

Abstract
Background:
Profilaggrin belongs to the S100 fused-type protein family expressed in keratinocytes and is important for skin barrier integrity. Its N-terminus contains an S100 (“A”) domain and a unique “B” domain with a nuclear localization sequence.
Objective:
To determine whether profilaggrin B domain cooperates with the S100 domain to bind macromolecules. To characterize the biochemical and structural properties of the profilaggrin N-terminal “AB” domain and compare it to other S100 fused-type proteins.
Methods:
We used biochemical (protease protection, light scattering, fluorescence spectroscopy, pull-down assays) and computational techniques (sequence analysis, molecular modeling with crystallographic structures) to examine human profilaggrin and S100 fused-type proteins.
Results:
Comparing profilaggrin S100 crystal structure with models of the other S100 fused-type proteins demonstrated each has a unique chemical composition of solvent accessible surface around the hydrophobic binding pocket. S100 fused-type proteins exhibit higher pocket hydrophobicity than soluble S100 proteins. The inter-EF-hand linker in S100 fused-type proteins contains conserved hydrophobic residues involved in binding substrates. Profilaggrin B domain cooperates with the S100 domain to bind annexin II and keratin intermediate filaments in a calcium-dependent manner using exposed cationic surface. Using molecular modeling we demonstrate profilaggrin B domain likely interacts with annexin II domains I and II. Steric clash analysis shows annexin II N-terminal peptide is favored to bind profilaggrin among S100 fused-type proteins.
Conclusion:
The N-terminal S100 and B domains of profilaggrin cooperate to bind substrate molecules in granular layer keratinocytes to provide epidermal barrier functions.
Keywords: S100 protein, calcium binding protein, epidermis, skin disease, protein structure, filaggrin
1. Introduction
Over 20 human proteins contain a specialized S100 EF-hand calcium-binding domain with target binding functions [1, 2]. In differentiating epidermis there exists a S100 “fused-type” protein (SFTP) family defined by its 7 members having an N-terminal S100 domain fused to additional C-terminal sequence (Fig. 1). SFTP sequence composition and organization beyond the N-terminal S100 domain varies significantly. Mammalian SFTP genes colocalize with the epidermal differentiation complex locus (chromosome 1q21) [2, 3] and are expressed in epidermis and other tissues like breast. The link between increased hornerin expression and breast cancer progression [4] exemplifies why it is important to investigate the biochemical and structural properties of SFTPs: what are the differences among SFTP target binding sites, and what are the biochemical and structural mechanisms of action of non-S100 residues?
Figure 1. Comparing the protein domain organization of the seven S100 fused-type protein family members.
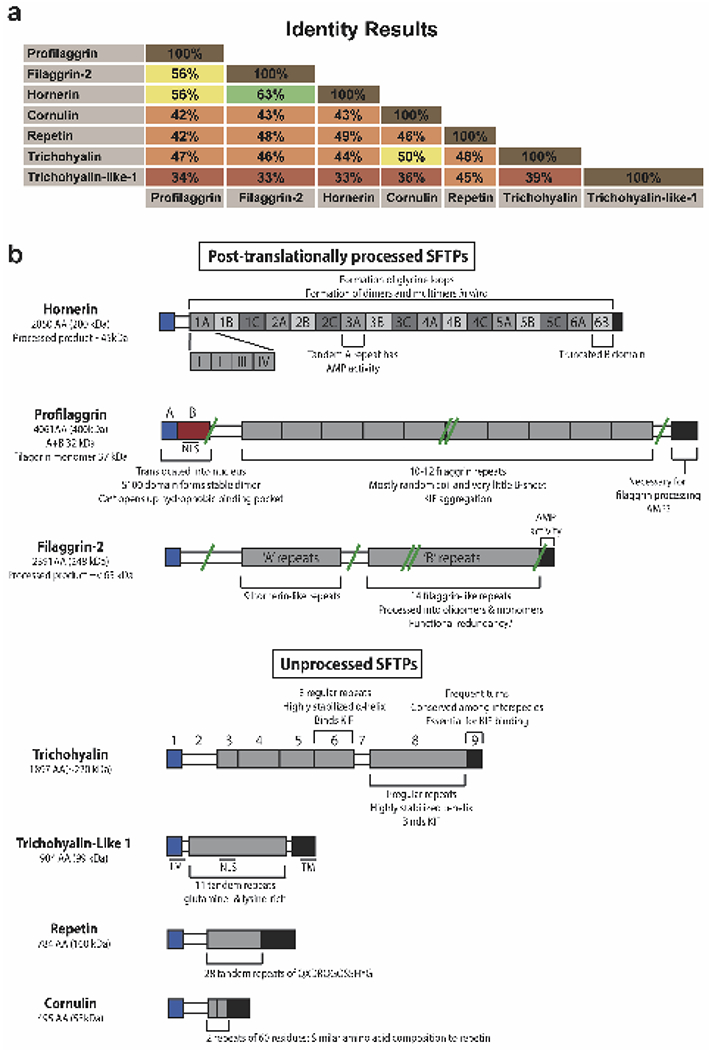
a) Protein sequence identities between SFTP members calculated using the Sequence Identities and Similarities (SIAS) algorithm. The 7 SFTP proteins are profilaggrin, filaggrin-2 (ifapsoriasin), hornerin (S100A18), cornulin (C1orf10), trichohyalin, and trichohyalin-like 1 (S100A17). b) Schematic of the protein domain organization of the SFTPs. SFTPs are divided into post-translationally processed and unprocessed groups. Important structural and molecular features of the SFTPs are annotated. S100 domains are colored blue, central repeat domains colored shades of gray, and the C-terminal domains colored black. Select proteolytic sites are indicated by green lines. Double green lines represent processing into monomers and smaller products. NLS, nuclear localization signal; TM, transmembrane domain.
Profilaggrin is a SFTP named because of its strong cationic ability to aggregate intermediate filaments (IFs) [5]. We previously determined the structure of the N-terminal S100 domain (the “A” domain or PF-A) of human profilaggrin [6] and found it forms a stable dimer and contains a hydrophobic pocket. We suggested PF-A dimerization could facilitate keratohyalin granule (KG) formation [6]. Only ~20% of the residues of the hydrophobic pocket are conserved among SFTPs. This finding raised questions on how SFTP pockets differ chemically and structurally across the family.
Adjacent to PF-A is the B domain (PF-B), whose structure and function is poorly understood. After profilaggrin is dephosphorylated and released from KGs, proteolytic cleavage separates an A-B domain fusion (PF-AB) from the filaggrin repeats. PF-AB then translocates to the nucleus of a differentiating keratinocyte within the transition layer (stratum granulosum to stratum corneum) due to a nuclear localization signal (NLS) within the B domain [7, 8]. PF-AB is proposed to regulate epidermal homeostasis [9] and nuclear envelope collapse, apoptosis, and desquamation. We showed PF-AB interacts with annexin II (ANXA2), hsp27, and stratifin, while other studies indicate PF-B could have cytosolic protein binding partners [6, 10].
Over 7,000 mutations exist in profilaggrin, with more than 300 in PF-AB. Several of these PF-AB mutations are associated with atopic dermatitis and ichthyosis vulgaris (Supplementary Fig. S1), skin diseases also linked to mutations in the filaggrin repeat region of profilaggrin [11–16].
This study examines how: 1) SFTP target binding sites differ chemically and structurally, and 2) the biophysical properties of PF-B affect target interactions. Using the PF-A structure, we generated S100 domain homology models for each SFTP member to characterize the solvent accessible surface around the hydrophobic pocket. Yeast two-hybrid studies identified ANXA2 interacts with PF-A [6]; here, we demonstrate PF-B cooperates with PF-A to bind ANXA2. Furthermore, binding studies show PF-AB interacts with IFs and ANXA2 through exposed hydrophobic surface on PF-A and cationic surface on PF-B. Lastly, we demonstrate ANXA2 N-terminal peptide (ANXA2Npep) is structurally favored to bind profilaggrin.
2. Materials and Methods
Protein Production and Purification.
pET-based plasmids of PF-A wild-type (WT) and Ile43Ala/Leu44Ala double mutant (res. 1-92), PF-AB WT (res. 1-293 or 1-257) and Ile43Ala/Leu44Ala double mutant (res. 1-257), annexin-II (res. 1-339), K1-1B (res. 226-331) and K10-1B (res. 195-296) were purchased from GenScript (Piscataway, NJ). Proteins were expressed and purified individually except PF-AB WT and annexin-II proteins, which were co-expressed/purified. Proteins were expressed in Escherichia coli strain BL21(DE3) (Agilent Technologies, Santa Clara, CA) and isolated using established methods [6]. Keratin 1B heterocomplex was prepared as described [17].
Multi-angle Light Scattering.
PF-AB WT (1 mg/ml) in 20 mM Tris-HCl buffer (pH 7.4) containing 200mM NaCl was applied at 0.5ml/min to Superdex200 16/600 (GE, Boston, MA) gel filtration column in-line with DAWN HELEOS II light scattering instrument (Wyatt Technology, Santa Barbra, CA; laser wavelength 658nm). Software was as described [17]. Procedure was repeated using PF-AB-WT-ANXA2 complex (1 mg/ml).
Fluorescence spectroscopy.
Fluorescence measurements were made using a Fluorolog®-3 spectrofluorometer (HORIBA Scientific, Albany, NY) following established protocol [18].
Structural modeling and surface analysis.
S100 domain models for SFTPs besides profilaggrin were made using our PF-A structure (PDB ID 4PCW) as a template, Swiss-Model [19], and UCSF Chimera (Resource for Biocomputing, Visualization, and Informatics, University of California, San Francisco) for energy minimization. ANXA2Npep used to create a Hex-docked ANXA2Npep-PF-A structural model [6] was superposed into each SFTP homology-model using PyMOL Molecular Graphics System (Version 2.0 Schrodinger, LLC). Structural analysis used UCSF Chimera. MolProbity generated clash scores for SFTP-S100+ANXA2Npep models. MSAs used Clustal Omega.
Proteolysis assay.
Filaggrin (1mg/ml) with and without ANXA2 was stored at 4°C for one week. SDS-PAGE samples were collected at 0, 1, 3, and 7 days.
Intermediate filament binding assay.
His6-filaggrin was used as bait to pulldown human recombinant K1/K10-1B and K1/K10-2B heterocomplexes. 0.3mg of His6-filaggrin was incubated with 100μL of pre-equilibrated nickel beads (0.1M Tris-HCl buffer (pH 7.4) containing 0.2M NaCl either with 5mM CaCl2 or 1mM ethylenediaminetetraacetic acid (EDTA)) (Goldbio, St Louis, MO) and mixed 1:1 with untagged K1/K10-1B or K1/K10-2B complex, gently rocked for 1h at 4°C, followed by centrifugation at 700xg for 5 minutes to pellet beads and associated proteins. Supernatants containing unbound proteins were removed and pelleted beads resuspended with 600μL wash buffer (0.1M Tris-HCl (pH 7.4) containing 0.2M NaCl, 20mM imidazole, and either 5mM CaCl2 or 1mM EDTA) followed by centrifugation again (wash step repeated 3 times). All steps performed at 4°C.
3. Results
3.1. S100 fused-type proteins have different domain organizations
To evaluate differences between SFTPs, we performed primary sequence and domain organization analysis on all seven SFTPs. SFTPs range from 33 to 63% sequence identity (Fig. 1a). The lowest identity is between trichohyalin-like 1 and filaggrin-2 or hornerin. The highest identity is between filaggrin-2 and hornerin. Multiple sequence alignment (MSA) revealed high conservation of the N-terminal S100 domains and lower conservation elsewhere. Computational analysis using domain prediction algorithms and published data indicate SFTPs can be divided into post-translationally processed and unprocessed groups (Fig. 1b). The former are large, multimeric precursor proteins with an N-terminal S100 calcium-binding domain, a central repeat region that is either hornerin-like or filaggrin-like, and a C-terminal domain of unknown function, possibly containing anti-microbial peptide (AMP) sequence (Supplementary Figs. S2 & S3). Post-translationally processed SFTPs are expressed in differentiated keratinocytes and other epithelial tissues; they colocalize within epidermal granular layer KGs [3]. Upon translation these precursor proteins are phosphorylated for storage, then when needed are proteolytically cleaved into smaller oligomers. For profilaggrin, “histidine-rich” filaggrin units perform keratinbinding functions [20] and ultimately breakdown into urocanic and pyrrolidone carboxylic acids (natural moisturizing factor) [21]. Unprocessed SFTP members share a S100 domain but have fewer and/or smaller centralized repeats (Fig. 1b).
3.2. Structural and chemical variation at the hydrophobic target binding site of SFTPs
To analyze structural differences between SFTP S100 domains, we used our PF-A structure to build homology models of the other six SFTP domains. Superposition of the models with the PF-A structure had 0.433 Å root-mean-square-deviation (RMSD), demonstrating the conserved S100 domain sequences dictate a four-helix bundle fold (Fig. 2a). Hydrophobic pocket residues differ between the SFTP S100 domains–some pockets are more acidic (comulin and trichohyalin), some are more basic (filaggrin-2 and hornerin), some are more polar (repetin and trichohyalin-like 1), and some are more hydrophobic (trichohyalin-like 1) than other SFTPs (Fig. 2b). To quantitate this variance in surface properties, solvent accessible surface area (SASA) for the residues at the putative hydrophobic target binding site was calculated (Fig. 2c) and compared to other calcium-binding proteins (CaBPs) previously analyzed [1]. The target interaction sites of SFTPs tend to be more hydrophobic and less acidic than soluble CaBPs, whereas polar and basic character vary. This data emphasizes that S100 proteins, whether soluble or fused-type, have unique surface properties at the target binding site. Variation in surface chemistry at the target binding site may regulate which macromolecules SFTPs bind.
Figure 2. Structural differences between SFTPs at the target binding pocket.
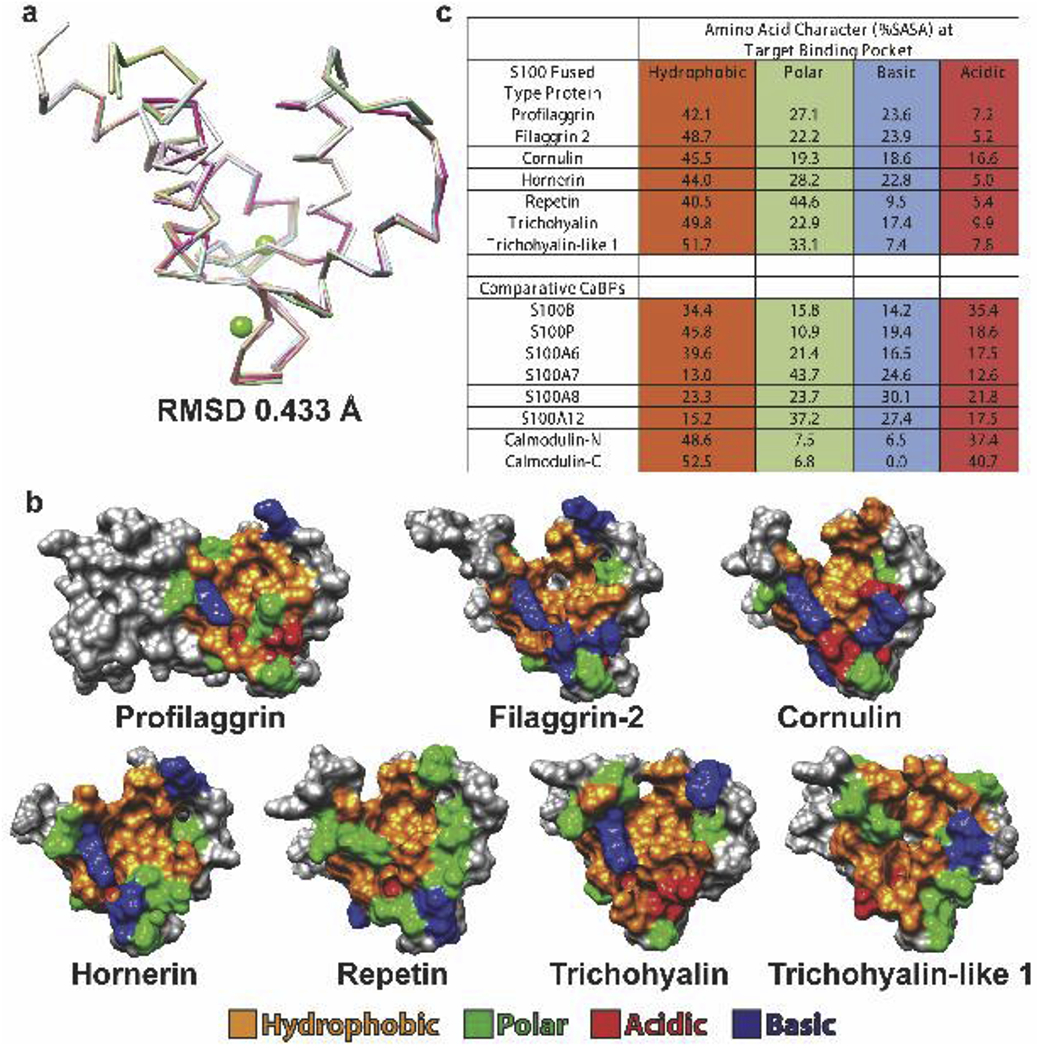
a) Superposition of the profilaggrin A domain crystal structure (PDB ID 4PCW) and homology models of the other six SFTPs demonstrated a root-mean-square deviation of 0.433 Å. b) Molecular surface representation of the profilaggrin A domain dimer and the S100 domains of the other six SFTP monomers; they are oriented to view the hydrophobic pocket. The molecular surface is colored by amino acid property: hydrophobic, orange; basic, blue; acidic, red; polar, green. Residues that do not contribute to the pocket are colored gray. c) Quantification of the solvent-accessible surface area (SASA) for hydrophobic, acidic, basic and polar residues surrounding the pocket in all SFTP family members. The SASAs are compared to those from other soluble calcium-binding proteins (CaBPs). Definitions of residues were: acidic (D, E), basic (R, K), polar (Q, N, H, S, T, Y, C, G) and hydrophobic (A, I, L, F, V, P, M, W).
3.3. Hydrophobic residues are conserved in the SFTP inter-EF hand linker
The PF-A structure showed the inter-EF-hand linker adopts different conformations (Fig. 3a), leading us to postulate SFTPs could facilitate target binding by using the linker to trap substrate in the hydrophobic pocket. Two of four inter-EF-hand linkers in the crystal asymmetric unit were in an extended conformation rather than a closed conformation (Fig. 3a). Conformational differences occurred because polyethylene glycol (PEG) in the crystallization solution bound the PF-A hydrophobic pocket, mimicking substrate binding. Ile43 and Leu44 of the linker bound and anchored PEG in the pocket. To further investigate the inter-EF-hand linker in SFTPs, we performed MSA of the linker region and identified conserved hydrophobic residues in positions 43 and 44 (Fig. 3b). This data suggests that all SFTPs have the potential to bind target molecules using, in part, hydrophobic residues within the linker. The structure of S100A10 dimer bound to ANXA2 N-terminus demonstrated F41 in the inter-EF-hand linker interacts with leucines from ANXA2 (Fig. 3c).
Figure 3. SFTPs conserve hydrophobic residues in the inter-EF-hand linker.
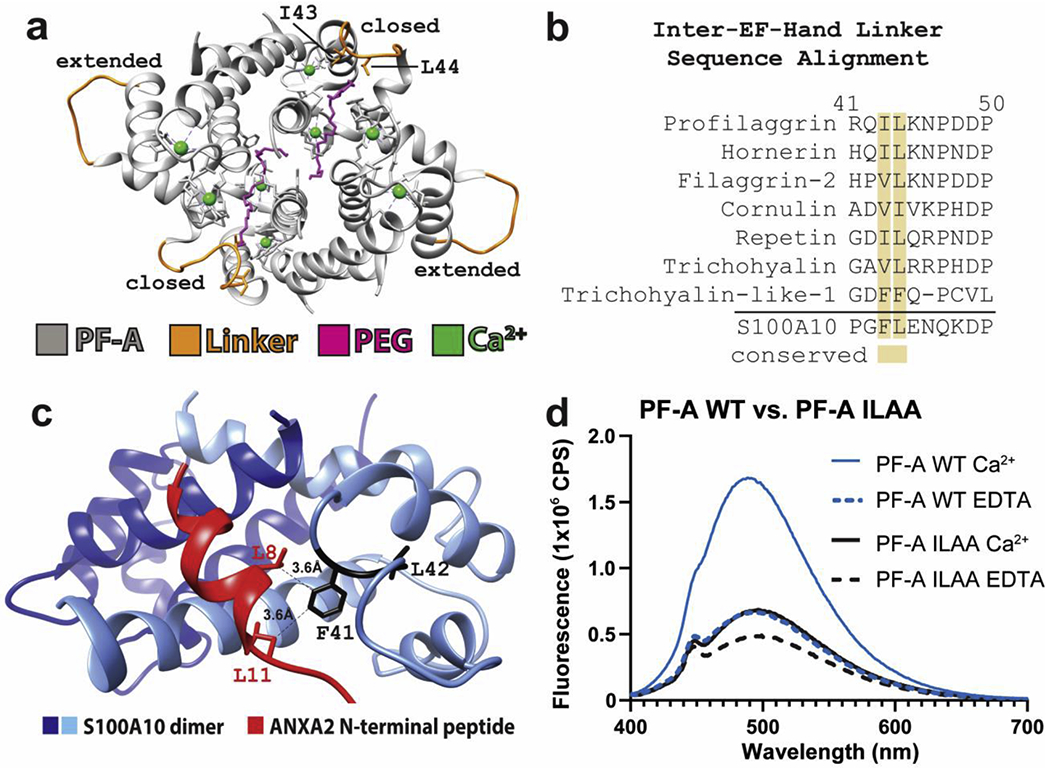
a) Ribbon diagram of the PF-A crystal structure asymmetric unit containing four profilaggrin S100 domain copies, with the inter-EF-hand linkers colored orange. Two linkers are in a “closed” conformation due to their binding of a PEG molecule (magenta), whereas the other two linkers are in an “extended” conformation. b) Multiple sequence alignment of the SFTP inter-EF-hand linker region demonstrates conservation of hydrophobic residues at positions equivalent to I43 and L44 of profilaggrin. The SFTP family is compared to soluble S100A10, which also conserves F41 and L42 in the inter-EF-hand linker. c) Structure of S100A10 dimer bound to ANXA2 (PDB ID 4HRE) shows the N-terminal ANXA2 peptide is anchored into the S100 hydrophobic pocket by interactions between F41 in the inter-EF-hand linker and leucine residues in the ANXA2 N-terminal peptide. d) Comparison of ANS fluorescence between WT PF-A and mutant PF-AI43A+L44A in the presence of Ca2+ or EDTA. WT PF-A undergoes the expected Ca2+-dependent conformational opening to expose hydrophobic surface for ANS to bind. Mutant PF-AI43A+L44A demonstrates a milder Ca2+-dependent conformational response, however, it may have an overall decreased capacity to bind ANS due to the I43A and L44A mutations. Abbreviations: Ca2+, buffer contained 10mM calcium chloride; EDTA, buffer contained 10mM ethylenediaminetetraacetic acid; PF-A, profilaggrin A (S100) domain; WT, wild-type; ILAA, double mutant Ile43Ala and Leu44Ala.
To determine whether profilaggrin inter-EF-hand linker substitutions I43A and L44A alter Ca2+-dependent conformational change (a reorientation of helices that exposes the hydrophobic pocket for substrate binding [22]), we used binding probe 8-anilino-1-naphthalenesulfonic acid (ANS) to compare fluorescence between wild-type PF-A and PF-AI43A+L44A. Wild-type PF-A exhibited a robust Ca2+-induced increase in ANS fluorescence compared to the protein in EDTA, whereas PF-AI43A+L44A demonstrated a more modest increase (Fig. 3d). The decreased fluorescence maximum observed for PF-AI43A+L44A in Ca2+ or EDTA solutions suggests Ile43 and Leu44 plays a role in the binding of substrate molecules.
3.4. Profilaggrin B domain contains exposed cationic surface
The B domain (PF-B) is unique to profilaggrin (Fig. 1b) and extends to the furin/proprotein convertase (PC) cleavage site in human and mouse proteins [7]. The PF-A structure showed helix IV ends at residue 88 [6]; therefore, we propose human profilaggrin residues 1-88 and 89-293 comprise the A and B domains, respectively (Fig. 4a). Using this we designed our recombinant PF-AB construct for this study (Methods).
Figure 4. Fusion of B domain to PF-A significantly increases ANS fluorescence.
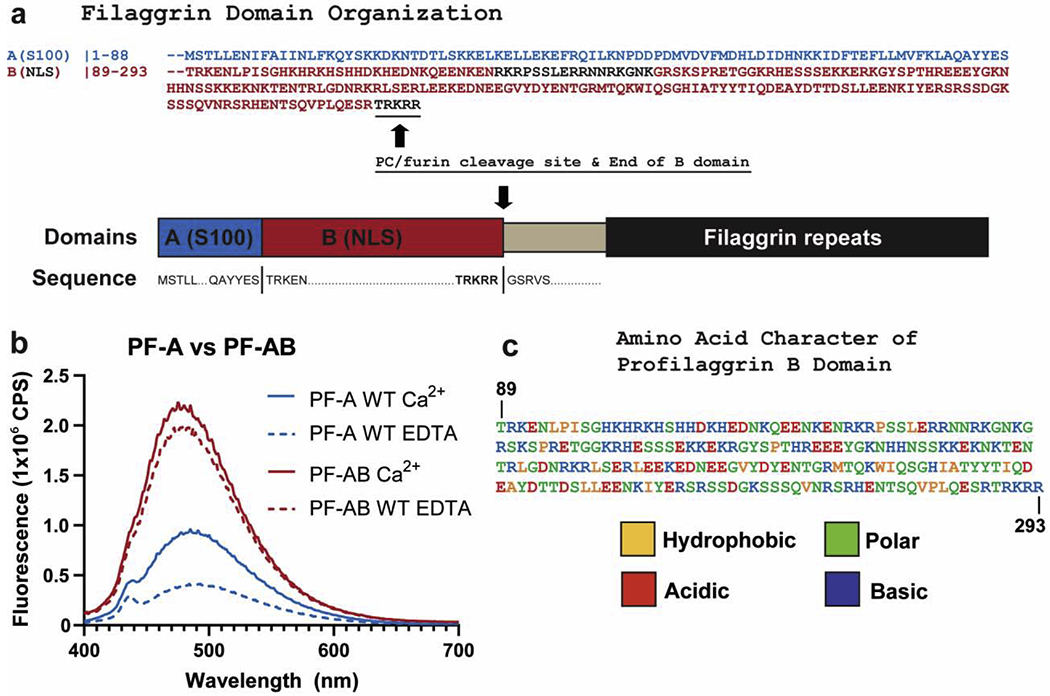
a) Schematic of the domains of the N-terminus of human profilaggrin with amino acid sequences listed for the beginning and termination of each domain. The B domain ends at the PC/furin cleavage site “RTRKRR.” b) Comparison of ANS fluorescence between wild-type PF-A and PF-AB in the presence of Ca2+ or EDTA. Wild-type PF-A undergoes the expected Ca2+-dependent conformational opening and increase in ANS fluorescence (blue). PF-AB, however, demonstrates a markedly robust binding and fluorescence of ANS (red), which is slightly stronger in the presence of Ca2+. c) PF-B amino acid sequence colored to reflect the hydrophobic, polar, acidic, and basic character of this domain as determined by ProtParam analysis. Abbreviations: NLS, nuclear localization signal; NTR, N-terminal truncated repeat.
To determine whether PF-B influences the ability of PF-A to bind Ca2+ and/or undergo conformational change, we used ANS fluorescence assay to compare PF-A with PF-AB (Fig. 4b). PF-AB showed a Ca2+-dependent increase in ANS fluorescence, which similar to other CaBPs [18, 22], represents a conformational opening that exposes the A domain hydrophobic pocket. PF-AB also displayed increased total fluorescence in both Ca2+ and EDTA conditions compared to PF-A at the same protein concentration, indicating PF-B contributes to ANS binding. ANS has affinity for positively charged residues in addition to hydrophobic ones [23]. PF-B is composed of 12% hydrophobic, 40% polar, and 48% charged (18% negative, 30% positive) residues (Fig. 4c). We suggest positively-charged residues of PF-B are the main contributor to its ability to bind ANS.
3.5. Profilaggrin B domain works cooperatively with A domain to bind target proteins
Despite PF-A being sufficient to bind ANXA2 in yeast-two-hybrid studies [6], we hypothesized PF-B participates in substrate binding given our fluorescence data and that PF-B is physiologically adjacent to PF-A. To investigate PF-AB target-binding functions, we analyzed PF-AB using protease protection, multi-angle light scattering (MALS), and pull-down assays. Recombinant wild-type (WT) PF-AB undergoes marked degradation during protein isolation/purification (Fig. 5a, PF-AB WT lane 0); moreover, degradation continues rapidly such that intact PF-AB is absent after 4 days at 4 °C (Fig. 5a, PF-AB WT lane 4d). The degradation occurs within the PF-B domain, likely due to intrinsic disorder; this is consistent with PF-A being stable for months at 4° C [6]. When ANXA2 is co-expressed with PF-AB, there is minor proteolysis initially (Fig. 5a, PF-AB WT+ANXA2 lane 0); however, degradation does not continue in contrast to PF-AB WT alone. Intact PF-AB is preserved 7 days post-isolation when co-expressed with ANXA2 (Fig. 5a, PF-AB WT+ANXA2 lane 7d). This data demonstrates ANXA2 binding to PF-AB protects PF-B from proteolysis. Thus, PF-B may work cooperatively with PF-A to bind ANXA2 and other protein targets.
Figure 5. Human profilaggrin B domain participates with the A domain to bind protein targets.
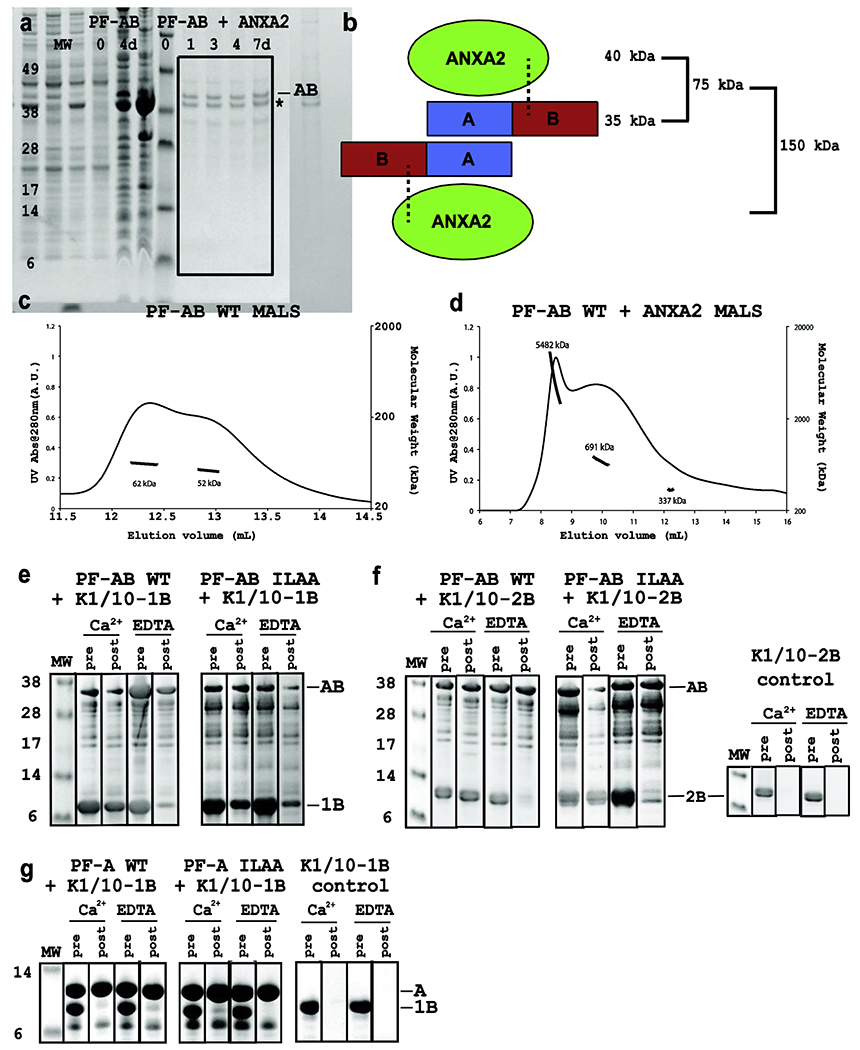
a) Protease protection assay comparing the degradation of PF-AB in the absence (left) and presence (right) of annexin-II (ANXA2) for up to 7 days at 4°C by SDS-PAGE. Whereas PF-AB is heavily degraded at day 0 and intact PF-AB gone by day 4, PF-AB in the presence of ANXA2 (asterix) is intact for 7 days indicating a stable, protected PF-AB-ANXA2 complex. Coexpression of PF-AB with ANXA2 led to reduced PF-AB concentration in the complex at day 0; however, ending up with more PF-AB at day 7 despite starting with less emphasizes the protective effect of ANXA2. b) Schematic showing one PF-AB dimer binding two ANXA2 molecules (based on PF-A dimer structure). Dotted lines represent potential stabilizing interactions between profilaggrin B domain and ANXA2. c) Multi-angle light scattering (MALS) of purified PF-AB shows two peaks that represent PF-AB dimers with minor protein degradation (62 and 52 kDa). d) MALS of PF-AB co-expressed and co-purified with ANXA2 in 10 mM CaCl2. All peaks correspond to several orders of magnitude above the expected 1:1 stoichiometric binding (see panel b), indicating PF-AB-ANXA2 complex forms high MW complexes/aggregates. e, f) Ni2+ pulldown assays using His6-tagged PF-AB as bait protein for keratin 1/10-1B (e) and keratin 1/10-2B (f) heterocomplex. WT and mutant PF-ABI43A+L44A exhibit Ca2+-dependent pulldown of K1/K10-1B and K1/K10-2B. In the presence of EDTA the ability for PF-AB or PF-ABI43A+L44A to pull down K1/K10-1B or K1/K10-2B is significantly diminished compared to that in the presence of calcium. Lanes are designated either “pre” or “post” the washing away of unbound proteins. Panels e and f used PF-AB (res. 1-257) corresponding to the major stable proteolyzed ~37 kDa fragment observed in panel a. g) In contrast, Ni2+ pulldown assays using His6-tagged PF-A as bait protein for the keratin 1/10-1B heterocomplex showed no keratin pulldown for either WT or mutant PF-AI43A+L44A in the presence of calcium or EDTA. Abbreviations: PF-AB, profilaggrin A and B fusion domain; MW, molecular weight; UV, ultra-violet; IF, intermediate filament; WT, wild-type; ILAA, double mutant Ile43Ala and Leu44Ala; Ca2+, buffer contained 10mM CaCl2; EDTA, buffer contained 10mM ethylenediaminetetraacetic acid; PF-A, profilaggrin A (S100) domain.
To characterize aggregation properties of PF-AB and PF-AB-ANXA2 in solution, we used MALS. From our PF-A structure we expected PF-AB to form a dimer in solution with calculated molecular weight (MW) 70 kDa. PF-AB was expected to bind one ANXA2 (~40 kDa) per A domain, generating a total MW of ~150 kDa (Fig. 5b). MALS on PF-AB alone revealed two overlapping peaks corresponding to ~52-62 kDa (Fig. 5c). These peaks are consistent with a PF-AB dimer that experienced mild proteolysis. In contrast, Ca2+-activated PF-AB-ANXA2 complex produced MALS peaks corresponding to large aggregates orders of magnitude higher than the expected 150 kDa (Fig. 5d). This data is consistent with our biochemical observation that PF-AB and ANXA2 aggregate and precipitate out of solution when lowering the NaCl concentration below 0.5 M in the presence of Ca2+. A soluble ANXA2 MALS control dataset was not possible due to lack of ANXA2 solubility without PF-AB (even with EDTA).
Another protein complex abundantly present in the stratum granulosum is keratin 1/keratin 10 (K1/K10), the predominant suprabasal cytoskeletal IF. K1/K10 filament networks are aggregated by filaggrin monomers (released by profilaggrin processing in KGs), facilitating cytoskeletal collapse of differentiated keratinocytes–a step critical in forming stratum corneum [24, 25]. Since profilaggrin N-terminus (PF-AB) is released simultaneously as filaggrin monomers are liberated from KGs, we reasoned PF-AB may bind an epidermal transition-layer protein-for example, keratin-for stability before nuclear translocation. We examined whether His6-tagged PF-A or PF-AB binds untagged K1/K10 (IB and 2B subdomains) using pulldown assay. There was significant Ca2+-dependent pulldown of untagged K1/K10-1B (Fig. 5e) and K1/K10-2B (Fig. 5f) using two His6-PF-AB proteins: WT and mutant (I43A+L44A). With EDTA, however, there was markedly diminished binding of K1/K10-1B or K1/K10-2B by PF-AB and PF-ABI43 V L44 \ The interaction between PF-AB and K1/K10 was also abrogated by elevated NaCl (500mM) (Supplementary Fig. S4). Mutating inter-EF-hand linker residues Ile43 and Leu44 to alanine did not alter K1/K10 binding. In comparison, neither His6-PF-A nor His6-PF-AI43A+L44A demonstrated binding to K1/K10-1B (Fig. 5g). These data demonstrate PF-B contributes protein binding function to PF-AB, and that PF-AB binds keratin in vitro.
3.6. Structural model for profilaggrin AB binding to annexin II
To investigate the structural role PF-B might play in PF-AB target binding, we superposed the structure of the S100A10 dimer complexed to ANXA2 with that of PF-A [6] (Fig. 6a). We illustrate PF-B as a green sphere and attached to the PF-A C-terminus (helix IV) where PF-B would exist in vivo. This sphere demonstrates how PF-B might exist spatially with respect to PF-A and ANXA2. Modeling predicts the ANXA2 molecular surfaces critical for PF-B binding are the helical I and II domains.
Figure 6. Structural modeling of human profilaggrin B domain interactions with ANXA2.
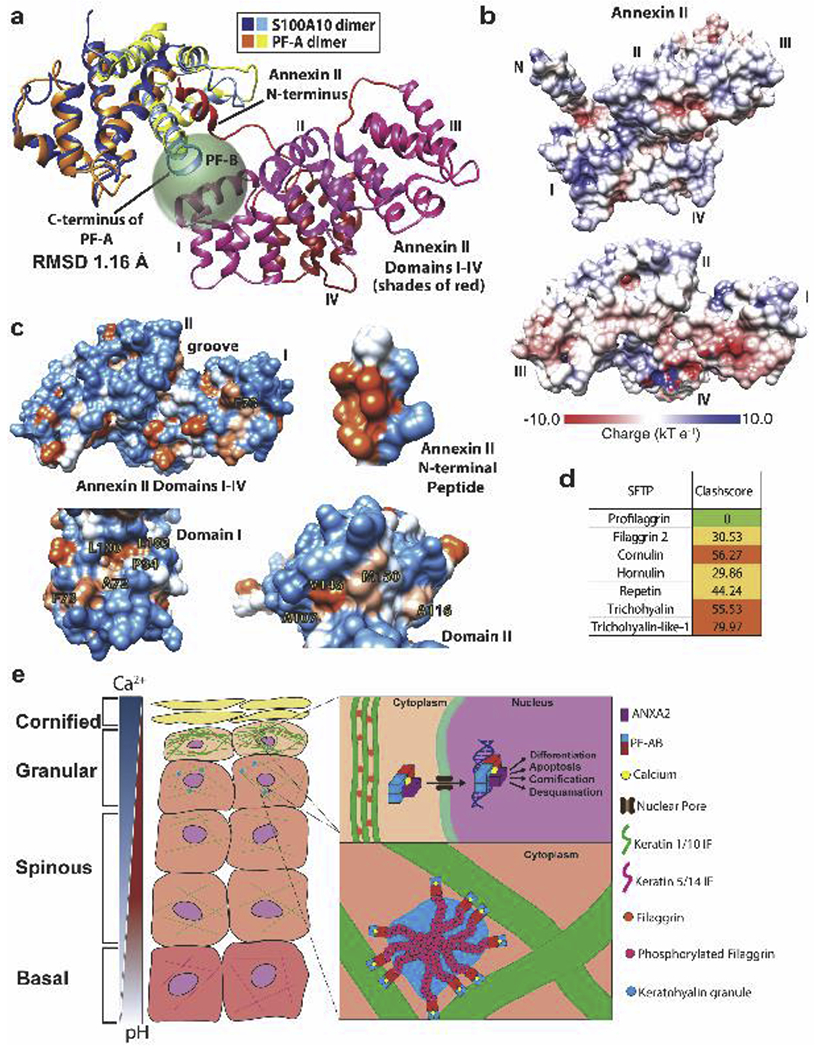
a) Superposition of S100A10+ANXA2 (PDB ID 4HRE) with PF-A (PDB ID 4PCW) demonstrated a root-mean-squared-deviation of 1.16 Å. The prediction of where the B domain (205 residues, green sphere) would reside in spatial relation to a PF-A-ANXA2 complex is depicted. PF-B begins at the end of the PF-A C-terminus. ANXA2 domains I and II are structurally positioned bind the profilaggrin B domain. b) Molecular surface of ANXA2 with the electrostatic potential (calculated using PDB2PQR and APBS software [50]) mapped onto the surface (acidic, red; basic, blue). ANXA2 in the upper panel is oriented exactly as in (a). Domains I and II have primarily a basic surface character where PF-B is most likely to bind. In the bottom panel, the viewpoint is from the N-terminal peptide looking back onto domains I-IV, with the N-terminal peptide and linker removed for clarity. There exists an acidic patch at the interface of domains I and IV. c) Molecular surface of ANXA2 with the hydrophobic potential (calculated using Chimera) mapped onto the surface (orange, hydrophobic; blue, polar). Specific hydrophobic residues with solvent-accessible surface in ANXA2 domains I and II are illustrated and may participate in PF-B binding. The hydrophobic nature of the PF-binding face of ANXA2 N-terminal peptide is shown for comparison. d) Using our PF-A structure docked with ANXA2Npep [6], we superposed the SFTP S100 domain homology models (see Fig. 2) onto the PF-A-ANXA2Npep complex and analyzed whether any other SFTP S100 domains have the capacity to bind ANXA2Npep using MolProbity clash scores. Despite high conservation of the S100 hydrophobic pocket, clash scores suggest unlikely binding of ANXA2Npep by any SFTP member except profilaggrin (due to steric hindrance). This data reinforces the concept each SFTP member has unique surface properties at the hydrophobic pocket dictating its molecular function. e) Proposed model for PF and PF-AB function in keratinocytes.
Enhanced ANS fluorescence by PF-AB compared to PF-A alone (Fig. 4b) indicated PF-B contains extensive solvent accessible cationic residues on its surface [23]; PF-B contains 30% basic and 18% acidic residues (Fig. 4c). To examine the molecular surface properties of ANXA2 at the predicted PF-B interaction site, we mapped ANXA2 electrostatic potential onto its molecular surface (Fig. 6b). ANXA2 domain II has overall basic surface character. In contrast, ANXA2 domains I, III and IV have also acidic surface (Fig. 6b). Together, these data suggest PF-B interactions with ANXA2 may be driven by electrostatic interactions.
We next asked whether solvent-accessible hydrophobic residues existed in ANXA2 domains I and II that could interact with PF-B. While the N-terminal linker (residues 13-33) contains several hydrophobic residues (L13, P20, P21, A23, V27, F33), they are either buried or far from the suggested PF-B binding site. We identified 5 hydrophobic residues in ANXA2 domain I (A72, F73, P84, L100, L103) and 4 in domain II (A107, A116, V146, M150) that are solvent-accessible and can potentially bind to the B domain (Fig. 6c).
To demonstrate the molecular selectivity and specificity of the SFTP hydrophobic pocket in binding substrate, we used our model of PF-A bound to the ANXA2 N-terminal 14-amino acid peptide (ANXA2Npep) [6] to superpose ANXA2Npep into the hydrophobic pocket of each of our SFTP structural models and examine them for steric clashes (Fig. 6d). The higher number of residue clashes between ANXA2Npep and each SFTP S100 domain other than profilaggrin suggests ANXA2 binding is unlikely for any SFTP other than profilaggrin.
4. Discussion
The PF-A structure and identification of PF-A target molecules [6] provided a foundation for understanding profilaggrin structure and function. This study further elucidates structural differences between SFTPs and characterizes the molecular basis for how PF-B binds proteins. Soluble S100 proteins are implicated in multiple diseases and interact with numerous molecules [26, 27]. SFTPs, via their common N-terminal S100 domain, likely have a similar diversity of molecular targets and biological roles. The data presented here demonstrate structural differences in the binding center of SFTPs, which may define specificity towards the binding target. Ultimately, defining the structural and functional differences between SFTPs and identifying the target proteins they interface with is critical to elucidating the role of SFTPs in cellular activity and human disease pathogenesis. We show the surface chemistry of the S100 hydrophobic pocket differs among SFTPs, as for soluble S100 proteins [1]. Work here identifies three components of the profilaggrin S100 substrate binding mechanism: i) unique chemical composition at the hydrophobic pocket; ii) conserved hydrophobic inter-EF-hand linker residues; and iii) cooperative binding effort from adjacent domains (e.g. PF-B).
There are currently no structures of PF-B, likely because it is intrinsically disordered (Supplementary Fig. S5a). This is consistent with secondary structure prediction, which indicates PF-B is random coil except for a group of charged C-terminal residues that may adopt beta-sheet conformation, and our data showing PF-B is susceptible to proteolysis in the absence of a binding partner (Fig. 5a; Supplementary Fig. S5b). This raises questions as to how PF-AB reaches the nucleus intact if it cannot maintain cytosolic stability independently.
Advances have been made in understanding the capacity of low-complexity proteins like PF to undergo liquid-liquid phase separation within cells [28, 29]. Both the S100 domain and tandem filaggrin units facilitated KG formation by phase separation [28]. The S100 domain specifically reduced the critical concentration of PF necessary for phase separation, demonstrating an important cellular function for PF-A in optimizing KG formation as we hypothesized from the PF-A structure [6]. KGs are membraneless biomolecular condensates [30, 31] that protect PF from proteolysis just as they protect keratinocytes from premature death and cornification [25]. PF-A and PF-B localize to KGs [32], meaning proteolytic processing of PF occurs after KG formation [33]. During terminal epidermal differentiation (TED), as granular layer keratinocytes become more acidic, lower pH stimulates over ~ 2h KG dissolution, chromatin compaction and loss, and nuclear destruction [28]. Dephosphorylation and processing of profilaggrin into PF-AB and filaggrin units occur during this time, causing cytoplasmic keratin IF bundling and PF-AB nuclear translocation [7] (Fig. 6e). The latter is believed to be a critical step in keratinocyte TED [7, 9]; therefore, protecting PF-AB against degradation appears vital for completing nuclear translocation in vivo. Lower pH failed to cause chromatin compaction and enucleation in skin lacking KGs, demonstrating KGs as pH-sensitive and important for TED [28]. We propose PF-AB must bind a target or chaperone protein in the cytoplasm, outside of KGs, to achieve stability before and during nuclear translocation. Candidate proteins include a nuclear import protein, hsp27/HspB1 (found in KGs) [34, 35], or ANXA2 (some ANXA2 exists in the nucleus [36–38]). Notably, HspB1 stabilizes actin scaffolds associated with KGs. In an AKT1-dependent mechanism, HspB1 switches to facilitate filaggrin processing and promote TED [39]. ANXA2, which has phospholipid and actin binding properties, could help regulate cytoskeleton (actin and IF) as well as keratinocyte membrane changes during TED [40, 41].
Previous studies demonstrated monomeric filaggrin binds and aggregates keratin and other IF types [5, 20, 42], but not F-actin or microtubules. We show human PF-AB binds human keratin 1/10 in vitro, indicating PF-AB has binding specificity towards types I and II IFs. This agrees with data showing K1/K10 IFs form networks around KGs contributing to their stability and reduced fusion rate [28]. It also agrees with data showing PF-AB binds loricrin, K10 (mapped to coil 2B and C-terminus residues 410-566), and small proline-rich protein [10]. We demonstrate PF-AB binds K1/K10 coiled-coil regions lacking the C-terminal tail in the presence of calcium (Figs. 5e,f). Keratin structures demonstrated the coiled-coil domains have acidic molecular surface [17, 43–45], which explains why positively-charged filaggrin units must be phosphorylated and stored in KGs to prevent premature aggregation of the keratin cytoskeleton in granular layer keratinocytes [46, 47]. ANS fluorescence suggests the PF-B surface is cationic, indicating PF-AB within KGs could provide stabilizing contacts to caged and fenced keratin IFs [28] while the phosphorylated filaggrin unit is inert to keratin. PF-AB exhibited its strongest activity in a high calcium environment in vitro, suggesting it utilizes cytoplasmic calcium in differentiated keratinocytes to function in vivo.
The IF-binding capacity of PF-AB suggests that in the nucleus it could bind lamin to either directly or indirectly promote the dissolution of the nuclear envelope. Lamins (type V IFs) contribute to the nuclear lamina which stabilizes the inner nuclear membrane. If PF-AB reaches the nucleus with any intact or partial filaggrin units attached, then filaggrin has the capability to aggregate lamins [48].
Four decades of work make profilaggrin the best characterized SFTP [49]. To better understand in vivo functions of profilaggrin, it is essential to advance our knowledge of the biochemical and structural mechanisms regulating its formation of KGs, interaction with binding proteins, and activities in the keratinocyte nucleus.
Supplementary Material
Highlights.
S100 fused-type proteins have distinct surface chemistries at target binding site
Inter-EF-hand linker residues help S100 fused-type proteins anchor substrate
Profilaggrin B domain works with A domain to bind and stabilize protein targets
Profilaggrin AB complex binds coiled-coil region of keratin intermediate filaments
Annexin 2 domains I and II are positioned to interact with profilaggrin B domain
ACKNOWLEDGEMENTS
We thank the late Professor Thomas A. Steitz (Dept. of Molecular Biophysics and Biochemistry, Yale Univ.) for providing a collegial environment, laboratory space, equipment, and supplies for this project (to C.G.B.). We thank Professors Leonard Milstone and Richard Presland for constructive feedback. This work was supported by the Dermatology Foundation through a Dermatologist Investigator Research Fellowship and a Career Development Award (to C.G.B.), and by NIH/NIAMS (Grant K08AR070290 to C.G.B.).
Funding Sources: This work was supported by the Dermatology Foundation through a Dermatologist Investigator Research Fellowship and a Career Development Award (to C.G.B.), and by NIH/NIAMS (Grant K08AR070290 to C.G.B.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest: The authors have no conflicts of interest to declare.
Aspects of this work were presented as abstract #353 at the 2015 Society for Investigative Dermatology 74th Annual Meeting (Atlanta, GA).
REFERENCES:
- 1.Bhattacharya S, Bunick CG, and Chazin WJ, Target selectivity in EF-hand calcium binding proteins. Biochim Biophys Acta, 2004. 1742(1-3): p. 69–79. [DOI] [PubMed] [Google Scholar]
- 2.Kizawa K, et al. , S100 and S100 fused-type protein families in epidermal maturation with special focus on S100A3 in mammalian hair cuticles. Biochimie, 2011. 93(12): p. 2038–47. [DOI] [PubMed] [Google Scholar]
- 3.Kypriotou M, Huber M, and Hohl D, The human epidermal differentiation complex: cornified envelope precursors, S100 proteins and the ‘fused genes’ family. Exp Dermatol, 2012. 21(9): p. 643–9. [DOI] [PubMed] [Google Scholar]
- 4.Choi J, et al. , Hornerin Is Involved in Breast Cancer Progression. J Breast Cancer, 2016. 19(2): p. 142–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steinert PM, et al. , Characterization of a class of cationic proteins that specifically interact with intermediate filaments. Proc Natl Acad Sci U S A, 1981. 78(7): p. 4097–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bunick CG, et al. , Crystal Structure of Human Profilaggrin S100 Domain and Identification of Target Proteins Annexin II, Stratifin and hsp27. J Invest Dermatol, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pearton DJ, Dale BA, and Presland RB, Functional analysis of the profilaggrin N-terminal peptide: identification of domains that regulate nuclear and cytoplasmic distribution. J Invest Dermatol, 2002. 119(3): p. 661–9. [DOI] [PubMed] [Google Scholar]
- 8.Ishida-Yamamoto A, et al. , Translocation of profilaggrin N-terminal domain into keratinocyte nuclei with fragmented DNA in normal human skin and loricrin keratoderma. Lab Invest, 1998. 78(10): p. 1245–53. [PubMed] [Google Scholar]
- 9.Aho S, et al. , Regulatory role for the profilaggrin N-terminal domain in epidermal homeostasis. J Invest Dermatol, 2012. 132(10): p. 2376–85. [DOI] [PubMed] [Google Scholar]
- 10.Yoneda K, et al. , Interaction of the profilaggrin N-terminal domain with loricrin in human cultured keratinocytes and epidermis. J Invest Dermatol, 2012. 132(4): p. 1206–14. [DOI] [PubMed] [Google Scholar]
- 11.Brown SJ, et al. , Intragenic copy number variation within filaggrin contributes to the risk of atopic dermatitis with a dose-dependent effect. J Invest Dermatol, 2012. 132(1): p. 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akiyama M, FLG mutations in ichthyosis vulgaris and atopic eczema: spectrum of mutations and population genetics. Br J Dermatol, 2010. 162(3): p. 472–7. [DOI] [PubMed] [Google Scholar]
- 13.Sandilands A, et al. , Comprehensive analysis of the gene encoding filaggrin uncovers prevalent and rare mutations in ichthyosis vulgaris and atopic eczema. Nat Genet, 2007. 39(5): p. 650–4. [DOI] [PubMed] [Google Scholar]
- 14.Palmer CN, et al. , Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet, 2006. 38(4): p. 441–6. [DOI] [PubMed] [Google Scholar]
- 15.Sandilands A, et al. , Prevalent and rare mutations in the gene encoding filaggrin cause ichthyosis vulgaris and predispose individuals to atopic dermatitis. J Invest Dermatol, 2006. 126(8): p. 1770–5. [DOI] [PubMed] [Google Scholar]
- 16.Smith FJ, et al. , Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat Genet, 2006. 38(3): p. 337–42. [DOI] [PubMed] [Google Scholar]
- 17.Eldirany SA, et al. , Human keratin 1/10-1B tetramer structures reveal a knob-pocket mechanism in intermediate filament assembly. EMBO J, 2019. 38(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bunick CG, et al. , Designing sequence to control protein function in an EF-hand protein. J Am Chem Soc, 2004. 126(19): p. 5990–8. [DOI] [PubMed] [Google Scholar]
- 19.Waterhouse A, et al. , SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res, 2018. 46(W1): p. W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lynley AM and Dale BA, The characterization of human epidermal filaggrin. A histidine-rich, keratin filament-aggregating protein. Biochim Biophys Acta, 1983. 744(1): p. 28–35. [DOI] [PubMed] [Google Scholar]
- 21.Scott IR, Harding CR, and Barrett JG, Histidine-rich protein of the keratohyalin granules. Source of the free amino acids, urocanic acid and pyrrolidone carboxylic acid in the stratum corneum. Biochim Biophys Acta, 1982. 719(1): p. 110–7. [DOI] [PubMed] [Google Scholar]
- 22.Zhang M, Tanaka T, and Ikura M, Calcium-induced conformational transition revealed by the solution structure of apo calmodulin. Nat Struct Biol, 1995. 2(9): p. 758–67. [DOI] [PubMed] [Google Scholar]
- 23.Gasymov OK and Glasgow BJ, ANS fluorescence: potential to augment the identification of the external binding sites of proteins. Biochim Biophys Acta, 2007. 1774(3): p. 403–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsui T and Amagai M, Dissecting the formation, structure and barrier function of the stratum corneum. Int Immunol, 2015. 27(6): p. 269–80. [DOI] [PubMed] [Google Scholar]
- 25.Candi E, Schmidt R, and Melino G, The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol, 2005. 6(4): p. 328–40. [DOI] [PubMed] [Google Scholar]
- 26.Santamaria-Kisiel L, Rintala-Dempsey AC, and Shaw GS, Calcium-dependent and - independent interactions of the S100 protein family. Biochem J, 2006. 396(2): p. 201–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hermann A, et al. , S100 calcium binding proteins and ion channels. Front Pharmacol, 2012. 3: p. 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quiroz FG, et al. , Liquid-liquid phase separation drives skin barrier formation. Science, 2020. 367(6483). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hughes MP, et al. , Atomic structures of low-complexity protein segments reveal kinked β sheets that assemble networks. Science, 2018. 359(6376): p. 698–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Banani SF, et al. , Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol, 2017. 18(5): p. 285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shin Y and Brangwynne CP, Liquid phase condensation in cell physiology and disease. Science, 2017. 357(6357). [DOI] [PubMed] [Google Scholar]
- 32.Presland RB, et al. , Evidence for specific proteolytic cleavage of the N-terminal domain of human profilaggrin during epidermal differentiation. J Invest Dermatol, 1997. 108(2): p. 170–8. [DOI] [PubMed] [Google Scholar]
- 33.Pearton DJ, et al. , Proprotein convertase expression and localization in epidermis: evidence for multiple roles and substrates. Exp Dermatol, 2001. 10(3): p. 193–203. [DOI] [PubMed] [Google Scholar]
- 34.O’Shaughnessy RF, et al. , AKT-dependent HspB1 (Hsp27) activity in epidermal differentiation. J Biol Chem, 2007. 282(23): p. 17297–305. [DOI] [PubMed] [Google Scholar]
- 35.Kayser J, et al. , The small heat shock protein Hsp27 affects assembly dynamics and structure of keratin intermediate filament networks. Biophys J, 2013. 105(8): p. 1778–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vishwanatha JK, Jindal HK, and Davis RG, The role of primer recognition proteins in DNA replication: association with nuclear matrix in HeLa cells. J Cell Sci, 1992. 101 (Pt 1): p. 25–34. [DOI] [PubMed] [Google Scholar]
- 37.Eberhard DA, et al. , Control of the nuclear-cytoplasmic partitioning of annexin II by a nuclear export signal and by p11 binding. J Cell Sci, 2001. 114(Pt 17): p. 3155–66. [DOI] [PubMed] [Google Scholar]
- 38.Liu J and Vishwanatha JK, Regulation of nucleo-cytoplasmic shuttling of human annexin A2: a proposed mechanism. Mol Cell Biochem, 2007. 303(1-2): p. 211–20. [DOI] [PubMed] [Google Scholar]
- 39.Gutowska-Owsiak D, et al. , Orchestrated control of filaggrin-actin scaffolds underpins cornification. Cell Death Dis, 2018. 9(4): p. 412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma AS and Ozers LJ, Annexins I and II show differences in subcellular localization and differentiation-related changes in human epidermal keratinocytes. Arch Dermatol Res, 1996. 288(10): p. 596–603. [DOI] [PubMed] [Google Scholar]
- 41.Hayes MJ, et al. , Regulation of actin dynamics by annexin 2. EMBO J, 2006. 25(9): p. 1816–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mack JW, Steven AC, and Steinert PM, The mechanism of interaction of filaggrin with intermediate filaments. The ionic zipper hypothesis. J Mol Biol, 1993. 232(1): p. 50–66. [DOI] [PubMed] [Google Scholar]
- 43.Lee CH, et al. , Structural basis for heteromeric assembly and perinuclear organization of keratin filaments. Nat Struct Mol Biol, 2012. 19(7): p. 707–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lomakin IB, et al. , Crystal structure of keratin 1/10(C401A) 2B heterodimer demonstrates a proclivity for the C-terminus of helix 2B to form higher order molecular contacts. Yale J Biol Med, 2020. 93(1). [PMC free article] [PubMed] [Google Scholar]
- 45.Bunick CG and Milstone LM, The X-Ray Crystal Structure of the Keratin 1-Keratin 10 Helix 2B Heterodimer Reveals Molecular Surface Properties and Biochemical Insights into Human Skin Disease. J Invest Dermatol, 2017. 137(1): p. 142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sandilands A, et al. , Filaggrin in the frontline: role in skin barrier function and disease. J Cell Sci, 2009. 122(Pt 9): p. 1285–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lonsdale-Eccles JD, Teller DC, and Dale BA, Characterization of a phosphorylated form of the intermediate filament-aggregating protein filaggrin. Biochemistry, 1982. 21(23): p. 5940–8. [DOI] [PubMed] [Google Scholar]
- 48.Dale BA, et al. , Transient expression of epidermal filaggrin in cultured cells causes collapse of intermediate filament networks with alteration of cell shape and nuclear integrity. J Invest Dermatol, 1997. 108(2): p. 179–87. [DOI] [PubMed] [Google Scholar]
- 49.Brown SJ and McLean WH, One remarkable molecule: filaggrin. J Invest Dermatol, 2012. 132(3 Pt 2): p. 751–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dolinsky TJ, et al. , PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res, 2004. 32(Web Server issue): p. W665–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.