outbreak occurred in the Pacific Islands and the recent epidemic in South America [35–37]. ZIKV has spread to over 60 countries [38–40]. The majority of the ZIKV-infected people are asymptomatic or have only mild symptoms. ZIKV-infected pregnant women, however, can pass the virus to their fetuses, leading to severe congenital microcephaly and neurological deficiency [41,42]. Also, ZIKV infection is associated with Guillain-Barre syndrome (GBS), a rare but serious autoimmune disorder [43–46].
Phylogenetic studies group ZIKV strains into two distinct lineages: Asian and African [47]. ZIKV infects neural precursor cells derived from pluripotent stem cells and causes apoptotic cell death and cell-cycle dysregulation [48,49]. ZIKV also infects human neural stem cells, inhibits their differentiation into neuroprogenitor, and causes the death of early differentiating neuroprogenitor cells [50]. The Asian lineage ZIKV is shown to infect the brains of the embryonic mice and cause microcephaly [51–53].
ZIKV is enveloped with a single-stranded, positive-sense RNA genome of about 10.7 kb in length. It encodes a single polyprotein that is produced and cleaved co-translationally into three structural proteins (capsid [C], precursor membrane [prM], and envelope [E]) and seven non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) [54]. Neither an effective treatment nor a vaccine is available for the control and prevention of ZIKV infection.
The recent outbreak in the central and south Americas has drawn wide public attention to ZIKV infection and led to the latest research on ZIKV. However, ZIKV-cell interactions are not well understood. Our earlier study demonstrated that ZIKV infection induces the elevation of KPNA6, another KPNA isoform, and that KPNA6 knockout dampens ZIKV replication [55]. During the study of KPNA6, we wondered if ZIKV had any effect on other KPNA isoforms. We conducted a preliminary test on KPNA1 and KPNA2 protein levels in ZIKV-infected cells and noticed the reduction of KPNA2. The objective of this study was to confirm the observation and determine the mechanism of ZIKV-induced reduction of KPNA2. We discovered that KPNA2 was degraded via the CMA pathway and that ZIKV infection enhanced the KPNA2 turnover via the viral NS2A protein. Notably, KPNA2 knockdown led to elevation of ZIKV proliferation. ZIKV with NS2A mutation that failed to reduce KPNA2 had much weaker replication than the wild-type virus. These results indicate that ZIKV mediates the KPNA2 downregulation via NS2A, which interferes with the KPNA2 functions and potentially contributes to the ZIKV invasion and pathogenesis.
Results
ZIKV infection reduces the KPNA2 protein level
The effect of ZIKV infection on KPNA2 was determined in Vero cells, which were infected with the ZIKV PRVABC59 strain (Asian lineage, GenBank Accession Number KX377337) [56] at a multiplicity of infection (MOI) of 10. Western blotting (WB) was performed to determine the KPNA2 protein level. The results showed that the ZIKV infection at 24 hours post-infection (hpi) reduced KPNA2 by approximately 80% in comparison to the mock-infected cells, whereas the level of KPNA1, another isoform of KPNA, had minimal change (Figure 1A). To investigate how ZIKV infection induced the KPNA2 reduction, we first tested whether ZIKV had any effect on KPNA2 transcription. Reverse transcription and quantitative PCR (RT-qPCR) was conducted. The results showed that ZIKV infection had minimal effect on the KPNA2 mRNA level in comparison with the mock-infected cells (Figure 1B), which indicates that the reduction of KPNA2 protein level in ZIKV-infected cells is probably not due to interruption at the transcriptional level.
Figure 1.
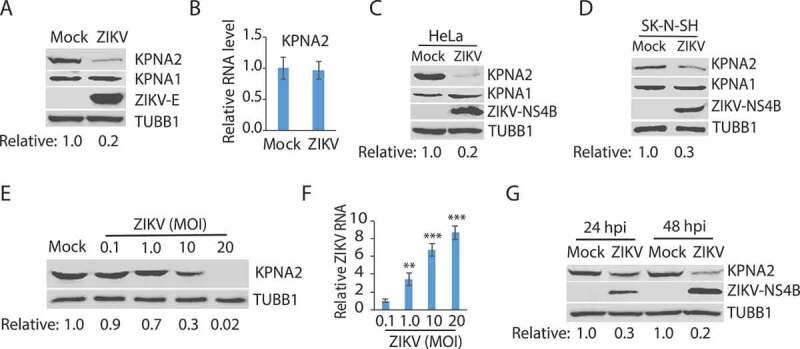
ZIKV infection reduces the KPNA2 protein level. (A) ZIKV infection of Vero cells leads to a lower KPNA2 protein level while having minimal effect on KPNA1. The cells were inoculated with ZIKV PRVABC59 strain at an MOI of 10 and harvested for western blotting (WB) at 24 h post-infection (hpi). Relative levels of KPNA2 are shown below the images after normalization with house-keeping gene TUBB1/tubulin beta. (B) ZIKV infection has minimal effect of KPNA2 mRNA level detected by RT-qPCR. The relative KPNA2 mRNA level is shown in comparison to the mock-infected Vero cells after normalization with the house-keeping gene RPL32. The cells were infected with ZIKV PRVABC59 strain at an MOI of 10 and harvested at 24 hpi. Error bars represent the standard errors of the means of three repeated experiments. (C and D) ZIKV infection of HeLa and SK-N-SH cells reduces KPNA2 level while having minimal effect on KPNA1. The cells were inoculated with ZIKV PRVABC59 strain at an MOI of 10 and harvested at 48 hpi. (E) The infection of Vero cells with incremental ZIKV inoculum leads to a dose-dependent reduction of KPNA2. The cells were harvested for WB at 24 hpi. Relative levels of KPNA2 are shown below the images. (F) ZIKV RNA levels in the infected cells at 24 hpi detected by RT-qPCR. The relative RNA levels in comparison with the MOI of 0.1 are shown. Error bars represent the standard errors of the means of three repeated experiments. Significant differences in RNA level from the sample of MOI 0.1 are denoted by asterisks (**, P < 0.01; ***, P < 0.001). G. ZIKV reduces KPNA2 in Vero cells at both 24 and 48 hpi
The results above showed KPNA2 reduction in ZIKV-infected Vero cells. We reasoned that ZIKV infection of other types of cells should have a similar effect on KPNA2. Indeed, ZIKV infection of HeLa and human neuroblastoma SK-N-SH cells led to lower KPNA2 but had minimal effect on KPNA1 level (Figure 1C,D).
As the ZIKV infection led to the KPNA2 reduction, we speculated that a higher rate of viral infection would induce a lower KPNA2 level. Indeed, Vero cells that were infected with ZIKV PR strain at the MOIs of 0.1, 1, 10, and 20 had KPNA2 protein level reduced further along with the incremental MOI (Figure 1E). ZIKV replication was confirmed by the ZIKV RNA level determined with RT-qPCR (Figure 1F). Moreover, the ZIKV-mediated reduction of KPNA2 level was verified at both 24 and 48 hpi (Figure 1G).
Lysosomal proteolysis accounts for ZIKV-mediated KPNA2 degradation
Proteasomes and lysosomes are the primary sites for protein degradation in cells [57,58]. We then tested whether the KPNA2 reduction was due to degradation by the ubiquitin-proteasome pathway or the lysosomal proteolysis. MG132, a proteasome inhibitor, and NH4Cl, a lysosomotropic compound that raises the lysosomal pH to block proteolysis [59], were added to the cells at 24 hpi. The cells were harvested 6 h later for Western blotting. The treatment with both MG132 and NH4Cl resulted in the restoration of the KPNA2 level in the ZIKV-infected Vero cells (Figure 2A). Compared with the DMSO solvent control, the 6-h treatment (from 24 to 30 hpi) with MG132 or NH4Cl had minimal effect on cell viability (Figure 2B) and ZIKV RNA level (Figure 2C). These results indicate that the restoration of the KPNA2 level by the two inhibitors was not due to their potential negative impacts on cell growth or virus replication. As MG132 also inhibits some lysosomal hydrolases [59], these results suggest that ZIKV-mediated KPNA2 reduction is via the lysosomal proteolysis instead of the ubiquitin-proteasome pathway.
Figure 2.
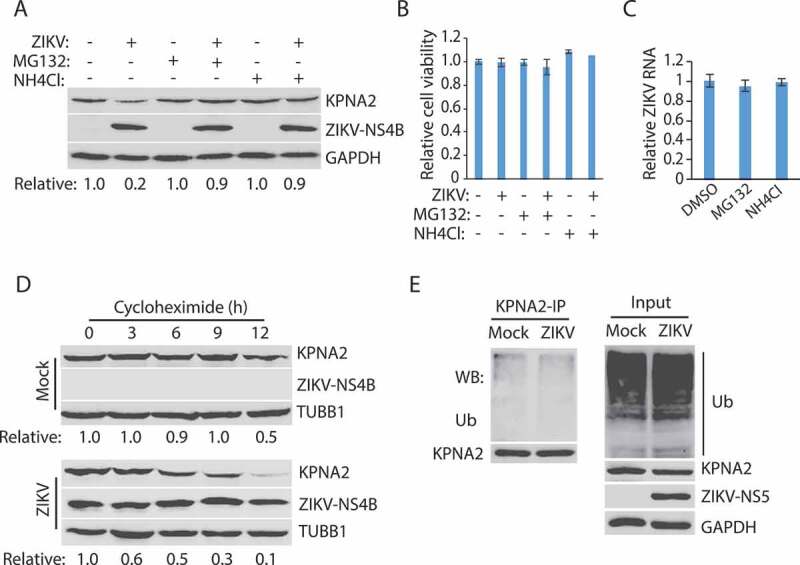
ZIKV infection induces KPNA2 degradation via the lysosomal pathway. (A) MG132 and NH4Cl treatment restore the KPNA2 level in ZIKV-infected cells. Vero cells were infected with ZIKV and 24 hpi, treated with MG132 or NH4Cl. The cells were harvested 6 h after the treatment. DMSO was included as a solvent control in the first two lanes. (B) The treatment with MG132 and NH4Cl for 6 h has minimal effect on cell viability. DMSO was included as a solvent control in the first two bars. (C) The treatment with MG132 and NH4Cl for 6 h has minimal effect on the ZIKV RNA level. (D) KPNA2 half-life is shortened by ZIKV infection. Vero cells were infected with ZIKV at an MOI of 10 for 24 h before treated with cycloheximide. Mock-infected cells were included for control. (E) ZIKV infection has a minimal effect on KPNA2 ubiquitination. Vero cells were infected with ZIKV, treated with NH4Cl for 6 h, and harvested for immunoprecipitation (IP) with KPNA2 antibody. The IP precipitate was subjected to WB with antibodies against ubiquitin (Ub) and KPNA2. The input of cell lysate was included as a control
ZIKV shortens KPNA2 half-life
As ZIKV reduced the KPNA2 protein level, we reasoned that the KPNA2 half-life in the ZIKV-infected cells would be shortened. Vero cells were infected with ZIKV and, 24 h later, treated with cycloheximide, an inhibitor of protein translation. WB results showed that KPNA2 level was reduced much faster in the infected than the control cells (Figure 2D). Densitometry analysis indicated that ZIKV infection shortened the KPNA2 half-life from 12 to 6 h.
The results above showed that ZIKV infection possibly reduced the KPNA2 level via the lysosomal proteolysis. We reasoned that the ZIKV infection would not induce KPNA2 polyubiquitination, which is needed for ubiquitin-proteasome degradation. To test this, we conducted co-immunoprecipitation (co-IP) of KPNA2, followed by WB with antibodies against ubiquitin and KPNA2. Treatment of the cells with NH4Cl to restore the KPNA2 level was done for comparison of KPNA2 ubiquitination in the ZIKV-infected and the control cells. Results showed that ZIKV infection had minimal effect on the KPNA2 ubiquitination level (Figure 2E). WB of input cell extract showed a similar total ubiquitination level between the infected and mock-infected cells. These results indicate that ZIKV-induced KPNA2 reduction does not depend on KPNA2 ubiquitination.
ZIKV induces KPNA2 reduction via selective autophagy
Protein degradation by lysosomes is mediated by autophagy, which includes macroautophagy, microautophagy, and chaperon-mediated autophagy (CMA) [60]. As ZIKV did not induce a change of KPNA1 level, we reasoned that the lysosomal degradation of KPNA2 could be via the microautophagy or CMA as they are selective in cargos whereas macroautophagy is generally not. Both the microautophagy or CMA pathways need the interaction of cargo proteins with the cytoplasmic chaperone, HSPA8/HSC70 (heat shock protein family A (Hsp70) member 8), which targets the cargos to the lysosome [60]. The CMA pathway depends on the lysosomal membrane receptor, LAMP2A (lysosome membrane-associated protein type 2A), which is not required for endosomal microautophagy [60]. We surmised that KPNA2 interacted with LAMP2A if ZIKV induced the KPNA2 reduction via the CMA pathway. Co-IP results showed that KPNA2 precipitates contained LAMP2A, while isotype control IgG did not (Figure 3A). Similarly, the co-IP precipitates of LAMP2A contained KPNA2 (Figure 3B). These results indicate that KPNA2 interacted with LAMP2A in the CMA pathway.
Figure 3.
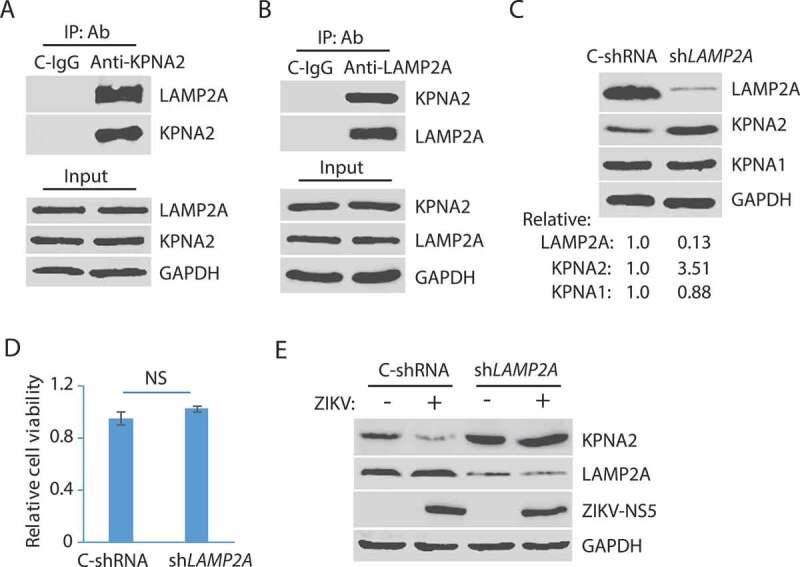
ZIKV-induced KPNA2 reduction is via the chaperon-mediated autophagy (CMA). (A) Co-immunoprecipitation (co-IP) of KPNA2 precipitates LAMP2A. Lysate of Vero cells was used for co-IP with KPNA2 antibody (anti-KPNA2) or an isotype control IgG (C-IgG). The input of the cell lysate was included in WB for control. (B) Co-IP of LAMP2A precipitates KPNA2. Lysate of Vero cells was used for co-IP with the LAMP2A antibody (anti-LAMP2A) or an isotype control IgG (C-IgG). (C) LAMP2A knockdown in Vero cells leads to the elevation of the KPNA2 level while having minimal effect on KPNA1. Vero cells stably transduced with recombinant retrovirus expressing control shRNA (C-shRNA) or shRNA against LAMP2A (shLAMP2A) were harvested for WB. Relative levels of LAMP2A, KPNA1, and KPNA2 are shown below the images. (D) LAMP2A knockdown in Vero cells has minimal effect on cell viability. NS: no significant difference. (E) ZIKV infection of Vero cells with LAMP2A knockdown has minimal effect on KPNA2. Vero cells stably transduced with retroviral shLAMP2A or C-shRNA were infected with ZIKV and harvested at 48 hpi for WB
The limiting factor in the CMA pathway is LAMP2A, as the knockdown of it blocks the CMA [61]. We speculated that LAMP2A knockdown would prevent KPNA2 degradation. To test this, we stably transduced Vero cells with recombinant retrovirus expressing shRNA against LAMP2A (shLAMP2A). An irrelevant shRNA (C-shRNA) was included as a control in the stable transduction. WB results showed that the KPNA2 level in the cells with LAMP2A knockdown was 3.51-fold higher than the cells with control shRNA, while KPNA1 had a minimal change (Figure 3C). LAMP2A level in the cells treated with shLAMP2A was reduced to 13% in comparison to the cells with control shRNA. Cell viability assay showed that the LAMP2A knockdown had minimal effect on cell growth (Figure 3D). ZIKV infection of the cells with the LAMP2A knockdown had minimal effect on the KPNA2 level while reducing the KPNA2 in the cells with the control shRNA (Figure 3E). These results demonstrate that the CMA pathway is responsible for KPNA2 degradation and that ZIKV infection accelerates the KPNA2 turnover.
ZIKV NS2A induces KPNA2 reduction
ZIKV encodes a polyprotein that is cleaved into individual structural or non-structural proteins: C, prM, E, NS1, NS2A, NS2B, NS2B3, NS3, NS4A, NS4B, and NS5 [54]. To determine which of these ZIKV proteins induces the KPNA2 reduction, we cloned sequences encoding these individual proteins for transient expression. HEK293 cells were co-transfected with plasmids encoding KPNA2 and one of the ZIKV proteins. WB results showed that compared with the empty vector control, ZIKV NS2A induced a significant reduction of KPNA2 protein level, whereas the other proteins had a minimal effect (Figure 4A,B).
Figure 4.
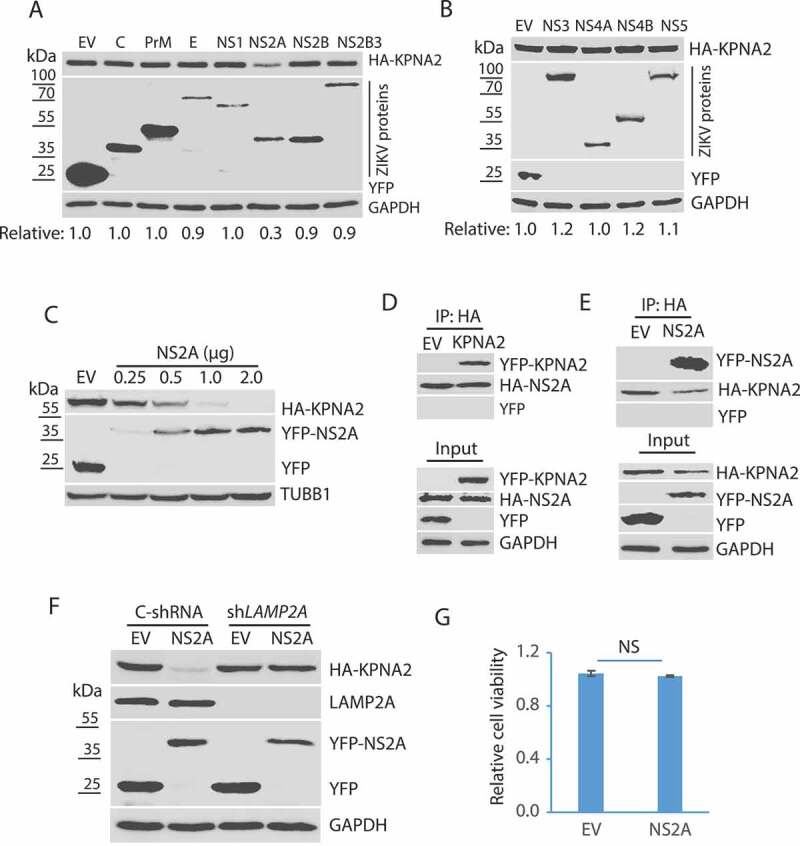
ZIKV NS2A protein induces KPNA2 reduction. (A and B). Identification of the ZIKV protein that causes KPNA2 reduction. HEK293 cells were co-transfected with the plasmids encoding the individual ZIKV proteins and HA-KPNA2. The cells were harvested for WB at 48 h post-transfection. Molecular weight markers are indicated on the left. Relative KPNA2 levels are shown below the images. EV: empty vector. (C) Dose-dependent reduction of KPNA2 by NS2A. HEK293 cells were co-transfected with HA-KPNA2 and NS2A plasmids. (D) KPNA2 is present in NS2A co-IP precipitates while YFP from the empty vector (EV) control is absent. HEK293 cells were co-transfected with HA-NS2A and YFP-KPNA2 plasmids. The input of cell lysate is included for control. (E) NS2A is present in KPNA2 co-IP precipitates. HEK293 cells were co-transfected with YFP-NS2A and HA-KPNA2 plasmids. (F) NS2A has minimal effect on the KPNA2 level in HEK293 cells with LAMP2A knockdown. HEK293 cells were transfected with control shRNA (C-shRNA) or shLAMP2A plasmids for 48 h and passaged. The transfection and passaging were repeated for three times before the cells were used for co-transfection with NS2A and KPNA2 plasmids. EV was included as a control. (G) NS2A expression in HEK293 cells has minimal effect on cell viability
To further confirm the effect of NS2A on KPNA2 reduction, we transfected HEK293 cells with an incremental amount of NS2A plasmid DNA and determined the KPNA2 level. WB result showed KPNA2 level was reduced in a dose-dependent manner along with the additional amount of NS2A plasmid (Figure 4C).
As NS2A induces the reduction of KPNA2, we speculated that the two proteins might interact. To test the speculation, we transfected HEK293 cells with the NS2A and KPNA2 plasmids for co-IP and WB. The results showed that NS2A precipitates contained KPNA2 but not YFP alone (Figure 4D). Similarly, NS2A was present in the KPNA2 precipitates, whereas YFP from the EV control was not (Figure 4E). To determine if NS2A reduces KPNA2 via the CMA pathway, we co-transfected HEK293 cells with NS2A and KPNA2 plasmids. WB results showed that NS2A reduced KPNA2 in the cells with control shRNA, whereas it had minimal effect on KPNA2 in the cells with the LAMP2A knockdown (Figure 4F). These results indicate that NS2A interacts with KPNA2 and induces its reduction via the CMA pathway. Cell viability assay excluded the possibility that NS2A might have cytotoxicity in HEK293 cells (Figure 4G).
The amino acid residue Thr100 of NS2A is indispensable for the NS2A-mediated reduction of KPNA2
NS2A is predicted to be a transmembrane protein with 226 amino acid residues. There are seven potential transmembrane domains in NS2A protein based on the Phobius prediction of transmembrane domains (http://phobius.sbc.su.se/poly.html). The analysis suggests there are four motifs oriented to expose to the cytoplasmic side: amino acids (aa) 24–35, 95–103, 144–164, and 220–226. Based on the sequence analysis, we constructed three truncation mutants of NS2A to determine the motif that correlates with KPNA2 reduction: NS2A-D1 (aa 1–100); NS2A-D2 (aa 90–180), and NS2A-D3 (aa 160–226) (Figure 5A).
Figure 5.
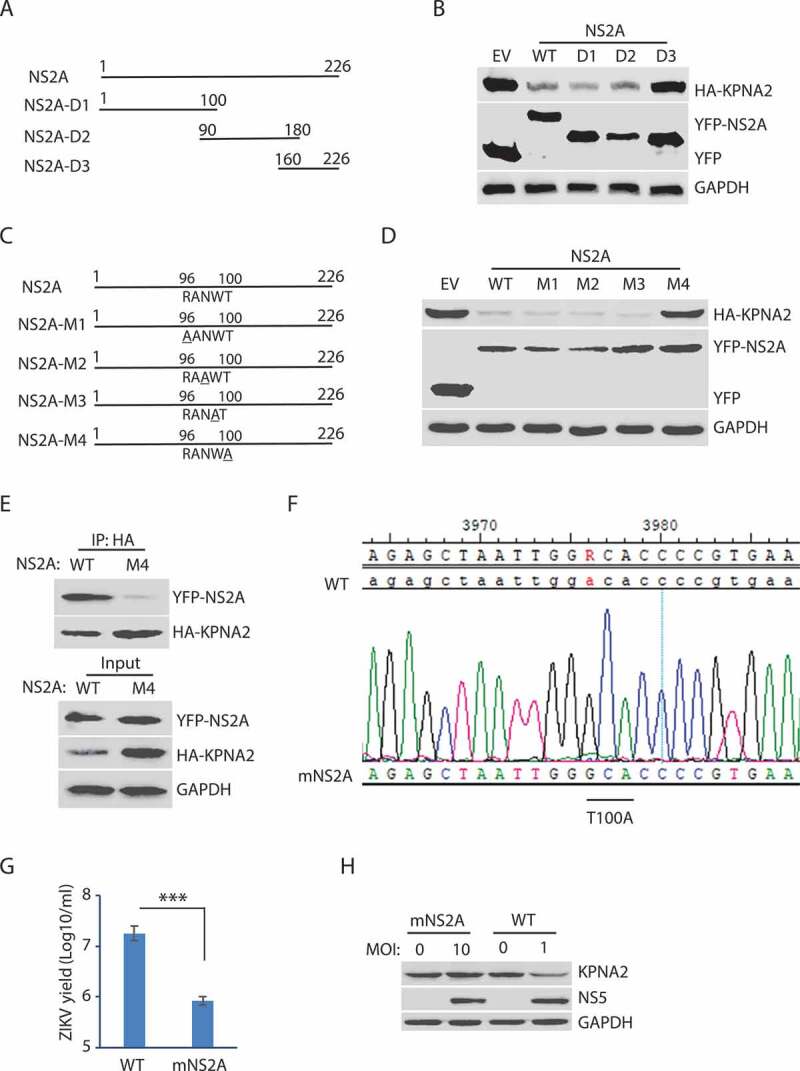
The amino acid residue T100 of NS2A is indispensable for the NS2A-mediated reduction of KPNA2. (A) Schematic illustration of the NS2A truncation constructs. The numbers above the lines indicate NS2A amino acid residues. (B) WB of KPNA2 in HEK293 cells co-transfected with plasmids of NS2A truncation constructs and HA-KPNA2. Wild-type (WT) NS2A and EV were included as controls. (C) Schematic illustration of mutant NS2A with point mutations. The amino acid residues of 96–100 are shown below the top line. Underlined residues indicate point mutations. M1: R96A, M2: N98A, M3: W99A, and M4: T100A. (D) WB of KPNA2 in HEK293 cells co-transfected with plasmids of mutant NS2A with point mutations and HA-KPNA2. WT NS2A and EV were included as controls. (E) Mutant NS2A-M4 (T100A) has minimal interaction with KPNA2. HEK293 cells were co-transfected with plasmids of HA-KPNA2 and WT NS2A or NS2A-M4. The input of cell lysate was included in WB. (F) Sequencing of the cDNA of the mutant ZIKV with NS2AT100A confirms the presence of codon mutation in its genome. The numbers above the sequence indicate nucleotide positions in cDNA of the ZIKV genome. The bar below the chromatogram indicates the codon change of T100A in NS2A. (G) Mutant ZIKV ICD (mNS2A) has a much lower proliferation than the WT virus. Vero cells were inoculated with an MOI of 0.01 for ZIKV proliferation and harvested for virus titration at 72 hpi. A significant difference is denoted by *** for P< 0.001. (H) Mutant ZIKV has minimal effect on the KPNA2 level. Vero cells were inoculated with an MOI of 10 for the mutant mNS2A virus or with an MOI of 1 for the WT virus to ensure both mutant and WT viruses had a similar level of replication and harvested at 48 hpi for immunoblotting
Transfection of HEK293 cells with the full-length and truncated NS2A plasmids, followed by WB with the antibody against KPNA2, was done. The results showed that NS2A-D3 could not reduce KPNA2, while NS2A-D1 and NS2A-D2 reduced KPNA2 similarly to the wild-type NS2A (Figure 5B). The finding suggests that the residues aa 90–100 of NS2A contain the motif inducing KPNA2 reduction as both the NS2A D1 and D2 truncates were able to cause KPNA2 reduction, and they overlap in the ten residues. Within the ten residues, aa 96–100 are predicted to orient toward the cytoplasmic side and might have the potential to interact with KPNA2.
To identify the critical amino acid residues in the NS2A motif, we conducted site-directed mutagenesis of the residues aa 96–100 and generated NS2A-M1 to NS2A-M4 mutant plasmids (Figure 5C). Transfection of HEK293 cells was done with wild-type NS2A and these mutant plasmids, separately. The WB results showed that NS2A-M4 had minimal effect on KPNA2, while NS2A-M1, NS2A-M2, NS2A-M3, and the wild-type NS2A reduced KPNA2 similarly (Figure 5D).
Since the NS2A-M4 (T100A) lost the ability to reduce KPNA2, we reasoned that the mutation might affect the NS2A interaction with KPNA2. Indeed, NS2A-M4 had a minimal presence in the KPNA2 precipitates, while the wild-type NS2A was readily detected (Figure 5E). These results demonstrated that the residue Thr100 was critical for the NS2A-mediated reduction of KPNA2.
To verify the effect of this critical residue Thr100 in whole virus infection, we conducted site-directed mutagenesis for T100A in NS2A of ZIKV ICD cDNA infectious clone plasmid [62]. Recovery of the mutant virus in Vero cells was conducted. Virus recovery with the wild-type ICD plasmid was also done. The recovered virus was passaged three times. Sanger DNA sequencing of the cDNA of the recovered virus was done and confirmed the presence of the T100A mutation in NS2A of the mutant virus (Figure 5F). The virus yield titration showed that the mutant virus had much lower titer than the wild-type virus by 1.33 log10/ml, or 22-fold (Figure 5G). To test whether the mutant virus was unable to reduce KPNA2, we inoculated the Vero cells with the mNS2A virus at an MOI of 10 or with the wild-type virus at an MOI of 1 to ensure they have a similar replication level. The mutant virus, as expected, lost its ability to reduce KPNA2 (Figure 5H). These results indicate that the Thr100 residue of NS2A is critical for the ZIKV-mediated KPNA2 reduction and the virus replication.
KPNA2 contains a CMA motif, and the mutation of Gln109 residue abolishes NS2A-mediated KPNA2 reduction
The above results show that ZIKV induced KPNA2 degradation via the CMA pathway. For proteins that are degraded by the CMA, they have a characteristic motif with the consensus sequence KFERQ (Lys-Phe-Glu-Arg-Gln) to interact with HSPA8 [58]. The substrate protein is then unfolded by HSPA8 and translocated across the lysosome membrane through the action of HSPA8 and the lysosome membrane protein LAMP2A. The CMA motif is often referred to as the KFERQ-like motif, which contains up to two hydrophobic residues, two positively charged residues, a single negatively charged residue, and one glutamine at N- or C-terminus [58]. The KPNA2 amino acid sequence has the peptide LSREKQ (aa104-109), which might be a CMA motif. Site-directed mutagenesis of the potential CMA motif in KPNA2 was conducted to generate four KPNA2 mutants: M1 (L104A-S105A), M2 (R106A-K108A), M3 (E107A), and M4 (Q109A) (Figure 6A).
Figure 6.
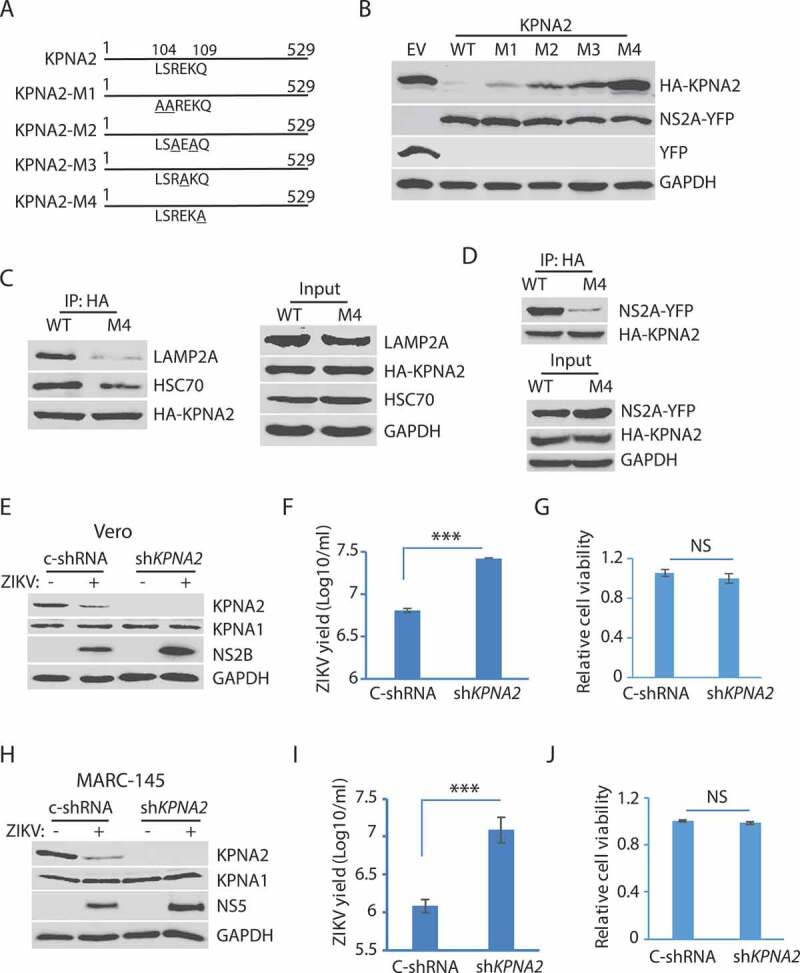
The residue Gln109 in the KPNA2 CMA motif is indispensable for NS2A-mediated reduction and ZIKV replication increases in the cells with KPNA2 knockdown. (A) Schematic illustration of KPNA2 mutation at the potential CMA motif. The numbers above the lines indicate KPNA2 amino acid residues. The underlined amino acid residues indicate point mutations. M1: mutation 1 (L104A and S105A), M2: R106A and K108A; M3: E107A, and M4: Q109A. (B) Effect of the point mutations in KPNA2 on NS2A-mediated KPNA2 reduction. WT KPNA2 and EV were included as controls. (C) Mutant KPNA2Q109A has much less interaction with LAMP2A than WT KPNA2. HEK293 cells were transfected with KPNA2-M4 or WT KPNA2 plasmids. The cells were treated with NH4CL for 6 h before harvested for co-IP. (D) The co-IP precipitate of mutant KPNA2Q109A contains much less NS2A than the precipitate of WT KPNA2. HEK293 cells were transfected with NS2A-YFP and KPNA2-M4 or WT KPNA2 plasmids. The cells were treated with NH4CL for 6 h before harvested for co-IP. (E and F). KPNA2 knockdown in Vero cells leads to higher ZIKV replication than control shRNA. Vero cells stably transduced with recombinant retrovirus expressing control shRNA (C-shRNA) or shRNA against KPNA2 (shKPNA2) were infected with ZIKV and harvested 48 hpi. ZIKV titers are shown as Log10/ml on Y-axis. A significant difference from cells with C-shRNA is denoted with *** for P< 0.001. (G) KPNA2 knockdown has minimal effect on Vero cell viability. NS: no significant difference. (H, I and J). KPNA2 knockdown in MARC-145 cells leads to significantly higher ZIKV replication than control shRNA in MARC-145 cells, whereas the cell viability remains stable
HEK293 cells were co-transfected with plasmids of NS2A and one of the four KPNA2 mutants. Compared to the empty vector control, the KPNA2-M4 level was stable, while KPNA2-M1, KPNA2-M2, and KPNA2-M3, as well as the wild-type KPNA2, were lower (Figure 6B). This finding indicates that the Q109A mutation in KPNA2-M4 abolishes the NS2A-mediated reduction of KPNA2. We speculated that KPNA2-M4 should have much less interaction with LAMP2A than the wild-type KPNA2. Indeed, the co-IP result showed that the KPNA2-M4 precipitate contained much less LAMP2A and HSPA8/HSC70 than the precipitate of wild-type KPNA2 (Figure 6 C). We also speculated that KPNA2-M4 would have less interaction with NS2A. Indeed, NS2A in the KPNA2-M4 precipitate was much less than in the precipitate of wild-type KPNA2 (Figure 6D). These results demonstrate that KPNA2 has a CMA motif, in which the residue Gln109 is critical to the CMA-mediated KPNA2 degradation.
KPNA2 knockdown leads to an elevation of ZIKV proliferation
To assess the role of KPNA2 in ZIKV replication, we conducted an RNAi-mediated knockdown of KPNA2 in Vero cells. The cells were stably transduced with recombinant retrovirus expressing shRNA against KPNA2. KPNA2 in the cells with shKPNA2 was significantly reduced in comparison to the cells with C-shRNA (Figure 6E). ZIKV replication in the cells with KPNA2 knockdown was higher than the cells with C-shRNA, shown by the level of NS2B (Figure 6E). Virus yield titration result showed that the Vero cells with KPNA2 knockdown had over 4-fold higher virus than the control cells (Figure 6F). The KPNA2 knockdown had minimal effect on the cell viability (Figure 6G).
As Vero cells are deficient in type-I interferon production, we verified the effect of KPNA2 knockdown in interferon-competent MARC-145 cells. MARC-145 cells were stably transduced with shKPNA2. KPNA2 knockdown was confirmed with WB in comparison with the cells of control shRNA (Figure 6H). ZIKV yield in the cells with KPNA2 knockdown was much higher than that in the control cells by ten-fold (Figure 6I). Cell viability assay showed the knockdown had little effect on cell growth (Figure 6J). These results indicate that KPNA2 plays a role in dampening ZIKV replication and that, conversely, ZIKV downregulates KPNA2 via the CMA pathway to maintain a conducive environment in the infected cells.
Discussion
KPNA2 is an essential nucleocytoplasmic transporter and has been identified as a prognostic indicator in multiple types of human cancers. Its turnover pathway and its role in ZIKV replication remain unknown. In this study, we discovered that KPNA2 is degraded via the CMA pathway and that ZIKV infection enhanced KPNA2 degradation. ZIKV NS2A protein was shown to be responsible for inducing the KPNA2 reduction. Verification of the ZIKV enhancement of KPNA2 degradation via the CMA was conducted in several aspects.
First, ZIKV infection leads to a reduction of KPNA2 protein level while having a minimal effect on KPNA1. ZIKV infection of several cell lines, including Vero, HeLa, and SK-N-SH, had a similar impact on KPNA2 reduction. The virus-induced KPNA2 reduction in SK-N-SH cells is more physiologically relevant to ZIKV-induced neurological disorders. The KPNA2 reduction, however, was not due to a change in the KPNA2 transcript level. The data shows that ZIKV specifically reduces KPNA2 but not KPNA1. In contrast, ZIKV induces elevation of KPNA6 protein level, and KPNA6 knockout impairs ZIKV replication [55]. This suggests that ZIKV perturbs certain karyopherins to generate a conducive milieu for its own replication.
Second, the ZIKV-induced KPNA2 reduction is due to the lysosomal proteolysis as treatment with both MG132 and NH4Cl restores the KPNA2 level in ZIKV-infected cells. Though MG132 is frequently used as a proteasome inhibitor, it also inhibits lysosomal proteases [59]. Therefore, the lysosomal inhibition by either MG132 or NH4Cl led to the blockage of KPNA2 protein degradation. The treatment with the two chemicals lasted for 6 h, which did not lead to a detectable negative impact on cell growth and viral replication. Further evidence is that ZIKV infection shortened KPNA2 half-life from 12 to 6 h but not alter the KPNA2 ubiquitination level. Polyubiquitination of a substrate protein occurs in the ubiquitin-proteasomal pathway. This further excludes the possibility of this pathway in the ZIKV-induced KPNA2 degradation. This is different from KPNA1 or KPNA6, both of which are degraded via the ubiquitin-proteasomal pathway [55,63]. Our data, however, does not completely exclude the possibility of the ubiquitin-proteasomal pathway in the degradation of KPNA2. We focused on the lysosomal proteolysis of KPNA2 in this study, as its inhibition minimized the ZIKV-mediated reduction of KPNA2.
Third, KPNA2 is degraded via the CMA pathway. The HSPA8 interaction with cargo proteins and LAMP2A are critical components of the CMA pathway [60]. However, HSPA8 has been discovered to involve in the microautophagy pathway too. To confirm the CMA pathway is responsible for the KPNA2 reduction, we conducted co-IP, which indicated that KPNA2 interacted with LAMP2A. RNAi-mediated knockdown of LAMP2A led to a much higher level of KPNA2 and abolished ZIKV-induced KPNA2 reduction, which demonstrates that the CMA pathway is responsible for KPNA2 degradation. The knockdown of LAMP2A did not affect the cell viability, which is consistent with the published finding that LAMP2A reduction has no effect on lysosomal stability and functions beyond the CMA [61].
Fourth, ZIKV protein NS2A mediates the KPNA2 reduction. Among all the ZIKV proteins, NS2A was found to reduce the KPNA2 protein level. The reduction was confirmed to be dose-dependent. NS2A enhances the KPNA2 turnover via the CMA as LAMP2A knockdown abolished NS2A-mediated KPNA2 reduction. NS2A is a small transmembrane protein with three loops that are potentially oriented in the cytosolic side. With the constructs of truncated NS2A, we identified the motif in NS2A that correlates with KPNA2 reduction. Further studies on site-directed mutagenesis led to the identification of the critical residue Thr100 in NS2A in the reduction of KPNA2. The mutant NS2A with T100A alteration failed to reduce KPNA2 and had minimal interaction with the later. The Thr100 residue is critical for the ZIKV-mediated KPNA2 reduction, and ZIKV replication as the mutant ZIKV with T100A alteration in NS2A had minimal effect on KPNA2 and produced much lower viral yield than the wild-type virus. These results suggest that NS2A enhances KPNA2 degradation via the CMA pathway.
Fifth, KPNA2 contains a CMA motif. Mutagenesis of the residues in the motif demonstrates that the residue Gln109 is indispensable for the KPNA2 degradation via the CMA. Mutations of the two basic and one acidic residue also minimized KPNA2 degradation, albeit at a much less scale than the mutation Q109A. Notably, the mutant KPNA2 with Q109A mutation had minimal interaction with LAMP2A and NS2A, shown by co-IP. KPNA2 knockdown led to a significant increase of ZIKV proliferation in both Vero and MARC-145 cells, which suggests that KPNA2 could mediate an antiviral effect and that ZIKV reduces KPNA2 for a conducive environment in the infected cells. It is known that KPNA2 transports NFKB into the nucleus to activate myriad genes, including antiviral factors [64,65]. Indeed, our NFKB luciferase reporter assay showed significantly lower reporter expression in KPNA2 knockdown cells than in the wild-type cells (data not shown). This might be one of the reasons for the KPNA2-mediated antiviral effect against ZIKV infection.
ZIKV NS2A is recently shown to have a single segment traversing the ER and six segments associating peripherally with the ER membrane [66]. NS2A plays a central role in the viral assembly. Our data is consistent with the proposed topological model of NS2A that the loop of aa 97–104 is oriented on the cytoplasmic side [66]. The residue Thr100 of NS2A is indispensable for the ZIKV-mediated KPNA2 degradation. Our data indicate that NS2A interacts with KPNA2 shown by co-IP, which suggests that NS2A probably increases the KPNA2 interaction with HSPA8 and transportation to the lysosome to enhance the CMA-mediated degradation of KPNA2. The NS2A interaction with KPNA2, however, possibly enhances HSPA8 binding as mutant Q109A KPNA2 had much less interaction with both HSPA8 and NS2A. It is known that HSPA8 binds cargo proteins via the CMA motif [60]. Mutation Q109A in KPNA2 led to minimal interaction with LAMP2A, which suggests that the CMA motif is essential for the NS2A-enhanced KPNA2 degradation.
KPNA2 plays a crucial role in ESC proliferation and differentiation by controlling the intracellular distribution of transcription factors that are essential for these activities [18]. ZIKV infection interferes with human neural stem cell growth and differentiation into neuroprogenitor cells [50,51]. Therefore, ZIKV-mediated KPNA2 degradation may contribute to the dysregulation of the growth and differentiation of neural stem cells.
Due to the importance of KPNA2 in cell signaling, some viruses target KPNA2. For example, vaccinia virus dysregulates NFKB signaling via A55 protein interrupting the interaction of RELA and KPNA2 [67]. Enterovirus 71 (EV71) infection upregulates KPNA2 expression by reducing MIR302 C (microRNA 302 c) [68]. Severe acute respiratory syndrome coronavirus (SARS-CoV) ORF6 protein, localized to the ER/Golgi membrane in infected cells, tethers KPNA2 and KPNB1 to the membrane to block the nuclear translocation of STAT1 [69].
In conclusion, our results demonstrate that KPNA2 is degraded via the CMA pathway and that ZIKV infection enhances KPNA2 degradation. Mechanistically, ZIKV NS2A interacts with KPNA2 and leads to the reduction of the KPNA2 protein level. The amino acid residue Thr100 of NS2A is indispensable for the ZIKV-mediated KPNA2 reduction. The residue Gln109 of KPNA2 is essential for CMA-mediated degradation. KPNA2 could involve in an antiviral role against ZIKV. Conversely, ZIKV induces the KPNA2 degradation to maintain efficient replication in the infected cells. Interestingly, ZIKV has been found to have oncolytic activity against human glioblastoma [70,71]. Our data of this study also suggest that ZIKV NS2A has translational potential as KPNA2 is upregulated in multiple cancers.
Materials and methods
Cells, virus, and chemicals
Vero (ATCC CCL-81), HEK293 (ATCC CRL-1573), HeLa (ATCC CCL-2), SK-N-SH (ATCC HTB-11) and MARC-145 [72] cells were cultured in Dulbecco Modified Eagle Medium (DMEM; Corning Life Sciences, 10–013-CV) supplemented with 10% fetal bovine serum (FBS) (Tissue Culture Biologicals, 101) at 37°C and 5% CO2. C6/36 (ATCC, CRL-1660), an Aedes albopictus cell line, was maintained using DMEM supplemented with 10% FBS at 28°C.
ZIKV strain PRVABC59 (ATCC, VR-1843) was used to inoculate Vero cells at the multiplicity of infection indicated in figure legends or results. Virus titers were determined in Vero cells by 10-fold serial dilutions and shown as the median tissue culture infectious dose (TCID50) as Log10/ml [73].
Proteasome inhibitor MG132 (Enzo Life Sciences, BML-PI102-0025) was used to treat cells at a final concentration of 10 µM for 6 h prior to harvesting. NH4Cl, a lysosome inhibitor, was used to treat cells at a final concentration of 10 mM for 6 h prior to harvesting. Cycloheximide (Fisher Scientific, AC357420010), a protein translation inhibitor, was added to ZIKV-infected and mock-infected cells at a final concentration of 50 μg/ml to determine the half-life of KPNA2.
Cell viability was determined with the CellTiter-Glo® Luminescent Cell Viability Assay Kit according to the manufacturer’s instruction (Promega, G7570).
Plasmids
The NS2B, NS2B3, NS3, NA4A, and NS5 of ZIKV PRVABC59 were cloned into the pCDNA3-VenusC1 vector [74] using the following primers: NS2B-F1, NS2B-R1, NS3-F1, NS3-R1, NS4A-F1, NS4A-R1, NS5-F1, and NS5-R1 (Table 1). NS2A was cloned into the pCAGEN-HA vector [74]. The C, prM, E, NS1, NS2A, NS4B with leader sequences [75] were cloned into the pCDNA3-VenusN1 vector (modified from pCDNA3 (a vector from Invitrogen, discontinued) by inserting YFP gene as a tag) using the following primers: C-F1, C-R2, PrM-F2, PrM-R2, E-F2, E-R2, NS1-F2, NS1-R2, SPG-C16-NS2A-F1, NS2A-R2, NS4B-F2, NS4B-R2. The NS2A truncation fragments NS2A-D1, NS2A-D2 and NS2A-D3 were cloned into the pCDNA3-VenusN1 vector using the following primers: NS2A-F2 and PR-NS2A-R3 for NS2A-D1, PR-NS2A-F3, and PR-NS2A-R4 for NS2A-D2, PR-NS2A-F4, and NS2A-R2 for NS2A-D3 (Table 1). NS2A mutants NS2AR96A, NS2AN98A, NS2AN98A, NS2AW99A, NS2AW99A, NS2AT100A were cloned into pCDNA3-VenusC1 vector using the following primers: NS2A-F2, NS2A-R96A-F, NS2A-R96A-R and NS2A-R2 for NS2A-R96A; NS2A-F2, NS2A-N98A-F, NS2A-N98A-R and NS2A-R2 for NS2A-N98A; NS2A-F2, NS2A-W99A-F, NS2A-W99A-R and NS2A-R2 for NS2A-W99A; NS2A-F2, NS2A-T100A-F, NS2A-T100A-R and NS2A-R2 for NS2A-T100A (Table 1).
Table 1.
List of primers used in this study
| Primera | Sequences (5ʹ to 3ʹ)b | Target gene/vector |
|---|---|---|
| C-F1 | CGAATTCATGAAAAACCCAAAAAAGAAATC | C |
| C-R2 | GCTCGAGTGCCATGGCTGTGGTCAGCAG | C |
| PRM-F2 | TGAATTCGCCACCATGGGCACAGATACTAGTGTCGGAAT | prM |
| PRM-R2 | GCTCGAGGCTGTATGCCGGGGCAATCAG | prM |
| E-F2 | TGAATTCGCCACCATGCAAAAAGTCATATACTTGGTC | E |
| E-R2 | GCTCGAGAGCAGAGACGGCTGTGGATAAG | E |
| NS2A-F2 | TGAATTCCACCATGGGATCAACTGATCACATGGAC | NS2A |
| SPG-C16-NS2A-F1 | CGAATTCGCCACCATGGGAGTCAAAGTTCTGTTTGCCCTGATCTGCATCGCTGTGGCCGAGCCCAGGAAAGAACCAGAAAG | NS2A |
| NS2A-R2 | TCTCGAGCCGCTTCCCACTCCTTGTGAG | NS2A |
| NS2B-F1 | TGAATTCAGCTGGCCCCCTAGCGAAGTAC | NS2B |
| NS2B-R1 | GCTCGAGTCACCTTTTTCCAGTCTTCACG | NS2B |
| NS3-F1 | TGAATTCAGTGGTGCTCTATGGGATGTG | NS3 |
| NS3-R1 | TCTCGAGTCATCTTTTCCCAGCGGCAAACTC | NS3 |
| NS4A-F1 | TGAATTCGGAGCGGCTTTTGGAGTGATG | NS4A |
| NS4A-R1 | TCTCGAGTCATCTTTGCTTTTCTGGCTCAGG | NS4A |
| NS4B-F2 | TGAATTCGCCACCATGTCTCCCCAGGACAACCAAATG | NS4B |
| NS4B-R2 | GCTCGAGACGTCTCTTGACCAAGCCAGC | NS4B |
| NS5-F1 | TGAATTCGGGGGTGGAACAGGAGAGA | NS5 |
| NS5-R1 | TCTCGAGTCACAGCACTCCAGGTGTAGAC | NS5 |
| ZIKV-RR-F | AARTACACATACCARAACAAAGTGGT | NS5 |
| ZIKV-RR-R | TCCRCTCCCYCTYTGGTCTTG | NS5 |
| KPNA2-RR-F1 | TGCTGGGCTATTTCCTACCT | KPNA2 |
| KPNA2-RR-R1 | GCTCTTAGGGCAGGAGTCAC | KPNA2 |
| PR-NS2A-R3 | ACTCGAGTGTCCAATTAGCTCTGAAGATGAAAG | NS2A |
| PR-NS2A-R4 | ACTCGAGAAACCCCCCGCAAGTAGCAAG | NS2A |
| PR-NS2A-F3 | TGAATTCGCCATGCTGGTATCTTTCATCTTCAGAG | NS2A |
| PR-NS2A-F4 | TGAATTCGCCATGCCACTGGCCCGGGGCACACTG | NS2A |
| NS2A-R96A-F | TTCATCTTCGCAGCTAATTGGACACCCCGTGAAAGC | NS2A-M1 |
| NS2A-R96A-R | TGTCCAATTAGCTGCGAAGATGAAAGATACCAGCAACG | NS2A-M1 |
| NS2A-N98A-F | TCTTCAGAGCTGCTTGGACACCCCGTGAAAGCATGC | NS2A-M2 |
| NS2A-N98A-R | CGGGGTGTCCAAGCAGCTCTGAAGATGAAAGATACCAG | NS2A-M2 |
| NS2A-W99A-F | TCAGAGCTAATGCGACACCCCGTGAAAGCATGCTGC | NS2A-M3 |
| NS2A-W99A-R | TCACGGGGTGTCGCATTAGCTCTGAAGATGAAAGATAC | NS2A-M3 |
| NS2A-T100A-F | AGCTAATTGGGCACCCCGTGAAAGCATGCTGCTGGC | NS2A-M4 |
| NS2A-T100A-R | GCTTTCACGGGGTGCCCAATTAGCTCTGAAGATGAAAG | NS2A-M4 |
| KPNA2-F1 | CGAATTCGCCATGTCCACCAACGAGAATGCTAATA | KPNA2 |
| KPNA2-R1 | GCTCGAGCTAAAAGTTAAAGGTCCCAGGAGCC | KPNA2 |
| K2-LS-M-F1 | TACTCAAGCTGCCAGGAAACTAGCAGCAAGAGAAAAACAGCCCCCCATAG | KPNA2-M1 |
| K2-LS-M-R1 | CTATGGGGGGCTGTTTTTCTCTTGCTGCTAGTTTCCTGGCAGCTTGAGTA | KPNA2-M1 |
| K2-RK-M-F1 | AGCTGCCAGGAAACTACTTTCCGCAGAAGCACAGCCCCCCATAGACAACATAA | KPNA2-M2 |
| K2-RK-M-R1 | TTATGTTGTCTATGGGGGGCTGTGCTTCTGCGGAAAGTAGTTTCCTGGCAGCT | KPNA2-M2 |
| K2-E-M-F1 | TGCCAGGAAACTACTTTCCAGAGCAAAACAGCCCCCCATAGACAACA | KPNA2-M3 |
| K2-E-M-R1 | TGTTGTCTATGGGGGGCTGTTTTGCTCTGGAAAGTAGTTTCCTGGCA | KPNA2-M3 |
| K2-Q-M-F1 | GAAACTACTTTCCAGAGAAAAAGCGCCCCCCATAGACAACATAATCC | KPNA2-M4 |
| K2-Q-M-R1 | GGATTATGTTGTCTATGGGGGGCGCTTTTTCTCTGGAAAGTAGTTTC | KPNA2-M4 |
| LAMP2A-ShRNA1F | GATCCGCACCATCATGCTGGATATTTCAAGAGAATATCCAGCATGATGGTGCTTTTTTACGCGTG | shLAMP2A |
| LAMP2A-ShRNA1R | AATTCACGCGTAAAAAAGCACCATCATGCTGGATATTCTCTTGAAATATCCAGCATGATGGTGCG | shLAMP2A |
| LAMP2A-ShRNA2F | GATCCGCAGGAGTACTTATTCTAGTTCAAGAGACTAGAATAAGTACTCCTGCTTTTTTACGCGTG | shLAMP2A |
| LAMP2A-ShRNA2R | AATTCACGCGTAAAAAAGCAGGAGTACTTATTCTAGTCTCTTGAACTAGAATAAGTACTCCTGCG | shLAMP2A |
| KPNA2-shRNA1F | GATCCGCTTAATTGAGAAGTATTATCAAGAGATAATACTTCTCAATTAAGCTTTTTTACGCGTG | shKPNA2 |
| KPNA2-shRNA1R | AATTCACGCGTAAAAAAGCTTAATTGAGAAGTATTATCTCTTGATAATACTTCTCAATTAAGCG | shKPNA2 |
| KPNA2-shRNA2F | GATCCGTGTCAAAGGCATAAATAATCAAGAGATTATTTATGCCTTTGACACTTTTTTACGCGTG | shKPNA2 |
| KPNA2-shRNA2R | AATTCACGCGTAAAAAAGTGTCAAAGGCATAAATAATCTCTTGATTATTTATGCCTTTGACACG | shKPNA2 |
| ICD-NS2A-M-F1 | CACGTGGAGGAAACATGTGGAACAAGAGGA | NS2A |
| ICD-NS2A-M-R1 | CCTCCACCAGGGAGAAATCACCACTCTCATCT | NS2A |
| NS2A-ICD-SEQ-R | CATCAGGTCGCCTTCCAAGGCGGAGATCGC | NS2A |
aF: forward primer, R: reverse primer.
bThe italicized alphabets indicate restriction enzyme cleavage sites for cloning.
KPNA2 and KPNA2 mutants were cloned into the pCAGEN-HA vector with the following primers: KPNA2-F1 and KPNA2-R1 for KPNA2; KPNA2-F1, K2-LS-M-F1, K2-LS-M-R1 and KPNA2-R1 for KPNA2-M1; KPNA2-F1, K2-RK-M-F1, K2-RK-M-R1 and KPNA2-R1 for KPNA2-M2; KPNA2-F1, K2-E-M-F1, K2-E-M-R1 and KPNA2-R1 for KPNA2-M3; KPNA2-F1, K2-Q-M-F1, K2-Q-M-R1 and KPNA2-R1 for KPNA2-M4 (Table 1) [55]. KPNA2 was also cloned into the pCDNA3-VenusC1 vector. The resulting recombinant plasmids were confirmed by Sanger DNA sequencing.
For shRNA against KPNA2 and LAMP2A, the oligos for the shRNA were cloned into the pSIREN-RetroQ-ZsGreen vector (Clontech, 632,455) according to the manufacturer’s instruction. The resulting recombinant plasmids were co-transfected into GP2-293 cells (Clontech, 631,458) with VSV-G vector (Addgene, 138,479; depositing lab Akitsu Hotta) for retrovirus packaging. The culture supernatant containing the recombinant retrovirus was used to transduce Vero or MARC-145 cells.
The construction of mutant ZIKV infectious clone with T100A of NS2A was done with overlapping PCR as described [76]. The round one PCR was done with primers ICD-NS2A-M-F1 and NS2A-T100A-R for fragment 1, and with NS2A-T100A-F and ICD-NS2A-M-R1 for fragment 2. The round two PCR was done with both fragment 1 and 2 as a template and with primers ICD-NS2A-M-F1 and ICD-NS2A-M-R1. The overlapping PCR product was ligated into the ZIKV-ICD plasmid [62] that was digested with PmlI and EcoNI. The resulting plasmid of the mutant ZIKV cDNA clone was confirmed by Sanger DNA sequencing. For virus recovery, the mutant plasmid was transfected into Vero cells, with the wild-type ZIKV-ICD plasmid included as a control. The cell culture supernatant at 48 h post-transfection was collected and used to inoculate fresh Vero cells. The virus passaging was repeated for three times. For passaging the mutant virus, Vero cells with KPNA2 knockdown were used. RNA was isolated from the recovered mutant virus for cDNA synthesis and DNA sequencing to confirm the presence of the mutant nucleotide.
RNA isolation and real-time PCR
Total RNA was isolated from Vero cells with TRIzol reagent (Thermo Fisher Scientific, 15,596,026) in accordance with the manufacturer’s instructions. Reverse transcription and real-time PCR (RT-qPCR) with SYBR Green detection (Thermo Fisher Scientific, 4,334,973) were performed as described previously [55,74,77]. The transcripts of RPL32 (ribosomal protein L32), a house-keeping gene, were also determined. The relative transcript levels were shown as folds in comparison to the control cells after RPL32 normalization. The real-time PCR primers used for KPNA2 and ZIKV were KPNA2-RR-F1, KPNA2-RR-R1, ZIKV-RR-F, and ZIKV-RR-R (Table 1). All experiments were repeated at least three times, with each conducted in triplicate.
Western blot (WB) analysis
Whole-cell lysates in Laemmli sample buffer were subjected to SDS-PAGE and WB as described previously [55,78]. The primary mouse monoclonal antibodies against KPNA1 (Santa Cruz Biotechnology, sc-101,292), KPNA2 (Santa Cruz Biotechnology, sc-55,537), hemagglutinin (HA) tag (ThermoFisher Scientific, 26,183), GFP (Biolegend, 75,818–584), HSPA8/HSC70 (Santa Cruz Biotechnology, sc-7298), ubiquitin (Santa Cruz Biotechnology, sc-8017), GAPDH (Santa Cruz Biotechnology, sc-365,062), and TUBB1/β-tubulin (Sigma, T7816), and rabbit polyclonal antibodies against ZIKV NS4B (GeneTex, GTX133311), ZIKV E (GeneTex, GTX133314), NS2B (GeneTex, GTX133318), NS5 (GeneTex, GTX133329) and LAMP2A (Boster, M01573) were used in this study. The secondary antibodies used in this study were goat anti-mouse or anti-rabbit IgG conjugated with horseradish peroxidase (Bio-Rad, 170–5046 and 170–5047). The chemiluminescence substrate was used to reveal the specific reactions, and the signal was recorded digitally using a ChemiDoc XRS imaging system using the QuantityOne Program, version 4.6 (Bio-Rad Laboratories, Hercules, CA). Densitometry analysis of the WB images was done with the QuantityOne Program, version 4.6 (Bio-Rad). All WB images were acquired in the linear range of digital intensity without saturated pixels.
Immunoprecipitation (IP)
IP was done as described previously [55,74]. Cell lysates were clarified and incubated with specific antibodies indicated in results or figure legends, followed by incubation with protein A/G-magnetic beads (Bimake.com, B23202). The IP complexes were subjected to WB for the detection of target proteins indicated in results or figure legends.
Statistical analysis
Differences in gene expression between the treatment group and control were assessed by using the Student t-test. A two-tailed P value of 0.05 was considered significant.
Acknowledgments
J. He and S. Lin are partially supported by the China Scholarship Council. This study was partially funded by a seed grant from the University of Maryland.
Abbreviations
- ARM
armadillo
- co-IP
co-immunoprecipitation
- CMA
Chaperone-mediated autophagy
- ESCs
Embryonic stem cells
- GBS
Guillain-Barre syndrome
- hpi
Hour post-infection
- HSPA8/HSC70
Heat shock protein family A (Hsp70) member 8
- IBB
Importin β binding
- IFA
Immunofluorescence assay
- KPNA2
karyopherin subunit alpha 2
- LAMP2A
lysosome membrane-associated protein type 2A
- MOI
Multiplicity of infection
- NLS
Nuclear localization sequence
- NPC
Nuclear pore complex
- NS
Non-structural
- POU5F1/OCT4
POU class 5 homeobox 1
- RT-qPCR
Reverse transcription and quantitative PCR
- SAMHD1
Sterile alpha motif and histidine/aspartic acid domain 1
- WB
Western blotting
- ZIKV
Zika virus
Disclosure statement
The authors declare no competing interests.
References
- [1].Mosammaparast N, Pemberton LF.. Karyopherins: from nuclear-transport mediators to nuclear-function regulators [Review]. Trends Cell Biol. 2004. October;14(10):547–556. [DOI] [PubMed] [Google Scholar]
- [2].Goldfarb DS, Corbett AH, Mason DA, et al. Importin alpha: a multipurpose nuclear-transport receptor [Review]. Trends Cell Biol. 2004. September;14(9):505–514. [DOI] [PubMed] [Google Scholar]
- [3].Stewart M. Molecular mechanism of the nuclear protein import cycle. Nat Rev Mol Cell Biol. 2007. March;8(3):195–208. [DOI] [PubMed] [Google Scholar]
- [4].Chook YM, Blobel G. Karyopherins and nuclear import [Review]. Current Opin Struct Biol. 2001. December; 11(6):703–715. [DOI] [PubMed] [Google Scholar]
- [5].Kelley JB, Talley AM, Spencer A, et al. Karyopherin alpha7 (KPNA7), a divergent member of the importin alpha family of nuclear import receptors. BMC Cell Biol. 2010. August 11;11(1):63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Pumroy RA, Cingolani G. Diversification of importin-alpha isoforms in cellular trafficking and disease states. Biochem J. 2015. February 15;466(1):13–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kohler M, Ansieau S, Prehn S, et al. Cloning of two novel human importin-alpha subunits and analysis of the expression pattern of the importin-alpha protein family. FEBS Lett. 1997. November 3;417(1):104–108. [DOI] [PubMed] [Google Scholar]
- [8].Tejomurtula J, Lee KB, Tripurani SK, et al. Role of importin alpha8, a new member of the importin alpha family of nuclear transport proteins, in early embryonic development in cattle. Biol Reprod. 2009. August;81(2):333–342. [DOI] [PubMed] [Google Scholar]
- [9].Hu J, Wang F, Yuan Y, et al. Novel importin-alpha family member Kpna7 is required for normal fertility and fecundity in the mouse. J Biol Chem. 2010. October 22;285(43):33113–33122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lott K, Cingolani G. The importin beta binding domain as a master regulator of nucleocytoplasmic transport [Review]. Biochim Biophys Acta. 2011. September;1813(9):1578–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Li X, Sun L, Jin Y. Identification of karyopherin-alpha 2 as an Oct4 associated protein. J Genet Genomics. 2008. December;35(12):723–728. [DOI] [PubMed] [Google Scholar]
- [12].Li J, Liu Q, Liu Z, et al. KPNA2 promotes metabolic reprogramming in glioblastomas by regulation of c-myc. J Exp Clin Cancer Res. 2018. August 16;37(1):194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wang CI, Chien KY, Wang CL, et al. Quantitative proteomics reveals regulation of karyopherin subunit alpha-2 (KPNA2) and its potential novel cargo proteins in nonsmall cell lung cancer. Mol Cell Proteomics. 2012. November;11(11):1105–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sandrock K, Bielek H, Schradi K, et al. The nuclear import of the small GTPase Rac1 is mediated by the direct interaction with karyopherin alpha2. Traffic. 2010. February;11(2):198–209. [DOI] [PubMed] [Google Scholar]
- [15].Schaller T, Pollpeter D, Apolonia L, et al. Nuclear import of SAMHD1 is mediated by a classical karyopherin alpha/beta1 dependent pathway and confers sensitivity to VpxMAC induced ubiquitination and proteasomal degradation. Retrovirology. 2014. April 8;11:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tao R, Xu X, Sun C, et al. KPNA2 interacts with P65 to modulate catabolic events in osteoarthritis. Exp Mol Pathol. 2015. October;99(2):245–252. [DOI] [PubMed] [Google Scholar]
- [17].Tseng SF, Chang CY, Wu KJ, et al. Importin KPNA2 is required for proper nuclear localization and multiple functions of NBS1. J Biol Chem. 2005. November 25;280(47):39594–39600. [DOI] [PubMed] [Google Scholar]
- [18].Yasuhara N, Yamagishi R, Arai Y, et al. Importin alpha subtypes determine differential transcription factor localization in embryonic stem cells maintenance. Dev Cell. 2013. July 29;26(2):123–135. [DOI] [PubMed] [Google Scholar]
- [19].Christiansen A, Dyrskjot L. The functional role of the novel biomarker karyopherin alpha 2 (KPNA2) in cancer. Cancer Lett. 2013. April 30;331(1):18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Grupp K, Habermann M, Sirma H, et al. High nuclear karyopherin alpha 2 expression is a strong and independent predictor of biochemical recurrence in prostate cancer patients treated by radical prostatectomy. Mod Pathol. 2014. January;27(1):96–106. [DOI] [PubMed] [Google Scholar]
- [21].Gao CL, Wang GW, Yang GQ, et al. Karyopherin subunit-alpha 2 expression accelerates cell cycle progression by upregulating CCNB2 and CDK1 in hepatocellular carcinoma. Oncol Lett. 2018. March;15(3):2815–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tsukagoshi M, Araki K, Yokobori T, et al. Overexpression of karyopherin-alpha2 in cholangiocarcinoma correlates with poor prognosis and gemcitabine sensitivity via nuclear translocation of DNA repair proteins. Oncotarget. 2017. June 27;8(26):42159–42172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yu L, Wang G, Zhang Q, et al. Karyopherin alpha 2 expression is a novel diagnostic and prognostic factor for colorectal cancer. Oncol Lett. 2017. March;13(3):1194–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhang Y, Zhang M, Yu F, et al. Karyopherin alpha 2 is a novel prognostic marker and a potential therapeutic target for colon cancer. J Exp Clin Cancer Res. 2015. December 1;34:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhou J, Dong D, Cheng R, et al. Aberrant expression of KPNA2 is associated with a poor prognosis and contributes to OCT4 nuclear transportation in bladder cancer. Oncotarget. 2016. November 8;7(45):72767–72776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zheng M, Tang L, Huang L, et al. Overexpression of karyopherin-2 in epithelial ovarian cancer and correlation with poor prognosis. Obstet Gynecol. 2010. October;116(4):884–891. [DOI] [PubMed] [Google Scholar]
- [27].Yang Y, Guo J, Hao Y, et al. Silencing of karyopherin alpha2 inhibits cell growth and survival in human hepatocellular carcinoma. Oncotarget. 2017. May 30;8(22):36289–36304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wang CI, Yu CJ, Huang Y, et al. Association of overexpressed karyopherin alpha 2 with poor survival and its contribution to interleukin-1beta-induced matrix metalloproteinase expression in oral cancer. Head Neck. 2018. August;40(8):1719–1733. [DOI] [PubMed] [Google Scholar]
- [29].Wang CI, Wang CL, Wang CW, et al. Importin subunit alpha-2 is identified as a potential biomarker for non-small cell lung cancer by integration of the cancer cell secretome and tissue transcriptome. Int J Cancer. 2011. May 15;128(10):2364–2372. [DOI] [PubMed] [Google Scholar]
- [30].Li C, Ji L, Ding ZY, et al. Overexpression of KPNA2 correlates with poor prognosis in patients with gastric adenocarcinoma. Tumour Biol. 2013. April;34(2):1021–1026. [DOI] [PubMed] [Google Scholar]
- [31].Huang L, Zhou Y, Cao XP, et al. KPNA2 is a potential diagnostic serum biomarker for epithelial ovarian cancer and correlates with poor prognosis. Tumour Biol. 2017. June;39(6):1010428317706289. [DOI] [PubMed] [Google Scholar]
- [32].Dahl E, Kristiansen G, Gottlob K, et al. Molecular profiling of laser-microdissected matched tumor and normal breast tissue identifies karyopherin alpha2 as a potential novel prognostic marker in breast cancer. Clin Cancer Res. 2006. July 1;12(13):3950–3960. [DOI] [PubMed] [Google Scholar]
- [33].Dick GW. Zika virus. II. Pathogenicity and physical properties. Trans R Soc Trop Med Hyg. 1952. September;46(5):521–534. [DOI] [PubMed] [Google Scholar]
- [34].Dick GW, Kitchen SF, Haddow AJ. Zika virus. I. Isolations and serological specificity. Trans R Soc Trop Med Hyg. 1952. September;46(5):509–520. [DOI] [PubMed] [Google Scholar]
- [35].Duffy MR, Chen TH, Hancock WT, et al. Zika virus outbreak on Yap Island, Federated States of Micronesia. N Engl J Med. 2009. June 11;360(24):2536–2543. [DOI] [PubMed] [Google Scholar]
- [36].Campos GS, Bandeira AC, Sardi SI. Zika Virus Outbreak, Bahia, Brazil. Emerg Infect Dis. 2015. October;21(10):1885–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Brooks T, Roy-Burman A, Tuholske C, et al. Real-Time Evolution of Zika Virus Disease Outbreak, Roatan, Honduras. Emerg Infect Dis. 2017. August;23(8):1360–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hennessey M, Fischer M, Staples JE. Zika Virus Spreads to New Areas - Region of the Americas, May 2015-January 2016. MMWR Morb Mortal Wkly Rep. 2016. January 29;65(3):55–58. [DOI] [PubMed] [Google Scholar]
- [39].Hills SL, Fischer M, Petersen LR. Epidemiology of Zika Virus Infection. J Infect Dis. 2017. December 16;216(suppl_10):S868–S874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Elliott R, Banerjee T, Santra S. Zika: an emerging disease requiring prevention and awareness. PLoS Negl Trop Dis. 2018. June;12(6):e0006486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Britt WJ. Adverse outcomes of pregnancy-associated Zika virus infection.Semin Perinatol. 2018. [cited 2018 March6];42(3):155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Counotte MJ, Meili KW, Taghavi K, et al. Zika virus infection as a cause of congenital brain abnormalities and Guillain-Barre syndrome: A living systematic review. F1000Res. 2019;8:1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Mlakar J, Korva M, Tul N, et al. Zika Virus Associated with Microcephaly. N Engl J Med. 2016. March 10;374(10):951–958. [DOI] [PubMed] [Google Scholar]
- [44].Panchaud A, Stojanov M, Ammerdorffer A, et al. Emerging role of Zika Virus in adverse fetal and neonatal outcomes. Clin Microbiol Rev. 2016. July;29(3):659–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Plourde AR, Bloch EM, Literature A. Review of Zika Virus. Emerg Infect Dis. 2016. July;22(7):1185–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Raposo-Amaral CE. Microcephaly: consequence of the Zika Virus Global Outbreak. J Craniofac Surg. 2016. September; 27(6):1383–1384. [DOI] [PubMed] [Google Scholar]
- [47].Wang L, Valderramos SG, Wu A, et al. From mosquitos to humans: genetic evolution of Zika Virus. Cell Host Microbe. 2016. May 11;19(5):561–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Garcez PP, Loiola EC, Madeiro da Costa R, et al. Zika virus impairs growth in human neurospheres and brain organoids. Science. 2016. May 13;352(6287):816–818. [DOI] [PubMed] [Google Scholar]
- [49].Shao Q, Herrlinger S, Yang SL, et al. Zika virus infection disrupts neurovascular development and results in postnatal microcephaly with brain damage. Development. 2016. November 15;143(22):4127–4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Devhare P, Meyer K, Steele R, et al. Zika virus infection dysregulates human neural stem cell growth and inhibits differentiation into neuroprogenitor cells. Cell Death Dis. 2017. October 12;8(10):e3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Li C, Xu D, Ye Q, et al. Zika virus disrupts neural progenitor development and leads to microcephaly in mice. Cell Stem Cell. 2016. November 3;19(5):672. [DOI] [PubMed] [Google Scholar]
- [52].Huang WC, Abraham R, Shim BS, et al. Zika virus infection during the period of maximal brain growth causes microcephaly and corticospinal neuron apoptosis in wild type mice. Sci Rep. 2016. October 7;6(6):34793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Li C, Wang Q, Jiang Y, et al. Disruption of glial cell development by Zika virus contributes to severe microcephalic newborn mice. Cell Dis. 2018;4(1):43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Lazear HM, Diamond MS. Zika virus: new clinical syndromes and its emergence in the Western hemisphere. J Virol. 2016;90 15(10):4864–4875. May. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Yang L, Wang R, Yang S, et al. Karyopherin Alpha 6 is required for replication of porcine reproductive and respiratory syndrome Virus and Zika Virus. J Virol. 2018. May 1;92(9):e00072–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Lanciotti RS, Lambert AJ, Holodniy M, et al. Phylogeny of Zika Virus in Western Hemisphere, 2015. Emerg Infect Dis. 2016;22(5):933–935. May. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ciechanover A. Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol. 2005. January;6(1):79–87. [DOI] [PubMed] [Google Scholar]
- [58].Jackson MP, Hewitt EW. Cellular proteostasis: degradation of misfolded proteins by lysosomes. Essays Biochem. 2016. October 15;60(2):173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. 2018. June;19(6):365–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Massey AC, Kaushik S, Sovak G, et al. Consequences of the selective blockage of chaperone-mediated autophagy. Proc Natl Acad Sci U S A. 2006. April 11;103(15):5805–5810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Tsetsarkin KA, Kenney H, Chen R, et al. A Full-Length Infectious cDNA Clone of Zika Virus from the 2015 epidemic in Brazil as a genetic platform for studies of virus-host interactions and vaccine development. MBio. 2016. August 23;7:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Wang R, Nan Y, Yu Y, et al. Porcine reproductive and respiratory syndrome virus Nsp1beta inhibits interferon-activated JAK/STAT signal transduction by inducing karyopherin-alpha1 degradation [Research Support, Non-U.S. Gov’t]. J Virol. 2013;87(9):5219–5228. May. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Cai Y, Shen Y, Gao L, et al. Karyopherin Alpha 2 promotes the inflammatory response in rat pancreatic acinar cells via facilitating NF-kappaB activation. Dig Dis Sci. 2016. March;61(3):747–757. [DOI] [PubMed] [Google Scholar]
- [65].Liang P, Zhang H, Wang G, et al. KPNB1, XPO7 and IPO8 mediate the translocation ofNF-kappaB/p65 into the nucleus. Traffic. 2013. November;14(11):1132–1143. [DOI] [PubMed] [Google Scholar]
- [66].Zhang X, Xie X, Zou J, et al. Genetic and biochemical characterizations of Zika virus NS2A protein. Emerg Microbes Infect. 2019;8(1):585–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Pallett MA, Ren H, Zhang RY, et al. Vaccinia virus BBK E3 ligase adaptor A55 targets importin-dependent NF-kappaB activation and inhibits CD8(+) T-Cell memory. J Virol. 2019. May 15;93: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Peng N, Yang X, Zhu C, et al. MicroRNA-302 Cluster downregulates enterovirus 71-induced innate immune response by targeting KPNA2. J Immunol. 2018. July 1;201(1):145–156. [DOI] [PubMed] [Google Scholar]
- [69].Frieman M, Yount B, Heise M, et al. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J Virol. 2007. September;81(18):9812–9824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Chen Q, Wu J, Ye Q, et al. Treatment of human glioblastoma with a live attenuated zika virus vaccine candidate. MBio. 2018. September 18;9:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Zhu Z, Gorman MJ, McKenzie LD, et al. Zika virus has oncolytic activity against glioblastoma stem cells. J Exp Med. 2017. October 2;214(10):2843–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Kim HS, Kwang J, Yoon IJ, et al. Enhanced replication of porcine reproductive and respiratory syndrome (PRRS) virus in a homogeneous subpopulation of MA-104 cell line. Arch Virol. 1993;133(3–4):477–483. [DOI] [PubMed] [Google Scholar]
- [73].Zhang YJ, Stein DA, Fan SM, et al. Suppression of porcine reproductive and respiratory syndrome virus replication by morpholino antisense oligomers. Vet Microbiol. 2006. October 31;117(2–4):117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Yang L, Wang R, Ma Z, et al. Porcine reproductive and respiratory syndrome virus antagonizes JAK/STAT3 signaling via nsp5, which induces STAT3 degradation. J Virol. 2017. February 1;91(3):e02087–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Shan C, Xie X, Muruato AE, et al. An Infectious cDNA clone of Zika virus to study viral virulence, mosquito transmission, and antiviral inhibitors. Cell Host Microbe. 2016. June 8;19(6):891–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Ma Z, Yu Y, Xiao Y, et al. The middle half genome of interferon-inducing porcine reproductive and respiratory syndrome virus strain A2MC2 is essential for interferon induction. J Gen Virol. 2017. July;98(7):1720–1729. [DOI] [PubMed] [Google Scholar]
- [77].Patel D, Opriessnig T, Stein DA, et al. Peptide-conjugated morpholino oligomers inhibit porcine reproductive and respiratory syndrome virus replication. Antiviral Res. 2008. February;77(2):95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Zhang YJ, Wang KY, Stein DA, et al. Inhibition of replication and transcription activator and latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus by morpholino oligomers. Antiviral Res. 2007. January;73(1):12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]