

Abstract
Bacteria are surrounded by a peptidoglycan cell wall that is essential for their survival1. During cell wall assembly, a lipid-linked disaccharide-peptide precursor called Lipid II is polymerized and crosslinked to produce mature peptidoglycan. As Lipid II is polymerized, nascent polymers remain membrane-anchored at one end and the other end becomes crosslinked to the matrix2–4. A longstanding question is how bacteria release newly synthesized peptidoglycan strands from the membrane to complete the synthesis of mature peptidoglycan. Here we show that a Staphylococcus aureus cell wall hydrolase and a membrane protein containing eight transmembrane helices form a complex that may function as a peptidoglycan release factor. The complex cleaves nascent peptidoglycan internally to produce free oligomers as well as lipid-linked oligomers that can undergo further elongation. The polytopic membrane protein, which is similar to a eukaryotic CAAX protease, controls the length of these products. A 2.6 Å resolution structure of the complex shows that the membrane protein scaffolds the hydrolase to orient its active site for cleavage of the glycan strand. We propose that this complex serves to detach newly-synthesized peptidoglycan polymer from the cell membrane to complete integration into the cell wall matrix.
The biosynthesis of the bacterial cell wall has been the focus of intense study for decades.1 The peptidoglycan precursor Lipid II is synthesized inside the cell, transported across the cytoplasmic membrane,5,6 and then assembled outside the cell into a crosslinked polymer that prevents osmotic lysis (Fig. 1a, left and middle panel)2. Two conserved families of peptidoglycan synthases carry out Lipid II polymerization and crosslinking2–4. These peptidoglycan synthases, particularly the transpeptidase (TP) components, have received a great deal of attention as targets for antibiotics. Indeed, transpeptidases are generically known as penicillin-binding proteins because they react covalently with beta-lactam antibiotics (PBPs)7. A necessary step in peptidoglycan biosynthesis that has received almost no attention is the release of newly synthesized peptidoglycan strands from the membrane (Fig. 1a, right panel). One way in which this might be achieved is with a glycosidase that cleaves within a glycan strand to separate the end that has been crosslinked into the cell wall from the lipid-linked nascent oligomer that is not yet crosslinked.8 Here, we describe a membrane protein complex comprising a glycosidase and a membrane protein that regulates its cleavage activity. This complex meets criteria for a peptidoglycan release factor and explains how nascent peptidoglycan is freed from the membrane in S. aureus.
Figure 1. The cell wall hydrolase SagB and the membrane protein SpdC form a complex.

a, Overview of the final steps in peptidoglycan assembly. Left panel: After translocation to the outer face of the cytoplasmic membrane, the peptidoglycan (PG) monomer Lipid II is polymerized into linear glycan strands by glycosyltransferases (GT). Middle panel: Transpeptidase domains (TPs) crosslink glycan strands into the cell wall. Right panel: The glycan strand must be released from the membrane through some type of cleavage process in order to be incorporated into the cell wall. b, A Staphylococcus aureus transposon library made by combining six sublibraries as previously reported9,10 was treated with a panel of beta-lactams (oxacillin, cefoxitin, mecillinam) that have different selectivities for the four native S. aureus penicillin-binding proteins (PBPs)11. Only two genes, sagB and spdC, displayed a response pattern in which transposon reads were depleted under oxacillin and mecillinam treatment but enriched under cefoxitin treatment. Corrected p-values are reported for this experiment in Supplementary Table 1. c, SagB is a membrane-anchored glucosaminidase12,13 and SpdC is an eight-pass membrane protein15. Myc-tagged SpdC was expressed in a ΔspdC S. aureus strain and co-immunoprecipitated from solubilized membranes. (“Co-IP”, see also Supplementary Fig. 1). SagB was identified by LC-MS-MS analysis (Supplementary Table 1). Tandem affinity purification of SagB-His6 and FLAG-SpdC from E. coli yielded a stable 1:1 complex (“co-purification”, see also Supplementary Figure 2).
To find genes important in cell wall assembly, we probed a S. aureus transposon library with sublethal concentrations of three different beta-lactams that have distinct PBP inhibition profiles (Fig. 1b)9–11. At those concentrations, oxacillin inhibits bifunctional PBPs 2 and 3, mecillinam inhibits PBP3, and cefoxitin selectively inhibits the monofunctional transpeptidase PBP411. For sagB, encoding a membrane-anchored glucosaminidase that affects peptidoglycan strand length12–14, we observed an unusual response pattern in that transposon reads were strongly depleted in the presence of oxacillin and mecillinam, but enriched in the presence of cefoxitin (Fig. 1b; Supplementary Table 1). Only one other gene displayed the same pattern: spdC, encoding a membrane protein similar to eukaryotic CAAX proteases, enzymes that cleave prenylated proteins C-terminal to the site of prenylation. CAAX protease homologs are widespread in bacteria, but their roles have been unclear because protein prenylation is a modification not found in bacteria. Notably, sagB and spdC knockouts were reported in separate studies to share several distinctive phenotypes12,13,15,16. Together with the shared Tn-seq profiles, these joint phenotypes led us to think SagB and SpdC may act in a complex.
To test whether SagB and SpdC form a complex, we expressed Myc-SpdC in S. aureus ΔspdC and immunoprecipitated the tagged protein from solubilized membranes. Polyacrylamide gel electrophoresis (PAGE) of the sample showed a band that contained SagB as a major component (Fig. 1c; Supplementary Figure 1, Supplementary Table 2). Based on this finding, we co-expressed SagB-His6 and FLAG-SpdC in E. coli and purified a complex containing SpdC and SagB in a 1:1 ratio (Fig. 1c, and also see Supplementary Figure 2). S. aureus contains only one other glucosaminidase with a transmembrane helix, SagA.12,13 The protein is homologous to SagB but did not have the same profile in our Tn-seq experiments, and we were unable to copurify SpdC with SagA (Supplementary Figure 3). Taken together, our experiments showed that SpdC and SagB form a stable, specific complex.
To determine whether SpdC affects SagB activity, we compared the cleavage activity of the complex and SagB alone. We incubated the enzyme or enzyme complex with uncrosslinked peptidoglycan prepared in vitro from synthetic [14C]-Lipid II17–19 and analyzed the reactions via PAGE-autoradiography (Fig. 2, and see Supplementary Fig. 4). Consistent with previous findings, SagB lacking its TM helix has low activity (Fig. 2b, lane 6; also Extended Data 1)13,20. In contrast, full-length SagB fully converted the peptidoglycan oligomers to diffuse bands high in the gel (Fig. 2b, lane 4). SagA produced similar product bands. These were found to be short cleavage products ranging from two to eight sugars in length (Supplementary Figure 5). The SagB-SpdC complex also produced short oligosaccharides, but they were longer on average than those produced by SagB alone (Supplementary Figure 5); moreover, we observed an accumulation of faster-migrating cleavage products not observed for SagB alone (Fig. 2b). We observed a similar accumulation of fast-migrating products when the native S. aureus substrate was used to make peptidoglycan polymer (Extended Data 2).
Figure 2. In vitro reconstitution shows that SagB-SpdC cleaves nascent peptidoglycan to short lipid-linked oligomers that can be elongated.

a, Chemical and cartoon representations of the synthetic Lipid II analog18 and native S. aureus Lipid II that were used to prepare peptidoglycan polymers in panels b-d. b, Radiolabeled peptidoglycan polymers were incubated with the SagB-SpdC complex, SagB alone, SagA, or SagB lacking its transmembrane helix. The signal towards the top of the autoradiograph in lanes 3–5 corresponds to short, lipid-free peptidoglycan fragments, but the distribution of lengths differs (Supplementary Figure 5). SagB-SpdC also produces a short ladder of radiolabeled peptidoglycan fragments (see also Supplementary Figure 4). c, Left: schematic of assay to determine whether SagB-SpdC product bands contained a lipid-anchor. Right: Radiolabeled SagB-SpdC products were incubated with unlabeled Lipid II and PBP2 and were extended to longer products. d, The bacteriocin Colicin M (ColM) de-lipidates Lipid II and peptidoglycan oligomers, but leaves the anomeric diphosphate (Supplementary Figures 6 and 7)23,24. Incubation of SagB-SpdC products (lane 3) with ColM (lane 4) resulted in the complete disappearance of fastmigrating bands and the appearance of slower-migrating products. Product characterization by LC-MS confirmed the indicated structure (Supplementary Figure 8). The faster migration of the SagB-SpdC products containing a lipid may be due to SDS binding to the lipid and increasing the net negative charge of these species. Experiments were performed at least three times in biological replicates.
We next sought to identify the cleavage fragments that uniquely accumulate for the SagB-SpdC complex. Their migration behavior suggested these fragments may still contain the diphospholipid anchor at the reducing end. Because peptidoglycan glycosyltransferases (GTs) add Lipid II to the reducing end of the growing polymer21–23, one way to test if the SagB-SpdC cleavage products retain the diphospholipid is to determine whether they are competent substrates for polymer extension (Fig. 2c). Therefore, we prepared radiolabeled peptidoglycan oligomers, cleaved them with SagB-SpdC, incubated the cleavage products with S. aureus PBP2 and cold Lipid II, and analyzed the products by PAGE autoradiography (Fig. 2c). The SagB-SpdC cleavage products shifted to higher molecular weight bands, showing they were competent substrates and implying the presence of a diphospholipid at the reducing end. Furthermore, when we treated the cleavage products with the bacteriocin colicin M, which removes the lipid, we observed that the cleavage products migrated more slowly by SDS-PAGE (Fig. 2d; Supplementary Figure 6–8).24,25 LC-MS analysis confirmed the presence of a diphosphate on a peptidoglycan fragment having an odd number of sugars, consistent with glucosaminidase cleavage to leave a terminal MurNAc (Supplementary Figure 8). Notably, SagB-SpdC’s cleavage activity in vitro depended on the conserved catalytic glutamate13,26,13,26,13,13,25,13,25,20 in SagB (E155), but not on an SpdC residue conserved in eukaryotic CAAX proteases and required for their proteolytic activity (Supplementary Figure 9). Moreover, cellular phenotypes of the SagB catalytic mutant resemble a sagB knockout, whereas SpdC mutants lacking putative catalytic residues resemble wild-type (Extended Data 3)15. These findings show that SagB plays a catalytic role in peptidoglycan cleavage while SpdC plays a noncatalytic role in controlling SagB. While we cannot exclude the possibility that SpdC has a catalytic function, its known in vitro and cellular phenotypes involve noncatalytic functions.
These results show that the SagB-SpdC complex satisfies criteria expected for a peptidoglycan release factor. First, the complex yields products that contain a reducing-end lipid; second, these products are capable of further elongation. SagB’s known effect on glycan strand length may also be due to its role as part of a release factor complex. If so, we would expect the loss of SpdC to similarly affect glycan strand length. By comparing glycan strands isolated from wild-type, ΔsagB, and ΔspdC cells, we found that the short glycan strands characteristic of wild-type S. aureus are lost in both the ΔsagB and ΔspdC mutants (Extended Data 4), showing that SpdC is also involved in glycan strand length control in cells. We would also expect SagB-SpdC to act on newly synthesized sections of peptidoglycan polymer that are not yet modified or crosslinked into the matrix because both proteins are anchored in the membrane. Consistent with this expectation, we observed a clear preference for cleavage of nascent, unmodified peptidoglycan over peptidoglycan that contained teichoic acid modifications or crosslinks (Extended Data 5 and 6). Our conclusion that SagB-SpdC acts early in the peptidoglycan maturation pathway is supported by recent studies that used atomic force microscopy to visualize SagB mutants14.
To understand how SpdC interacts with SagB to control cleavage of nascent polymer, we solved the structure of SagB complexed with a truncated form of SpdC (SpdC1−256) that lacks the cytoplasmic C-terminal domain (Fig. 3a–c; Extended Data 7)15. Removal of this apparently unstructured domain did not affect formation of the complex, change its in vitro activity, or impact cellular phenotypes of SpdC (Supplementary Figures 9–11). We refer hereafter to this truncated complex simply as SagB-SpdC. Our structure, obtained using lipidic cubic phase (LCP) crystallography27, resolves nearly all of SpdC, which contains 8 transmembrane helices linked by short extracellular and cytoplasmic loops, as well as the transmembrane helix and glucosaminidase domain of SagB (Fig. 3c; Supplementary Figure 12).
Figure 3. A 2.6 Å resolution crystal structure of the SagB-SpdC complex establishes that two interfaces are critical for its function.

a, A cartoon representation of the SagB-SpdC crystal structure. The extracellular domains of both proteins interact (blue box); a helix at the bottom of the active site cleft of SagB (violet) contacts an extracellular loop between TM3 and TM4 of SpdC (green). The approximate location of the membrane is denoted in gray. b, Several hydrogen bonds and a salt-bridge form at the interface between the SagB helix and the SpdC loop. c, A view from the extracellular face of the transmembrane helices shows that SagB closely contacts TM3 of SpdC. SagB lacking its TM helix does not co-purify with SpdC (Extended Data 8). d and e, Radiolabeled peptidoglycan oligomers were incubated with SagB-SpdC or with constructs containing mutations designed to either disrupt or stabilize the extracellular interface between SagB and SpdC. d, SagBinterface* denotes SagBN115S, K118N, R119Q, V122D, D123G, L127E, in which SagB residues at the interface were switched to the corresponding SagA residues. e, A variant of SagB-SpdC with two cysteinesubstituted residues, SagBK118C - SpdCD106C, was purified as the disulfide-linked complex (Supplementary Fig 12). Activity of the oxidized complex (lane 3) was compared to the activity of SagBK118C-SpdCD106C incubated with reducing agent (lane 4), and to wild-type SagB-SpdC without and with reducing agent (lanes 2 and 5). Unlabeled cleavage products were also treated with ColM and analyzed using LC-MS analysis. Extracted ion chromatogram (EIC) traces are shown for both wild-type SagB-SpdC and SagBK118C-SpdCD106C reactions, and further confirm that longer oligosaccharide products are preferred for a disulfide-restricted complex. Notably, short oligosaccharides ionize better relative to longer oligosaccharides. Experiments were performed at least three times.
SagB makes contacts to SpdC on both its extracellular face and in the membrane (Fig. 3a–c). A helix and loop in SagB’s glucosaminidase domain (residues 115 to 127) sit over the edge of the SpdC helical bundle and make extensive hydrogen bonding and ionic interactions with the surface-exposed loop between SpdC transmembrane helix 3 (TM3) and TM4 (Fig. 3b). Adjacent to this interface, the top of the SagB transmembrane helix also contacts the SpdC TM3-TM4 loop (Q33, Fig. 3b), and from there the SagB TM helix maintains tight hydrophobic contacts with SpdC TM3 across the membrane (Fig. 3c, and also see Extended Data 8). We found that the SagB TM helix is required to form the complex: SpdC did not co-purify with soluble SagB and swapping the TM helix of SagB with that of SagA greatly reduced stability of the complex (Extended Data 8). However, the SagB TM helix is not sufficient for robust complexation with SpdC; when we replaced the TM helix of SagA with that of SagB, they did not co-purify as a 1:1 complex (Extended Data 8). Taken together, our results show that the transmembrane interactions are necessary to form a complex, but are not sufficient for the activity displayed by SagB-SpdC.
We thought the contacts at the extracellular SagB-SpdC interface could affect cleavage function and made mutations predicted to disrupt key interactions. SagA and SagB share the same fold and are 53% similar (Supplementary Figure 3), but the extracellular residues in SagB that contact SpdC are not conserved in SagA. Replacing these residues on the extracellular SagB helix that contacts SpdC with the corresponding SagA residues (SagB115-SEVNQLLKG−123) did not prevent complex formation, which may be driven largely by the interactions between TM helices in the membrane; however, we observed an erosion of product length control (Fig. 3d, lane 5). We identified a possible salt bridge between SagB lysine 118 and SpdC aspartate 106 in the crystal structure (Fig. 3b), and found that replacing either of these amino acids with a residue having the opposite charge also resulted in a product distribution more closely resembling that of purified SagB alone (Fig. 3d, lane 4). These results suggested that the relative orientation of SpdC and the SagB glucosaminidase domain is important for determining the product distribution.
To test whether the interface conformation observed in the crystal structure is critical for product length control, we generated disulfide-linked SagB-SpdC complexes using the crystal structure as a guide. We mutated proximal interface residues in SagB and SpdC to cysteines and purified the corresponding complexes. Both SagBN115C-SpdCS107C and SagBK118C-SpdCD106C complexes formed disulfide linkages as judged by SDS-PAGE analysis (Supplementary Figure 12a). Both disulfide-linked complexes produced an altered distribution of lipid-linked peptidoglycan products compared to wild-type (Fig. 3e and Supplementary Figure 12), with a shift to longer products. By comparing the mobility of the lipid-linked SagB-SpdC cleavage products to Lipid II and short oligomers, we concluded that the cleavage products from the disulfide-bonded complexes have 9–13 sugars (n=4–6; Supplementary Figure 12). In an analogous experiment, unlabeled peptidoglycan oligomers were treated with wild-type or the disulfide-bonded complex, followed by ColM treatment and then LC-MS analysis. Similar to PAGE autoradiography, the predominant species were longer for the reaction with the disulfide-bonded complex (Fig. 3e). These results show that restricting the orientation of the two proteins to that present in the crystal structure results in tighter length control. Noting that the crystal structure was obtained in a membrane-like environment, we infer that this orientation is relevant to the mechanism of cleavage in cells.
Our results suggest a mechanism for how SagB-SpdC generates the observed product lengths (Fig. 4). The structure of SpdC is similar to that of the CAAX protease Rce1 (Extended Data 9), which cleaves prenylated proteins just after the modified cysteine residue. The resemblance of SpdC to a CAAX protease suggests that SpdC may bind part of the lipid pyrophosphate carrier of nascent peptidoglycan. SpdC contains a cavity that opens to the membrane between TM1 and TM5, a possible site of prenyl chain entry. A groove extends from this opening along the extracellular face of SpdC all the way to the active site pocket of SagB. Several lines of evidence have established the directionality of oligosaccharide binding for the family of glucosaminidases to which SagB belongs28, and this directionality is consistent with nascent peptidoglycan following the groove such that the non-reducing end of the polymer exits toward the cell wall (Supplementary Figure 14). Consistent with our model, we found that the distribution of nascent cleavage products was unchanged whether polymerase and SagB-SpdC were added simultaneously or sequentially (Supplementary Figure 15). These results imply that cleavage occurred after polymer release from the polymerase, leaving the substrate lipid tail accessible for SpdC to bind. The product lengths observed in vitro are in good agreement with the lengths that would be predicted from the physical dimensions of the complex if the polymer tracks along the grooves in SpdC and SagB (Fig. 4b, Extended Data 10). To determine if SagB-SpdC activity depends on the presence of the lipid portion of oligomers, we pre-treated peptidoglycan with ColM and incubated the mixture with SagB-SpdC. As analyzed by PAGE autoradiography, SagB-SpdC activity is abrogated when oligomers lack the lipid, consistent with a role for the lipid in interacting with the complex (Extended Data 10). Obtaining a structure of SagB-SpdC bound to a lipid-linked nascent oligomer is now a key goal.
Figure 4. SagB-SpdC is a peptidoglycan release factor that cleaves at a defined length from the reducing end to allow strands to be fully incorporated into the cell wall.

a, A cross-section of SpdC shows a groove that extends from the membrane and into the SagB catalytic groove (also see Extended Data 9). As depicted by electrostatic surface potential, SpdC provides a path for a glycan strand that extends from an “entry” point in the membrane and into the “cleavage” site as denoted by the catalytic glutamate (red). b, Measuring the distance along this path provides approximately equivalent lengths required for binding a glycan strand that represents the average lipid-linked product lengths observed for the SagB-SpdC complex. c, Scheme depicting the proposed mode by which SagB-SpdC could act as peptidoglycan release factor. To prevent wasteful release of lipid-free peptidoglycan fragments, SagB-SpdC cleaves short strands that are being or have been crosslinked into the cell wall matrix; lipid-linked peptidoglycan products can be further elongated by peptidoglycan polymerases (PGTs).
SagB-SpdC is likely to function as a release factor complex that cleaves nascent peptidoglycan from its membrane anchor, which would allow its full integration into the cell wall. As S. aureus can survive without SagB-SpdC, we infer that other peptidoglycan release factors exist, and some may also be membrane-anchored cell wall hydrolase complexes. We note that this first structure and biochemical analysis of a bacterial member of the CAAX protease family suggests that other CAAX protease homologs, which are widespread in bacteria, may also act to scaffold membrane proteins involved in cell envelope synthesis.
Methods and Materials
Materials
All reagents and chemicals were purchased from Sigma-Aldrich unless indicated otherwise. Lysostaphin was purchased from Ambicin. Staphylococcus aureus was grown in tryptic soy broth (TSB) with aeration or on TSB with 1.5% agar at 30 or 37°C. Antibiotics were used at the following concentrations for S. aureus strains: kanamycin (50 μg/mL), neomycin (50 μg/mL), tetracycline (3 μg/mL), chloramphenicol (10 μg/mL), and erythromycin (10 μg/mL). NovaBlue (DE3) Escherichia coli (E. coli) strains were grown in Lysogeny broth (LB). BL21(DE3) E. coli strains were grown in terrific broth at temperatures between 18°C and 37°C as described below. Native Lipid II was extracted from Staphylococcus aureus as previously described19. Synthetic Lipid II was prepared as previously described18. The radiolabeled wall teichoic acid (WTA) precursor, [14C]-LIIAWTA, was prepared as previously reported17. FMOC-Biotin-D-Lysine (BDL) was used to prepare BDL29. PBP2S398G, SgtBY181D, EfPBPX, and TagT were expressed and purified as described in previous methods19,30–32. Colicin M was expressed and purified as previously described5. LytA was expressed and purified as previously described33.
Methods
Beta-lactam treatment and sequencing of a methicillin-resistant Staphylococcus aureus transposon library
We created a high-density transposon library, which comprises six sublibraries, in S. aureus USA300 by phage-based transposition as previously described9,10,34. The transposon library was treated with beta-lactams (8 μg mL−1 for mecillinam, 0.4 μg mL−1 for cefoxitin, and 0.1 μg mL−1 for oxacillin) at 37°C. A low concentration of oxacillin was chosen to identify factors important for beta lactam resistance. For mecillinam and cefoxitin, the concentrations were chosen such that only one PBP should be significantly inhibited in each condition. For mecillinam, the concentration was that at which the treated cells phenocopied a pbp3 mutant at 43 °C. For cefoxitin, the concentration was that which sensitized cells to oxacillin to the same degree as a pbp4 mutant. Cultures were shaken until A280=1–2.0. Cells were spun down, and DNA was isolated in preparation for Tn-Seq as previously described. Using reported methods9,10, genes significantly enriched and depleted under cefoxitin, mecillinam, or oxacillin conditions were identified using a two-sided Mann-Whitney U test corrected for multiple hypothesis testing using the Benjamini-Hochberg method. A gene was considered to be enriched if the treated:control read ratio was greater than five and depleted if the treated:control read ratio was less than 0.1. For mecillinam, the cut-off for depletion was loosened to 0.3, as mecillinam was moderately selective. Scripts for this analysis can be found at https://github.com/SuzanneWalkerLab/TnSeqMOAPrediction.
Plasmid construction for S. aureus strains
pKFC_spdC_kan
The kanR marker (primers SM45 and SM46) and the 1-kb sequences upstream (primers SM43 and SM44) and downstream (primes SM47 and SM48) of the spdC open reading frame were amplified by PCR and stitched together by overlap PCR. The resulting fragment was cloned between the BamHI and SalI restriction sites of pKFC.35
pKFC_spdC
The 700-bp sequences upstream (primers SM1 and SM2) and downstream (primers SM3 and SM4) of the spdC open reading frame were amplified by PCR and stitched together by overlap PCR. The resulting fragment was cloned between the BamHI and SalI restriction sites of pKFC.
pJP47
The pTarKO vector was linearized with primers F_pKTarO and R_pKTarO using the plasmid pTD4736 as a template. The 1-kb sequences upstream (primers F_1kb+_sagB and R_1kb+_sagB) and downstream (F_1kb(−)_sagB and R_1kb(−)_sagB) of the sagB open reading frame were amplified by PCR. Overlap PCR was performed to assemble these fragments with the tetR marker, and then the resulting fragment was ligated into the plasmid backbone between the restriction sites BamHI and SalI. Then, the tetR marker (primers oJP51 and oJP52) and upper homology arm (primers oJP54 and oJP33) were sequentially replaced by Takara Bio In-Fusion seamless cloning after linearizing the plasmid with primers oJP49 and oJP50 and oJP53 and oTD145 respectively. The insert containing the sagB homology arms and tetR marker was amplified with primers oJP32 and oJP35 and cloned between the BamHI and SalI restriction sites in pKFC. To exchange the tetR marker for a kanR marker, this plasmid was linearized with primers oJP79 and oJP80. The linearized DNA was phosphorylated at the 5’ ends using T4 polynucleotide kinase, and the ends were ligated to produce circular DNA. The kanR marker (primers oTD73 and oTD74) was then cloned into the plasmid at the XbaI restriction site. Finally the insert containing the sagB homology arms and the kanR marker was amplified with primers oJP32 and oJP35 and cut into the pTarKO backbone between the BamHI and SalI restriction sites37.
pSM_spdC_myc, pJP17, and pJP42
For pSM_spdC_myc and pJP42, the full or truncated spdC gene sequence with its native ribosome-binding site (−17) was amplified from HG003 S. aureus genomic DNA and an amino-terminal cMyc tag appended by PCR using primers SM165 and SM166 for pSM_spdC_myc and primers oJP25 and oJP81 for pJP42. The fragments were then cloned between the KpnI and BlpI restriction sites of pTP63.38 For pJP17, the cMyc-spdC fragment with the native ribosome binding site was amplified from pSM_spdC_myc using primers oJP25 and oJP26 and cloned between the KpnI and BlpI restriction sites of pTP63.
pSM_spdC_his
The spdC gene and native ribosome-binding site was amplified from HG003 S. aureus genomic DNA and a carboxy-terminal hexa-histidine tag was appended by PCR with primers SM124 and SM125. This fragment was cloned between the KpnI and BlpI restriction sites of pTP63.
pSM_spdC_E135A, pSM_spdC_R139A, pSM_spdC_H210A
These three plasmids were constructed using QuikChange site-directed mutagenesis with primers SM130 and SM131, SM132 and SM133, and SM134 and SM135 respectively and pSM_spdC_his as a template.
pJP15 and pJP19
The sagB or sagB E155A gene sequence with a carboxy-terminal hexa-histidine tag was amplified from pspdC_sagB or pspdC_sagBE155A respectively and the native ribosome binding site appended by PCR with primers oJP21 and oJP22. The fragments were cloned between the KpnI and BlpI restriction sites in pTP63.
pJP22
A gBlock gene fragment was synthesized by IDT. The fragment was amplified with primers oJP30 and oJP31 and cloned between the KpnI and BlpI restriction sites in pTP63.
S. aureus strain construction
pKFC_spdC_kan and pKFC_spdC were used to construct SHM056 and SHM002 respectively using a previously published method35. pJP47 was used to construct JP132 using a previously published method37. The deletions, or in the case of SHM002 the integrated plasmid before recombination, were transduced to HG003 S. aureus. The final deletions were confirmed by colony PCR and sequencing. Phage transductions were performed using a previously published protocol39.
To construct JP012 and JP065, a phage lysate was prepared from SAUSA300 JE2 sagB::Tn-ermR from the Nebraska library and used to transduce HG003 S. aureus and SHM056 respectively.
To construct strains containing pTP6338 constructs, the plasmids were first electroporated into TD011, and transformants were selected on 10 μg/mL chloramphenicol at 30°C. The pTP63 constructs were transduced from these transformants into strain JP012 to produce strains JP051 and JP053 and into SHM056 to produce SHM226, JP054, JP064, and JP128. For JP061, JP062, and JP063, the pTP63 constructs were first transduced into SHM002, and from there transduced into SHM056.
Co-immunoprecipitation with Myc-tagged SpdC in Staphylococcus aureus
This protocol was adapted from previously published protocols40. An overnight culture of SHM226 was diluted 1:100 into 1 L of TSB. The culture was grown at 37°C with shaking at 200 r.p.m. until A600nm = 0.6 and then 0.2 μM anhydrotetracycline was added to induce plasmid expression. After a 3 h induction, cells were pelleted at 5000xg, 15 minutes, 4°C. Cell pellets were then resuspended in lysis buffer (1X PBS (pH 7.4), 20 μg mL−1 DNase and RNase, 10 μg mL−1 lysostaphin, 5 mM MgCl2) and incubated at 37°C for 1 h. In samples treated with a chemical crosslinker, 0.5 mM DSP was added to the mixture for 1 h and then quenched with 20 mM Tris (pH 7.5). After cooling on ice, cells were lysed with a French press two times at 20,000 psi on a high ration setting. Unbroken cells were then removed by centrifugation at 10,000xg, 4°C, 15 min. Membranes were pelleted by ultracentrifugation at 100,000xg, 4°C for 60 minutes in a Beckman 45Ti rotor. For solubilization, membrane pellets were resuspended in buffer B (1X PBS (7.4), 500 mM NaCl, 1% Triton X-100). Cell membranes were then rocked at 4°C overnight before insoluble cell debris was removed by ultracentrifugation at 100,000xg, 4°C, 30 minutes. Equilibrated magnetic anti-Myc beads (Clontech, Catalog #635699) were then added to the solubilized membranes and rocked at 4°C overnight. Equilibrated beads were then washed three times with wash buffer (1X PBS pH 7.4, 200 mM NaCl, 1% Triton X-100). Protein was eluted with elution buffer provided in the Myc Immunoprecipitation kit (Clontech, Catalog # 635698). Elution buffer was then neutralized with 1 N NaOH before running on a 4–20% SDS-PAGE gel. Protein bands were prepared for LC-MS-MS analysis, adapted from previous protocols. Protein bands were excised from the gel and stored in deionized H2O prior to submission for LC-MS-MS analysis at the Taplin Mass Spectrometry Facility, Harvard Medical School.
Spot dilution assay
Overnight cultures were diluted 1:100 into 3 mL TSB and grown at 30 °C with aeration until mid-log phase. Cultures were then diluted to OD600 = 0.5. Five 10-fold serial dilutions of the resulting cultures were prepared for each strain, and 5 μL of each dilution was spotted on TSA plates with or without 0.4 μM anhydrotetracycline inducer and, where indicated, 0.8 μg/mL tunicamycin or 1 μg/mL lysostaphin. Plates were imaged after approximately 16 hours of incubation at 30 °C. Strains HG003 wild-type, SHM056, JP012, JP051, JP053, JP054, JP061, JP062, JP063, JP064, JP065, and JP128 were used for these assays.
Cloning, expression, and purification of S. aureus glucosaminidases and SpdC variants
Cloning of S. aureus glucosaminidases and SpdC
Genes encoding SagA (SAV2307), SagB (SAOUHSC_01895), and SpdC (SAOUHSC_02611) were amplified by PCR from Staphylococcus aureus strain NCTC 8325 genomic DNA. For co-expression, SagB and SpdC were cloned into a pDUET containing an amino-terminal SUMO-fusion followed by a Flag epitope tag and a carboxy-terminal hexa-histidine tag in another site. SagB and SpdC were amplified using F_SagB/R_SagB and F_SpdC/R_SpdC, and ligated into the pDUET, using primers F_DUET_SagB/R_DUET_SagB and F_DUET_FLAG_SpdC/R_DUET_FLAG_SpdC in two steps using Gibson assembly (New England Biolabs, # E2611L). Similar methods were used for SagA and SpdC co-purification. Oligonucleotide primers were purchased from Eton Bio.
Expression and co-purification of full-length, wild-type S. aureus SagB and SpdC
For co-expression of S. aureus SagB and SpdC, overnight cultures of BL21(DE3) E. coli containing pDUET-SUMO-FLAG-SpdC and SagB-His6 and an arabinose-inducible Ulp1 protease plasmid3 (pAM174) were diluted 1:100 into terrific broth supplemented with 50 μg ml−1 carbenicillin and 35 μg ml−1 chloramphenicol. Cultures were grown at 37°C with shaking at 200 r.p.m. until A600nm = 0.6 and then shifted to 18°C. At an A600nm = 1.0, protein expression was induced by addition of 0.5 mM isopropyl-ß-D-thiogalactoside (IPTG) for SagB and SpdC expression, and 0.2% arabinose for Ulp1 expression. After a 18 h expression, cells were collected by centrifugation and resuspended in buffer containing 50 mM Tris-HCl (pH 7.4), 300 mM NaCl, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 cOmplete protease inhibitor table (Sigma-Aldrich), 50 μg/ml DNase 1. Resuspended cells were lysed by a 4x passage through an Emulsiflex C3 homogenizer (Avestin) at 15,000 p.s.i. Lysed cells were separated from unbroken cells by centrifugation at 12,000g, 4°C for 10 minutes. Membranes were pelleted by ultracentrifugation at 100,000g, 4°C for 60 minutes in a Beckman 45Ti rotor. For solubilization, membrane pellets were resuspended in buffer B (50 mM Tris-HCl (7.4), 300 mM NaCl, 10% glycerol), homogenized using an IKA T18 UltraTurrax, and then supplemented with 1% w/v dodecyl-maltoside (DDM; Anatrace). Cell membranes were rocked at 4°C for 1 hour before insoluble cell debris was removed by ultracentrifugation at 100,000xg, 4°C, 30 minutes. Equilibrated Ni-NTA agarose (equilibrated with buffer B supplemented with 10 mM imidazole; Qiagen) was resuspended with solubilized membranes and rocked at 4°C for 1 hour before gravity flow through a column. Following flow-through, resin was washed with 20 column volumes (cv) of buffer B supplemented with 10 mM imidazole and 0.05% DDM and then 20 cv of buffer B with 30 mM imidazole and 0.05% DDM. Protein was eluted with 4 cv buffer B with 200 mM imidazole, 0.05% DDM, and 2 mM CaCl2. Elution fractions were then loaded onto a 4 mL M1-anti-Flag antibody affinity resin using gravity flow twice. The resin was then washed with 100 ml buffer containing 50 mM HEPES (pH 7.5), 300 mM NaCl, 10% glycerol, 0.05% DDM, and 2 mM CaCl2. Protein was eluted in 20 mM HEPES (pH 7.5), 500 mM NaCl, 20% glycerol, 0.1% DDM supplemented with 5 mM EDTA and 0.2 mg ml−1 Flag peptide (Genescript). SagB-SpdC was concentrated using a 50 MWCO concentrator (Amicon) and further purified by size exclusion chromatography on a Sephadex S200 Increase 10/300 GL (GE Healthcare) in buffer (for biochemical reactions, buffer contained 50 mM Tris-HCl (7.4), 300 mM NaCl, 10% glycerol, 0.1% DDM; for crystallography, buffer contained 50 mM Tris-HCl (7.4), 300 mM NaCl, 3% glycerol, 0.02% DDM). For biochemical reconstitutions, SagB-SpdC was concentrated into approximately 2 mg ml−1 aliquots, flash-frozen with liquid nitrogen, and stored at −80°C. The attempted purification of SagA-SpdC used a similar protocol, with the expression plasmid containing SagA-His6 in place of SagB.
Expression and purification of S. aureus individual glucosaminidases and SpdC
Individual SagB-His6, SagA-His6, and FLAG-SpdC were expressed and purified in a similar manner as described above with the following modifications. Overnight culture of BL21(DE3) E. coli containing the plasmid with SagA, SagB, or SpdC was diluted 1:100 in terrific broth supplemented with 0.1% glucose and 50 μg ml−1 carbenicillin. Growth conditions and initial purification steps were similar to as described above, with the exception of using a Ni_NTA resin to bind and purify individual hydrolases and the M1-anti-FLAG resin to bind and purify SpdC. After elution from respective resin, protein was concentrated using a 30 MWCO concentrator and further purified using a Sephadex 200 10/300 GL column using buffer containing 50 mM Tris-HCl (pH 7.4), 300 mM NaCl, 0.1% DDM, 10% glycerol.
For the soluble SagB construct, protein was expressed and purified in the same manner with slight modifications. Overnight cultures of BL21 (DE3) E. coli containing a pET_28(b)+ with SagB lacking its transmembrane helix (32–284 aa) was diluted 1:100 in LB. Cells were grown at 37°C at 200 r.p.m. until A600nm=0.4 and then cooled to 18°C; at A600nm=0.6, protein expression was induced with 0.5 mM IPTG. After 18 hr expression, cells were collected by centrifugation and lysed as described above. After lysis, unbroken cell debris was removed by centrifugation at 12,000g, 4°C for 10 minutes. Supernatant was further clarified by ultracentrifugation at 100,000g, 4°C for 30 minutes. Ni-NTA resin was equilibrated with clarified supernatant for 1 hour, rocking at 4°C. Following flow-through, resin was washed with 20 column volumes (cv) of buffer B supplemented with 10 mM imidazole and then 20 cv of buffer B with 30 mM imidazole. Protein was concentrated using a 10 MWCO concentrator tube (Amicon) and then further purified using size exclusion chromatography with Sephadex 75 10/300 GL and a buffer containing 50 mM Tris-HCl (pH 7.4), 300 mM NaCl, 10% glycerol. Aliquots of concentrated soluble SagB at approximately 2–4 mg ml−1 were then flash-frozen with liquid nitrogen and stored at −80°C.
Expression and purification of S. aureus SagB with truncated SpdC (1–256 amino acids)
S. aureus SagB-SpdC (1–256 amino acids) was expressed and purified in the same manner with slight modifications. Overnight cultures of BL21(DE3) E. coli containing the pDUET with SUMO_Flag_SpdC (1–256 aa) and SagB-His6, and an arabinose-inducible Ulp1 protease plasmid (pAM174) were diluted 1:100 in terrific broth supplemented with 0.1% glucose. Cultures were grown at 30°C with shaking at 200 r.p.m. until A600nm = 0.6 and then shifted to 24°C. At an A600nm = 1.1, protein expression was induced by addition of 0.5 mM isopropyl-ß-D-thiogalactoside (IPTG) for SagB and SpdC expression, and 0.2% arabinose for Ulp1 expression and grown for 16 h. Purification of SagB-SpdC was similar to as described above.
Expression and purification of S. aureus SagB-SpdC cysteine mutants
The S. aureus SagB-SpdC cysteine mutants were expressed and purified in the same manner with slight modifications. Overnight cultures of BL21(DE3) E. coli containing the pDUET with SUMO_Flag_SpdC and SagB-His6 with the cysteine mutants, and an arabinose-inducible Ulp1 protease plasmid (pAM174) were diluted 1:100 in terrific broth supplemented with 0.1% glucose. Cultures were grown at 25°C with shaking at 200 r.p.m until A600nm = 0.6 and then shifted to 20°C. At an A600nm = 1.1, protein expression was induced by addition of 0.5 mM isopropyl-ß-D-thiogalactoside (IPTG) for SagB and SpdC expression, and 0.2% arabinose for Ulp1 expression and grown for 16 hours. Purification of SagB-SpdC was similar to as described above with the following modifications. Pelleted cells were resuspended in 50 mM Tris-HCl (pH 7.4), 300 mM NaCl, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 cOmplete protease inhibitor table (Sigma-Aldrich), 50 μg/ml DNase 1. To facilitate disulfide formation, 0.3 mM CuSO4 and 0.3 mM 1,10-phenanthroline were added to the mixture. The remaining purification steps followed those described above.
PAGE autoradiograph experiments with PG oligomers and S. aureus hydrolases
The protocol for analyzing and preparing peptidoglycan oligomers was adapted from similar methods previously reported17,41. A PBP2 construct19 (59–716 amino acids, 1 μM) was incubated with synthetic-[14C]-Lipid II analog18 (20 μM in DMSO; specific activity=300 μCi/μmol from UDP-[14C]-GlcNAc (American Radiolabeled Chemicals, Inc.) in reaction buffer (50 mM MES (6.5), 100 mM CaCl2) with 20% DMSO (v/v). After mixing the Eppendorf tube by flicking and spinning down the reaction mixture, polymerization proceeded at room temperature for 2 hours. To test oligomers prepared from SgtB* (SgtBY181D)30, similar conditions were set up with 800 nM SgtB*. Proteins were removed by precipitation by heating the reaction mixture at 95 °C for 10 minutes and then spinning down the precipitated protein. The PG oligomers were then aliquoted into separate Eppendorf tubes (10 μl mixture for each reaction) and a hydrolase (SagA, SagB, SagB-SpdC, or the ΔTM-SagB; 3 μM) was added. For reactions in which the individual hydrolase was incubated with SpdC, the hydrolase SagA or SagB (3 μM) were added to SpdC (3 μM) for 1 hour on ice before addition to the PG mixture. After overnight incubation, the reaction was quenched with heat inactivation (95 °C for 10 minutes) and the reactions were dried completely using a speed vacuum. Reactions were resuspended in 10 μl of SDS loading buffer and loaded onto a 10% acrylamide-Tris gel. The gel was run at 30 mA for 5 h at 4°C; an anode buffer consisted of 100 mM Tris (pH 8.8) and cathode buffer consisted of 100 mM Tris, 100 mM tricine (pH 8.25), 0.1% SDS41. Gels were dried on filter paper (19 × 18.5 cm; Biorad) and then exposed to a phosphor screen for at least 24 h. Phosphor screens were imaged using an Azure Sapphire Biomolecular Imager (Azure biosystems). Images were further analyzed using ImageJ.
Reactions to test hydrolase activities with concurrent PBP2 transglycosylase activity were set up and analyzed in a similar manner with some modifications. A reaction mixture was prepared with synthetic-[14C]-Lipid II analog18 (20 μM in DMSO; specific activity=300 μCi μmol−1 from UDP-[14C]-GlcNAc (American Radiolabeled Chemicals, Inc.), reaction buffer (50 mM MES (6.5), 100 mM CaCl2), 20% DMSO (v/v). PBP2 (59–716 amino acids; 5 μM) and the hydrolase (SagA, SagB, SagB-SpdC; 3 μM) was then added). Similar methods were used to test the activities of interface mutants of SagB-SpdC. To test the activities of disulfide-linked SagB-SpdC complexes, protein under reducing conditions were treated with 5 mM DTT on ice for 30 minutes before the addition to the reaction mixture that included the addition of 5 mM DTT. After overnight incubation, the reaction was quenched with heat inactivation (95 °C for 10 minutes) and the reactions were dried completely using a speed vacuum. Reactions were resuspended in 10 μl of 2x SDS loading buffer and loaded onto a 10% acrylamide-Tris gel. The gel was run and analyzed as described above.
Testing hydrolase activities with wall-teichoic acid labeled peptidoglycan oligomers was adapted from similar methods previously reported36. Peptidoglycan oligomers were prepared as described above although with unlabeled, synthetic-Lipid II (20 μM). After precipitation to remove the PGT, a portion of the mixture was incubated with TagT (1 μM) and [14C]-LIIAWTA (8 μM) for four hours at room temperature. The TagT ligase was then heat inactivated, precipitated, and removed from the reaction mixture. The resulting wall teichoic acid-labeled oligomers were then incubated with SagB (3 μM), SagB-SpdC (3 μM), or mutanolysin (2.5 U ml−1). The remaining portion of unlabeled PG oligomers was also incubated with the SagB-SpdC complex (3 μM). After an overnight incubation at room temperature, the reaction was quenched with heat inactivation (95 °C for 10 minutes). To test TagT ligation after SagB-SpdC incubation, TagT (1 μM) and [14C]-LIIAWTA (8 μM) was added to the respective reaction for four additional hours. Reactions were dried completely using a speed vacuum and the mixture was re-dissolved in 10 μl of 2x SDS loading buffer and loaded onto a 10% acrylamide-Tris gel. The gel was run and analyzed as described above.
Western blot analysis of hydrolase activities with peptidoglycan oligomers prepared from S. aureus Lipid II
The protocol for detecting peptidoglycan oligomers prepared from extracted S. aureus Lipid II was adopted from similar methods previously reported19,23,29. To generate uncrosslinked PG oligomers, S. aureus Lipid II (10 μM) was added to reaction buffer (50 mM MES (6.5), 10 mM CaCl2), 20% DMSO (v/v) and thetranspeptidase inactive construct PBP2S398G (5 μM) was added to the reaction. To test ongoing hydrolase and polymerase activity, the reaction mixture was aliquoted into separate tubes and the respective glucosaminidase was also added (SagA, SagB, or SagB-SpdC; 3 μM). For a mutanolysin-digest reaction, 2.5 U ml−1 of mutanolysin (Sigma-Aldrich) was added42. If testing hydrolase activity with pre-assembled PG oligomers, PBP2S398G was precipitated after heat inactivation at 95°C. PG oligomers were aliquoted into separate 10 μl reactions, and the hydrolase was added as described above. Reactions were inactivated at 95°C for 10 minutes. To label uncrosslinkedPG oligomers, BDL (4 mM in H2O) and E. faecalis PBPX31 (20 μM) was added to the reaction mixtures. After a 1 h incubation at room temperature, 2x loading buffer was added to quench the reactions. To generate crosslinked PG, S. aureus Lipid II (10 μM) was added to reaction buffer (50 mM MES (6.5), 10 mM CaCl2), 20% DMSO (v/v), BDL (4 mM), and a wild type PBP2 construct (5 μM). After the addition of PBP2, the respective glucosaminidase was also added (SagB or SagB-SpdC; 3 μM) and the reactions were incubated at room temperature for 5 hours. Reactions were split into two aliquots after heat inactivation at 95°C, and lysostaphin (100 μg ml−1) was added to one aliquot and then incubated at 37°C for 2 hours. 2x loading dye was added to each reaction, and these mixtures were loadedonto a 4–20% polyacrylamide gel which ran at 175 V for 1 h. The gel was transferred to PVDF membrane (Biorad) at 10 mV for 1 h. After incubating with starting block (Thermo Scientific, catalog number #37578), the membrane was rocked with HRP-streptavidin (1:5000) in TBS-T and then washed repetitively. The membrane was imaged using the chemiluminescence function on an Azure imager (Azure biosystems). Images were further analyzed using ImageJ.
LC-MS method for detecting digest products of S. aureus glucosaminidases and mutanolysin
The cleaved muropeptide products of glucosaminidase reactions (SagA, SagB, SagB-SpdC) were analyzed by adopting a previously reported LC-MS method for detecting mutanolysin-digested products42. Hydrolase reactions were prepared using synthetic Lipid II (20 μM), reaction buffer (50 mM MES (6.5), 10 mM CaCl2), 20% DMSO (v/v), PBP2S398 (5 μM), and then the respective hydrolase (SagA, SagB, SagB-SpdC, 3 μM) in a total volume of 50 μl. A control mutanolysin reaction was likewise set-up with 5 U ml−1 mutanolysin. After an overnight incubation, sodium borohydride (10 mg ml−1 in H2O; equal reaction volume) was added and incubated for 20 minutes at room temperature. The reaction was quenched with the addition of 20% phosphoric acid which also adjusted the pH to 3–4. Reactions were completely dried under a nitrogen stream and then resuspended in H2O. LC-MS analysis was conducted using an Agilent Technologies 1200 series HPLC in line with an Agilent 6210 TOF mass spectrometer with electrospray ionization (ESI) and operating in positive mode. Muropeptide cleavage products were separated using a Waters Symmetry Shield RP18 column (5 μm, 3.9 × 150 mM) with a matching guard column and the following method: 0.4 mL min−1 solvent A (water/0.1% formic acid) for 5 minutes followed by a linear gradient of 0 to 40% solvent B (acetonitrile/0.1% formic acid) over 25 min. Mass spectrometry data was analyzed using Agilent MassHunter Workstation Qualitative Analysis software version B.06.00 and Prism 7.0b.
LC-MS method for detecting lipid-linked PG oligomers
To character lipid-linked cleavage products, we adapted previously published methods23. Peptidoglycan oligomers were prepared by incubating Lipid II (40 μM) with PBP2S398G (5 μM) in 20% DMSO and reaction buffer (50 mM MES (6.5), 100 mM CaCl2) and 20% DMSO (v/v) for a total of 2 h. SagB-SpdC (3 μM) was added to the mixture and then incubated for approximately 8 hours. Enzymes were heat inactivated at 95°C for 5 minutes. After cooling reactions to room temperature, ColM (1 mg mL−1) was added to the mixture and incubated for approximately 3 hours at room temperature23. Protein was precipitated with equal volume methanol. Dried reactions were then resuspended in 20 μL of H2O.
Glycan strand assay
Overnight cultures of wild-type HG003 S. aureus, SHM056, and JP132 were diluted 1:100 into 1 L of TSB each and grown at 30 °C for 5 hours. Cells were harvested and sacculi were isolated following a previously published protocol43 with the modification that samples were boiled in SDS for 3 hours. Sacculi were resuspended to 5 mg/mL in 500 μL of 25 mM NaH2PO4 pH 7.0 and treated with 100 μg/mL lysostaphin at 37 °C with shaking for 9 hours. LytA was then added to 100 μg/mL and shaking continued at 37 °C for another 14 hours. Enzymes were heat inactivated at 95 °C for 5 min. Proteins were precipitated with an equal volume of MeOH and removed by centrifugation. To label glycans, we adapted methods previously published44. Dried samples were resuspended in 25 μL of 1 M 2-methylpyridine borane complex in DMSO and 25 μL of 200 mM 8-aminonaphthalene-1,3,6-trisulfonic acid in 15% acetic acid. Reactions were incubated at room temperature overnight protected from light. Reactions were quenched with 450 μL of H2O at room temperature for 1 hr, and an equal volume of MeOH was added. After spinning down, the supernatants were removed to new tubes and dried. Dried samples were resuspended in 50 μL of 1x loading buffer (125 mM Tris-tricine pH 8.2, 10% glycerol) and loaded on a 20% polyacrylamide gel, which ran for 9 hours at 25 mA. The gel was visualized under UV light at 365 nm.
Crystallization of SagB-SpdC1−256 and data collection
For crystallization trials, SagB-SpdC1−256 was purified as described above. Freshly purified SagB-SpdC1−256 was concentrated to 35–40 mg/ml and immediately reconstituted into lipidic cubic phase by mixing protein and monoolein (Hampton Research) at a 1:1.5 ratio by mass, using the coupled syringe method27. All samples were mixed at least 100 times prior to crystallization trials. The resulting mixture was dispensed onto glass plates in 35–50 nL drops, then overlaid with 600 nL precipitant solution using an NT8 robot (Formulatrix). Crystals of SagB-SpdC1−256 were grown in precipitant solution containing 24–32% PEG400, 500 mM (NH4)2SO4, and 100mM sodium acetate or sodium citrate pH 4.4–5.0; at higher pH crystallization required higher concentrations of PEG400. Most crystals appeared within 36 hours of drop setting and were full-grown in 3–7 days. Crystals were harvested using mesh loops and flash-frozen in liquid nitrogen.
Diffraction data were collected at Argonne National Laboratory using NE-CAT beamlines 24-ID-C and 24-ID-E. Two rounds of grid scanning with large and then small beam size were used to locate crystals on the mesh and then precisely determine their positions. All data were collected at 0.979 Å. Datasets were collected 0.2-s exposure and a 0.2° oscillation angle. The presence of multiple crystals on the mesh prevented the collection of full datasets from individual crystals. Data were indexed and integrated in XDS45; the SagB-SpdC crystals belonged to the C2 space group. Diffraction data was processed using structural biology software accessed through the SBGrid consortium46. Partial datasets from five well-diffracting crystals were then scaled and merged using the CCP4 suite47 program AIMLESS48.
Structure determination and refinement
The structure was determined by molecular replacement in Phaser using an unpublished structure of the soluble domain of SagB (PDB# 6FXP)20,49. We attempted to place SpdC by molecular replacement using models prepared from the structure of Methanococcus maripaludis CAAX protease Rce1 (PDB# 4CAD, 20% sequence identity to SpdC)50, but no good solutions were found. Following placement of soluble SagB, only weak density was visible for the transmembrane helices of the complex; those that were clearest were initially modeled as poly-alanine helices using Coot51,52. After one round of automated refinement using phenix.refine53, placement of all nine transmembrane helices was possible, but residue identities were not obvious. Initial assignment was made by sequence alignment and comparison to the structure of Rce1, which appeared to have a similarly threaded transmembrane domain. Density for sidechains in the transmembrane domain became much clearer after several rounds of manual building and automated refinement, and simulated annealing composite omit maps were used to check for model bias and correct errors in the register throughout the process. Towards the end of refinement, a region of clear density remained present at a crystallographic interface between two copies of SagB near residues 38–45 and appeared to be a peptide forming a continuous β-sheet with the two copies of SagB. This peptide is on the unit cell edge but does not appear to be part of either protein; we think it is likely FLAG peptide that carried through the purification and may orient either direction at this interface. Water molecules, sulfate ions, PEG molecules were also added near the end of refinement. Structure quality was assessed using MolProbity54, and figures were prepared using PyMOL.
Extended Data
Extended Data Fig. 1. Hydrolase activities are unaffected by acidic conditions.

Peptidoglycan oligomers were prepared at pH 5.5 in sodium citrate buffer, and then incubated with the respective hydrolase. Cleavage patterns are similar to those observed at pH 6.5 (see main text Figure 2).
Extended Data Fig. 2. The complex cleaves native PG oligomers to defined lengths similarly to synthetic PG oligomers.

To prepare linear glycan strands, extracted S. aureus Lipid II was incubated with transpeptidase-inactive PBP2S398G and then the SagB-SpdC complex or individual hydrolases (SagB or SagA). Reactions were quenched and products were then labeled with biotin-D-lysine (BDL) in the presence of the E. faecalis PBPX and visualized using a western blot method with HRP-streptavidin detection19,23,29.
Extended Data Fig. 3. The catalytic glutamate of SagB (E155), but not conserved CAAX protease residues in SpdC (E135, R139, H210)5, is necessary to complement shared cell wall phenotypes of sagB and spdC mutants.

Spot dilution series of S. aureus strains with mutations in sagB or spdC were plated on TSA or TSA containing tunicamycin or lysostaphin. Plates shown on the right are replicates of those on the left but the agar contains the inducer, anhydrotetracycline.
Extended Data Fig. 4. Glycan strand lengths are longer in the absence of spdC or sagB.

a, Sacculi were isolated from wild-type, ΔsagB, and ΔspdC S. aureus strains. The purified peptidoglycan was then treated with the endopeptidase lysostaphin, which cleaves the PG crosslinks, and the amidase LytA, which removes the remaining stem peptides. These denuded glycan strands were then labeled at the reducing end by reductive amination with the anionic fluorophore 8-aminonaphthalene-1,3,6-trisulfonic acid (ANTS), and separated by gel electrophoresis for in-gel imaging (UV excitation at 365 nm, visible emission). b, In lane 1, showing PG isolated from a wild-type strain, shorter glycan strands are visualized as a discrete ladder. In lanes 2 and 3, representing glycan strands of ΔsagB and ΔspdC respectively, this discrete ladder of short glycan strands is lost.
Extended Data Fig. 5. Nascent peptidoglycan is the preferred substrate of SagB-SpdC.
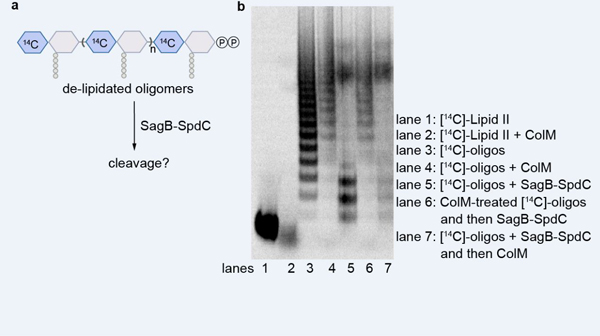
Cold glycan strands were prepared with radiolabeled wall teichoic acid (lanes 2–5) and incubated with a hydrolase. In the presence of either SagB (lane 3) or SagB-SpdC (lane 4), the ladder signal remained the same as a no treatment control (lane 2). There was reduced radiolabeled signal towards the top of the gel in both reactions; this signal represents a population of longer peptidoglycan oligomers and the fainter signal suggests less wall teichoic acid present. In contrast, mutanolysin was able to hydrolyze wall teichoic acid-modified glycan strands. To test whether SagB-SpdC cleavage interfered with wall teichoic acid ligation, cold oligomers were prepared and then incubated with radiolabeled wall teichoic precursor, LIIAWTA, and the wall teichoic acid ligase, TagT. The appearance of the radiolabeled ladder indicates that SagB-SpdC products were ligated with wall teichoic acid (lane 9).
Extended Data Fig. 6. Crosslinked PG is not a substrate for SagB-SpdC.

S. aureus Lipid II was co-incubated with wild-type PBP2, a hydrolase, and BDL for visualization. Reactions were quenched, halved, and then one portion was treated with lysostaphin. Crosslinked peptidoglycan oligomers do not readily enter the gel matrix (lane 1) and appear as a dark smear when crosslinks are cleaved by the endopeptidase lysostaphin (lane 4). In reactions containing SagB-SpdC, a low-molecular weight ladder of bands is present without lysostaphin (lane 2) or with lysostaphin (lane 5). This shows that these products, generated by SagB-SpdC, are not crosslinked as they were not changed with lysostaphin treatment. Notably, approximately 20% of peptidoglycan peptides are crosslinked by wild-type PBP2 in vitro36. The high molecular weight signals (smears) in lanes 5 and 6 are similar to the signal in lane 4, indicating that crosslinked PG is not cleaved by either SagB-SpdC or full-length SagB.
Extended Data Fig. 7. Representative electron density.
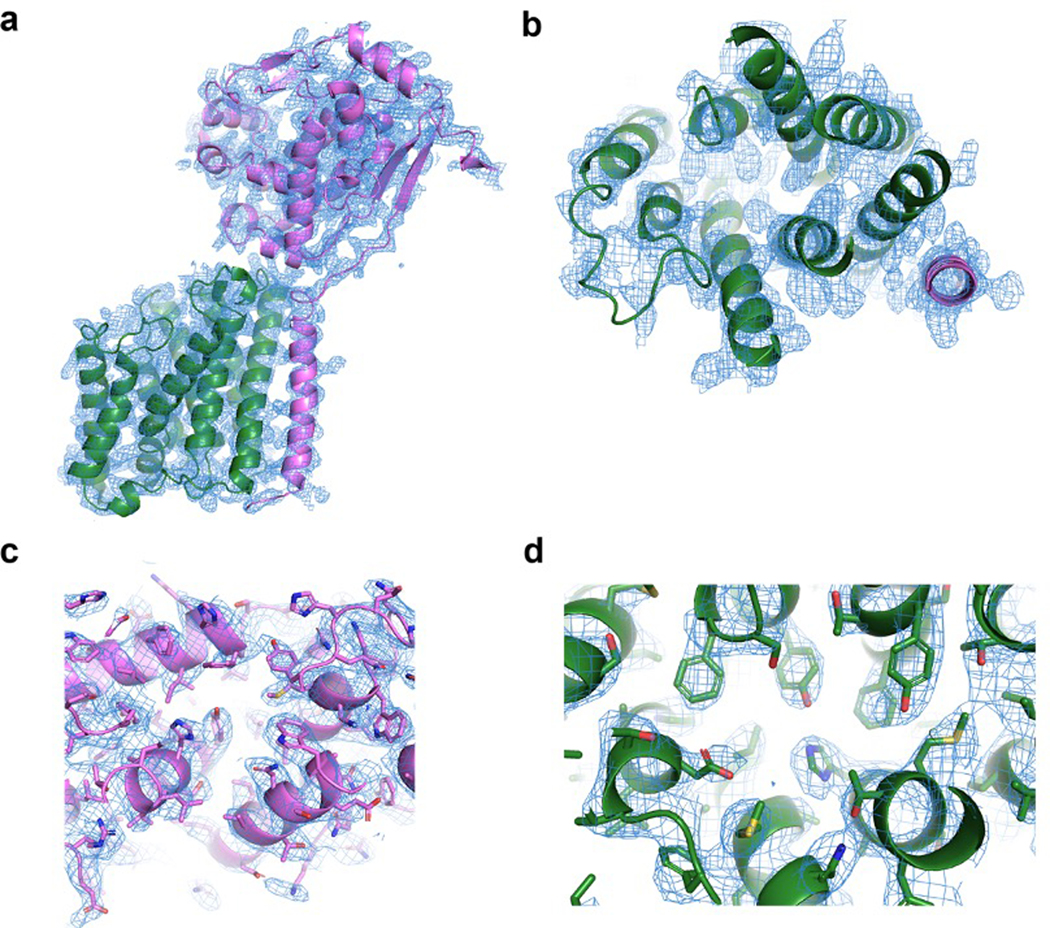
a-d, Simulated-annealing composite omit 2FO-FC electron density maps contoured at 1σ and carved 1.6Å from the model, with SagB colored violet and SpdC colored green. a, Overall structure of SagB-SpdC viewed in the plane of the membrane. b, View of the transmembrane helices from the extracellular face. c, Active site groove of SagB, with sidechains shown as sticks. d, Central region of SpdC between several transmembrane helices in the same orientation as shown in b.
Extended Data Fig. 8. The interface between the transmembrane helices of SagB and SpdC is crucial for complexation.

a, Cartoon representation of the SagB-SpdC complex shows that the transmembrane (TM) helix of SagB is making close contacts to the TM3 of SpdC. b, Close-up of the interface between the SagB TM helix and SpdC. Left panel: Surface representation, Right panel: the same view showing the side-chains of the SagB TM helix as sticks. c, An attempt to co-purify ΔTM-SagB with SpdC did not yield a complex, although the individual components could be recovered after their corresponding affinity purification steps, as shown in SDS-PAGE. d, Alignment of sequences corresponding to the TM helices of SagA and SagB shows significant sequence differences. A construct of SagB containing the TM helix of SagA was co-expressed with SpdC. A stable complex did not form between SpdC and SagB with the TM of SagA, and very little of this SagB variant co-purified along SpdC as shown in SDS-PAGE. Inversely, a construct of SagA containing the TM of SagB was co-expressed with SpdC and did not form a stable 1:1 complex (right gel). e, Radiolabeled peptidoglycan oligomers treated with SagB-SpdC, individual hydrolases, or hydrolases with the addition of SpdC. Unlike SagB, SagA activity is not significantly altered by the presence of SpdC. f, Swapping the TM of SagB onto SagA has only a minor effect on its product distribution.
Extended Data Fig. 9. Alignment of SpdC and Rce1 structures shows broad similarities in their transmembrane domains, but key differences in the predicted substrate binding site.

a, Cartoon representation of the transmembrane helices of SagB-SpdC (violet, green) aligned with the structure of Rce1 (blue, PDB# 4CAD), with arrows denoting the views in b. b, The aligned structures when viewed from within the plane of the membrane. Although the helices of Rce1 and SpdC are threaded in the same pattern, the helices differ in length and the loops between helices adopt significantly different conformations. c, Several Rce1 catalytic residues (blue sticks) are not present in SpdC (green sticks); conserved residues are labeled in black. d, Surface electrostatic maps reveal that Rce1 (left) and SpdC (right) both contain deep central cavities that open to the membrane (circled in black). Both Rce1 and SagB-SpdC are oriented as in a, showing that the cavities open to the membrane at different locations and that the rim of the SpdC cavity contains more positively charged residues.
Extended Data Fig. 10. Removal of the lipid carrier from nascent peptidoglycan inhibits SagB-SpdC cleavage.

a, Schematic of experiment shown in b. b, Radiolabeled peptidoglycan oligomers (lane 3) were treated with Colicin M (lane 4), which removes the lipid carrier, leaving a pyrophosphate on the reducing terminus of the glycan. Lipid-linked and de-lipidated oligomers were then treated with SagB-SpdC (lanes 5 and 6, respectively). Comparison of lane 3 to lane 5 shows changes to the sample characteristic of SagB-SpdC activity, whereas comparison of lane 4 to lane 6 shows no change, indicating that SagB-SpdC was not able to cleave oligomers lacking the lipid carrier.
Supplementary Material
Acknowledgements
We thank Dr. Samir Moussa for his preliminary experiments investigating the roles of SpdC and SagB. We also thank Dr. Andrew Kruse for helpful discussions on crystallography. This work used NE-CAT beamlines (GM103403), a Pilatus detector (RR029205), and an Eiger detector (OD021527) at the APS (DE-AC02-06CH11357). This research was supported by GM076710 and U19 AI109764 to D.K. and S.W. and T32GM007753 to J.E.P.
Footnotes
Competing interests
The authors declare no competing interests.
Data availability
Transposon sequencing data (BioProject accession number PRJNA573479) can be found in the NCBI BioProject database. The coordinates and structure factors for the SagB-SpdC structure have been deposited with the PDB under the accession number 6U0O.
References:
- 1.Silhavy TJ, Kahne D & Walker S. The bacterial cell envelope. Cold Spring Harb Perspect Biol 2, a000414, doi: 10.1101/cshperspect.a000414 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vollmer W, Blanot D & de Pedro MA Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 32, 149–167, doi: 10.1111/j.1574-6976.2007.00094.x (2008). [DOI] [PubMed] [Google Scholar]
- 3.Meeske AJ et al. SEDS proteins are a widespread family of bacterial cell wall polymerases. Nature 537, 634–638, doi: 10.1038/nature19331 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taguchi A et al. FtsW is a peptidoglycan polymerase that is functional only in complex with its cognate penicillin-binding protein. Nat. Microbiol. 4, 587–594, doi: 10.1038/s41564-018-0345-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sham LT et al. Bacterial cell wall. MurJ is the flippase of lipid-linked precursors for peptidoglycan biogenesis. Science 345, 220–222, doi: 10.1126/science.1254522 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruiz N. Bioinformatics identification of MurJ (MviN) as the peptidoglycan lipid II flippase in Escherichia coli. Proc. Natl. Acad. Sci. USA 105, 15553–15557, doi: 10.1073/pnas.0808352105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sauvage E, Kerff F, Terrak M, Ayala JA & Charlier P. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 32, 234–258, doi: 10.1111/j.1574-6976.2008.00105.x (2008). [DOI] [PubMed] [Google Scholar]
- 8.Yunck R, Cho H & Bernhardt TG Identification of MltG as a potential terminase for peptidoglycan polymerization in bacteria. Mol. Microbiol. 99, 700–718, doi: 10.1111/mmi.13258 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Santiago M et al. Genome-wide mutant profiling predicts the mechanism of a Lipid II binding antibiotic. Nat. Chem. Biol. 14, 601–608, doi: 10.1038/s41589-018-0041-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Santiago M et al. A new platform for ultra-high density Staphylococcus aureus transposon libraries. BMC Genomics 16, 252, doi: 10.1186/s12864-015-1361-3 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Georgopapadakou NH, Smith SA & Bonner DP Penicillin-binding proteins in a Staphylococcus aureus strain resistant to specific beta-lactam antibiotics. Antimicrob. Agents Chemother. 22, 172–175, doi: 10.1128/aac.22.1.172 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wheeler R et al. Bacterial Cell Enlargement Requires Control of Cell Wall Stiffness Mediated by Peptidoglycan Hydrolases. MBio 6, e00660, doi: 10.1128/mBio.00660-15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan YG, Frankel MB, Missiakas D & Schneewind O. SagB Glucosaminidase Is a Determinant of Staphylococcus aureus Glycan Chain Length, Antibiotic Susceptibility, and Protein Secretion. J. Bacteriol. 198, 1123–1136, doi: 10.1128/JB.00983-15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pasquina-Lemonche L et al. The architecture of the Gram-positive bacterial cell wall. Nature, doi: 10.1038/s41586-020-2236-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grundling A, Missiakas DM & Schneewind O. Staphylococcus aureus mutants with increased lysostaphin resistance. J. Bacteriol. 188, 6286–6297, doi: 10.1128/JB.00457-06 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poupel O, Proux C, Jagla B, Msadek T & Dubrac S. SpdC, a novel virulence factor, controls histidine kinase activity in Staphylococcus aureus. PLoS Pathog. 14, e1006917, doi: 10.1371/journal.ppat.1006917 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schaefer K, Matano LM, Qiao Y, Kahne D & Walker S. In vitro reconstitution demonstrates the cell wall ligase activity of LCP proteins. Nat. Chem. Biol. 13, 396–401, doi: 10.1038/nchembio.2302 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ye XY et al. Better substrates for bacterial transglycosylases. J. Am. Chem. Soc. 123, 3155–3156, doi: 10.1021/ja010028q (2001). [DOI] [PubMed] [Google Scholar]
- 19.Qiao Y et al. Lipid II overproduction allows direct assay of transpeptidase inhibition by beta-lactams. Nat. Chem. Bio.l 13, 793–798, doi: 10.1038/nchembio.2388 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pintar S, Borisek J, Usenik A, Perdih A & Turk D. Domain sliding of two Staphylococcus aureus N-acetylglucosaminidases enables their substrate-binding prior to its catalysis. Commun. Biol. 3, 178, doi: 10.1038/s42003-020-0911-7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perlstein DL, Zhang Y, Wang TS, Kahne DE & Walker S. The direction of glycan chain elongation by peptidoglycan glycosyltransferases. J. Am. Chem. Soc. 129, 12674–12675, doi: 10.1021/ja075965y (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang TS et al. Primer preactivation of peptidoglycan polymerases. J. Am. Chem. Soc. 133, 8528–8530, doi: 10.1021/ja2028712 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Welsh MA, Schaefer K, Taguchi A, Kahne D & Walker S. The direction of chain growth and substrate preferences of SEDS-family peptidoglycan glycosyltransferases. J. Am. Chem. Soc, doi: 10.1021/jacs.9b06358 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.El Ghachi M et al. Colicin M exerts its bacteriolytic effect via enzymatic degradation of undecaprenyl phosphate-linked peptidoglycan precursors. J. Biol. Chem. 281, 22761–22772, doi: 10.1074/jbc.M602834200 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Touze T et al. Colicin M, a peptidoglycan lipid-II-degrading enzyme: potential use for antibacterial means? Biochem. Soc. Trans. 40, 1522–1527, doi: 10.1042/BST20120189 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Alcorlo M, Martinez-Caballero S, Molina R & Hermoso JA Carbohydrate recognition and lysis by bacterial peptidoglycan hydrolases. Curr. Opin. Struct. Biol. 44, 87–100, doi: 10.1016/j.sbi.2017.01.001 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Caffrey M & Cherezov V. Crystallizing membrane proteins using lipidic mesophases. Nat. Protoc. 4, 706–731, doi: 10.1038/nprot.2009.31 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mihelic M et al. The mechanism behind the selection of two different cleavage sites in NAG-NAM polymers. IUCrJ 4, 185–198, doi: 10.1107/S2052252517000367 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qiao Y et al. Detection of lipid-linked peptidoglycan precursors by exploiting an unexpected transpeptidase reaction. J. Am. Chem. Soc.. 136, 14678–14681, doi: 10.1021/ja508147s (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rebets Y et al. Moenomycin resistance mutations in Staphylococcus aureus reduce peptidoglycan chain length and cause aberrant cell division. ACS Chem. Biol. 9, 459–467, doi: 10.1021/cb4006744 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Welsh MA et al. Identification of a Functionally Unique Family of Penicillin-Binding Proteins. J. Am. Chem. Soc. 139, 17727–17730, doi: 10.1021/jacs.7b10170 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schaefer K, Owens TW, Kahne D & Walker S. Substrate Preferences Establish the Order of Cell Wall Assembly in Staphylococcus aureus. J. Am. Chem. Soc. 140, 2442–2445, doi: 10.1021/jacs.7b13551 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flores-Kim J, Dobihal GS, Fenton A, Rudner DZ & Bernhardt TG A switch in surface polymer biogenesis triggers growth-phase-dependent and antibiotic-induced bacteriolysis. Elife 8, doi: 10.7554/eLife.44912 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coe KA et al. Comparative Tn-Seq reveals common daptomycin resistance determinants in Staphylococcus aureus despite strain-dependent differences in essentiality of shared cell envelope genes. bioRxiv (2019). [Google Scholar]
- 35.Kato F & Sugai M. A simple method of markerless gene deletion in Staphylococcus aureus. J. Microbiol. Methods 87, 76–81, doi: 10.1016/j.mimet.2011.07.010 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Do T et al. Staphylococcus aureus cell growth and division are regulated by an amidase that trims peptides from uncrosslinked peptidoglycan. Nat. Microbiol. 5, 291–303, doi: 10.1038/s41564-019-0632-1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee W et al. Antibiotic Combinations That Enable One-Step, Targeted Mutagenesis of Chromosomal Genes. ACS Infect. Dis. 4, 1007–1018, doi: 10.1021/acsinfecdis.8b00017 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pang T, Wang X, Lim HC, Bernhardt TG & Rudner DZ The nucleoid occlusion factor Noc controls DNA replication initiation in Staphylococcus aureus. PLoS Genet. 13, e1006908, doi: 10.1371/journal.pgen.1006908 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Do T et al. The cell cycle in Staphylococcus aureus is regulated by an amidase that controls peptidoglycan synthesis. bioRxiv, doi: 10.1101/634089 (2019). [DOI] [Google Scholar]
- 40.Bertsche U et al. Interaction between two murein (peptidoglycan) synthases, PBP3 and PBP1B, in Escherichia coli. Mol. Microbiol. 61, 675–690, doi: 10.1111/j.1365-2958.2006.05280.x (2006). [DOI] [PubMed] [Google Scholar]
- 41.Barrett D et al. Analysis of glycan polymers produced by peptidoglycan glycosyltransferases. J. Biol. Chem. 282, 31964–31971, doi: 10.1074/jbc.M705440200 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lebar MD et al. Forming cross-linked peptidoglycan from synthetic gram-negative Lipid II. J. Am. Chem. Soc. 135, 4632–4635, doi: 10.1021/ja312510m (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuhner D, Stahl M, Demircioglu DD & Bertsche U. From cells to muropeptide structures in 24 h: peptidoglycan mapping by UPLC-MS. Sci. Rep. 4, 7494, doi: 10.1038/srep07494 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jackson P. The use of polyacrylamide-gel electrophoresis for the high-resolution separation of reducing saccharides labelled with the fluorophore 8-aminonaphthalene-1,3,6-trisulphonic acid. Detection of picomolar quantities by an imaging system based on a cooled charge-coupled device. Biochem. J. 270, 705–713, doi: 10.1042/bj2700705 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kabsch W. Xds. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132, doi: 10.1107/S0907444909047337 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morin A et al. Collaboration gets the most out of software. Elife 2, e01456, doi: 10.7554/eLife.01456 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Winn MD et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol. Crystallogr. 67, 235–242, doi: 10.1107/S0907444910045749 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Evans PR & Murshudov GN How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214, doi: 10.1107/S0907444913000061 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCoy AJ et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674, doi: 10.1107/S0021889807021206 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Manolaridis I et al. Mechanism of farnesylated CAAX protein processing by the intramembrane protease Rce1. Nature 504, 301–305, doi: 10.1038/nature12754 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Emsley P & Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132, doi: 10.1107/S0907444904019158 (2004). [DOI] [PubMed] [Google Scholar]
- 52.Emsley P, Lohkamp B, Scott WG & Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501, doi: 10.1107/S0907444910007493 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221, doi: 10.1107/S0907444909052925 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21, doi: 10.1107/S0907444909042073 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.