

Abstract
Frustrated Lewis pairs (FLPs) are well known for their ability to activate small molecules. Recent reports of radical formation within such systems indicate single‐electron transfer (SET) could play an important role in their chemistry. Herein, we investigate radical formation upon reacting FLP systems with dihydrogen, triphenyltin hydride, or tetrachloro‐1,4‐benzoquinone (TCQ) both experimentally and computationally to determine the nature of the single‐electron transfer (SET) events; that is, being direct SET to B(C6F5)3 or not. The reactions of H2 and Ph3SnH with archetypal P/B FLP systems do not proceed via a radical mechanism. In contrast, reaction with TCQ proceeds via SET, which is only feasible by Lewis acid coordination to the substrate. Furthermore, SET from the Lewis base to the Lewis acid–substrate adduct may be prevalent in other reported examples of radical FLP chemistry, which provides important design principles for radical main‐group chemistry.
Keywords: frustrated Lewis pairs, radicals, reactivity, single-electron transfer, substrate coordination
The radical reactivity of archetypal P/B frustrated Lewis pair (FLP) systems is probed through utilization of the light dependency of direct single‐electron transfer from the phosphine to the borane to determine that reactivity with H2 does not proceed via a radical pathway. For radical FLP reactions with carbonyl‐containing substrates, direct SET is excluded in favor of borane coordination to the carbonyl, after which SET occurs.

Introduction
Frustrated Lewis pairs (FLPs) combine a Lewis acidic electron‐pair acceptor and a Lewis basic electron‐pair donor to activate small molecules, most notably H2 and CO2, granting access to fascinating main group chemistry and catalysis. [1] It is generally accepted that the FLP components cooperatively interact with the substrate to facilitate heterolytic bond cleavage; [2] however, recent reports suggest that radicals may play an important role too and, in some cases, provide alternative homolytic pathways.[ 2b , 2c , 3 ] Stephan et al. reported the detection of a weak radical signal by electron paramagnetic resonance (EPR) spectroscopy for the archetypal FLP PMes3/B(C6F5)3 (Mes=2,4,6‐triphenylmethyl), and after switching the Lewis acid to Al(C6F5)3 found a similar, yet much clearer, EPR signal that could be unambiguously attributed to the phosphine radical cation (PMes3 .+). [2c] Furthermore, reaction of PMes3/B(C6F5)3 with Ph3SnH resulted in formation of [Mes3PH][HB(C6F5)3] and Ph3SnSnPh3, instead of the [Mes3P‐SnPh3][HB(C6F5)3] product that would be expected for heterolytic cleavage of the Sn−H bond. [2c] In the case of PtBu3/B(C6F5)3, for which no radicals were detected in the reaction mixture, indeed [tBu3P‐SnPh3][HB(C6F5)3] was obtained, corresponding to nucleophilic substitution at tin (Scheme 1 a). As the difference in products was proposed to be caused by a change in mechanism (homolytic vs. heterolytic), this led to the use of Ph3SnH as a probe for determining the mechanistic nature of FLP reactions.[ 2c , 4 ] Furthermore, Stephan et al. determined that reacting PMes3/B(C6F5)3 with tetrachloro‐1,4‐benzoquinone (TCQ) leads to radical formation after observation of PMes3 .+ by EPR spectroscopy (Scheme 1 b).[ 2c , 5 ] Furthermore, Melen et al. recently reported that the PMes3/B(C6F5)3 pair can be utilized to facilitate C−C bond formation by coupling diarylmethyl groups to styrenes via a mechanism involving single‐electron transfer (SET; Scheme 1 c). [6] Ooi et al. also achieved C−C bond formation using an amine/B(C6F5)3 system to couple methylvinylketone to the amine employing catalytic amounts of borane (Scheme 1 d). [7] They also showed that the reaction requires light and proceeds via radical species which they postulated to be the result of photo‐induced SET from the amine directly to B(C6F5)3 yielding the corresponding radical ion pair [amine.+, B(C6F5)3 .−]. Subsequent addition of this species to the substrate was proposed, which then led to product formation.
Scheme 1.

a) Different pathways proposed by Stephan et al. for reactions of FLPs with Ph3SnH. b) Reactivity observed by Stephan et al. for Mes3P/B(C6F5)3 with tetrachloro‐1,4‐quinone (TCQ). c) Reactivity observed by Melen et al. (ArF=Ph, p‐F‐Ph or fluorene; Ar=variety of aryl groups, see Ref. [6]. d) Reactivity observed by Ooi et al. utilizing catalytic B(C6F5)3 (10 mol %) (R=Me or Br); e) Light dependence for radical ion pair generation from archetypal FLP systems observed by Slootweg et al. (For P: R=Mes or tBu, for N: R=Ph or p‐Me‐Ph).
Recently, we demonstrated the generality of SET for FLP type donor–acceptor systems[ 8 , 9 ] and showed that for common P/B FLPs (PMes3/B(C6F5)3 and PtBu3/B(C6F5)3) and analogous N/B systems visible light is required to induce SET to generate the corresponding transient radical ion pairs (Scheme 1 e). [10] This light dependence provides an excellent probe for determining whether an FLP reaction proceeds via a radical mechanism or via concerted, polar pathways, as carrying out the reaction in the absence of light precludes the formation of the radical ion pair. The work presented herein focuses on applying this notion to investigate the reaction of archetypal FLPs with the substrates H2, Ph3SnH and TCQ. In addition, we analyzed the nature of the initial single‐electron transfer event that is responsible for the radical chemistry observed by Melen et al. and Ooi et al. For all cases, we determine whether the boron Lewis acid is directly involved in SET, or plays a facilitating role by enhancing the oxidizing power of the substrate. [11]
Results and Discussion
First, we assessed the influence of light on the reaction of PMes3 and B(C6F5)3 with H2 (1 atm), which is known to generate the corresponding phosphonium borate [Mes3PH][HB(C6F5)3]. [12] We previously showed that this combination of donor (PMes3) and acceptor (B(C6F5)3) forms a violet charge‐transfer complex in solution from which the corresponding radical ion pair [PMes3 .+, B(C6F5)3 .−] is generated by irradiating this electron donor–acceptor (EDA) complex with visible light (534 nm). [10] Thus, if formation of this radical ion pair is a significantly contributing factor in hydrogen splitting, the reaction should exhibit a change in reaction rate depending on the absence or presence of light. Comparison of reaction samples kept in the dark or irradiated (534 nm, 2.2 W LEDs; Scheme 2) whilst stirring for 2.5 hours showed near‐identical conversions to the phosphonium borate [Mes3PH][HB(C6F5)3], and again after 4 hours, as determined by 31P‐NMR spectroscopy (Supporting Information, Figures S1, S2). [13] These data show that the reaction is not light dependent and therefore the formation of the radical ion pair does not significantly influence the reaction kinetics. This finding suggests that the photo‐stationary concentration of the radical ion pair [PMes3 .+, B(C6F5)3 .−] is too low and/or its lifetime is too short to significantly influence the reaction rate. Indeed, this charge‐separated state lies much higher in energy (54.4 kcal mol−1) than the neutral donor‐acceptor pair [PMes3, B(C6F5)3] and undergoes rapid back‐electron transfer (lifetime=237 ps) as determined by transient absorption spectroscopy to regenerate the FLP, [10] thus preventing build‐up of a concentration of radicals large enough to influence the reaction kinetics. This leads to the conclusion that the splitting of dihydrogen by PMes3 and B(C6F5)3 proceeds via a two‐electron, heterolytic pathway, even when the reaction is performed in ambient light.[ 2e , 2f , 2g ]
Scheme 2.

Reactivity of PMes3/B(C6F5)3 with H2 for which no light dependence was observed.
Next, we probed the reaction between PMes3/B(C6F5)3 and Ph3SnH (2 equiv) to analyse whether light affects the formation of phosphonium borate [Mes3PH][HB(C6F5)3] and Ph3Sn−SnPh3. We found that the reaction proceeds rapidly in both darkness and ambient light and, in both cases, within minutes full conversion to [Mes3PH][HB(C6F5)3] and Ph3Sn−SnPh3 was observed by multi‐nuclear NMR spectroscopy (δ31P=−28.6, δ11B=−26.1, δ119Sn=−131.7; Supporting Information, Figure S5–S9; Scheme 1 a). This suggests that also in this case radicals are not responsible for the reaction outcome.
But how is [Mes3PH][HB(C6F5)3] formed when using Ph3SnH instead of H2? For this, changing the phosphine to PtBu3 provided insight. Addition of 1 equiv of Ph3SnH to PtBu3/B(C6F5)3 in C6H5Cl at room temperature instantly led to heterolytic cleavage of the Sn−H bond and the formation of [tBu3P‐SnPh3][HB(C6F5)3] (δ31P=65.8, 1 J P‐Sn=90 Hz; Scheme 3; Supporting Information, Figure S10), supporting the observations by Stephan et al. [2c] We noted, however, that when more Ph3SnH (up to 2.5 equiv) was used, the reaction continued and after 20 hours [tBu3PH][HB(C6F5)3] (δ31P=58.1, 1 J P‐H=410 Hz; Supporting Information, Figure S12) as well as Ph3Sn‐SnPh3 (Scheme 3; Supporting Information, Figure S14) was observed. [13] We also noted that this reaction proceeds equally in the absence of light, in ambient light, or under direct irradiation of the charge‐transfer band of [PtBu3, B(C6F5)3] (400 nm, 2.2 W LED). These findings show that for both phosphines R3P (R=Mes and tBu) a polar, heterolytic mechanism is operative and that the initial product [R3P‐SnPh3][HB(C6F5)3] can convert into [R3PH][HB(C6F5)3] in the presence of Ph3SnH.
Scheme 3.

Reactivity of PtBu3/B(C6F5)3 with Ph3SnH.
To elucidate the heterolytic splitting of Ph3SnH in more detail, we first combined it with B(C6F5)3 in calculations and found the formation of an adduct with a bridging hydride [Ph3Sn−H−B(C6F5)3] (ΔE=−21.3, ΔG°298K=−1.9 kcal mol−1; Figure 1, left), which is analogous to the key, transient intermediate in the B(C6F5)3‐catalyzed hydrosilylation. [14] We also observed the [Ph3Sn−H−B(C6F5)3] adduct in C6H5Cl solution by 19F‐NMR spectroscopy that shows a decrease in resonance difference between the meta‐ and para‐fluorines (Δδ 18.2 to 13.7 ppm), which is indicative of a transition from a trigonal planar borane to a more tetrahedral geometry. [15] Furthermore, 119Sn‐NMR spectroscopy supports this notion, the clear downfield shift indicates a more electron deficient Sn nucleus (δ=165 to 130 ppm; Supporting Information, Figure S15, S16). [16] These observations evidence activation of the tin hydride by B(C6F5)3, making it more susceptible to nucleophilic attack by a phosphine in an SN2 fashion to produce the initial [R3P‐SnPh3][HB(C6F5)3] species.
Figure 1.

Computed structure for the adducts of Ph3SnH with B(C6F5)3 (left) and Ph3Sn+ (right) featuring a bridging hydride (DFT: ωB97X‐D/def2‐TZVP). Selected bond lengths and angles: Ph3Sn−H−B(C6F5)3: Sn−H 1.83 Å, B−H 1.37 Å; Sn‐H‐B 180°. [Ph3Sn−H−SnPh3]+: Both Sn−H 1.87 Å; Sn‐H‐B 147°.
To investigate the subsequent reaction steps and determine the influence of the P‐substituent (Mes vs. tBu), we again employed computational chemistry (ωB97X‐D/def2‐TZVP; [HB(C6F5)3]− anion omitted), [15] which highlighted a formal metathesis reaction of the [R3P−SnPh3]+ cation with Ph3Sn−H, reminiscent of reactions between tin hydrides and neutral stannyl phosphines. [17] When using PMes3 (Figure 2, in green), [Mes3P‐SnPh3]+ undergoes a facile reaction with Ph3SnH to afford the bridging hydride [Ph3Sn−H−SnPh3]+ (ΔE ≠ TS1≈9, ΔE=−4.9 kcal mol−1), [15] akin to the tin hydride‐B(C6F5)3 adduct (Figure 1). Subsequent deprotonation by the liberated phosphine, which induces Sn−Sn bond formation (ΔE ≠ TS2=19.9, ΔE=−13.1 kcal mol−1), affords [Mes3PH]+ and Ph3Sn−SnPh3. This reaction profile supports the notion that [Mes3P−SnPh3][HB(C6F5)3] is a transient, unobserved intermediate in the formation of [Mes3PH][HB(C6F5)3]. [2c]
Figure 2.
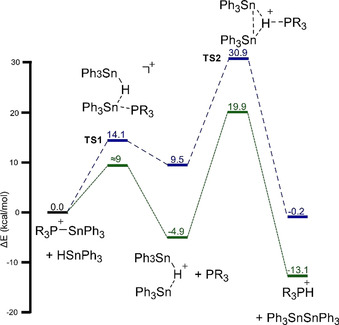
Proposed reaction mechanism based on DFT calculations at the ωB97X‐D/def2‐TZVP level of theory. R=tBu (blue, dashed) or Mes (green, dotted). [HB(C6F5)3]− anion has been omitted for clarity. Energies in kcal mol−1.
Changing the phosphine to PtBu3 has a significant impact. First, the formation the bridging hydride [Ph3Sn−H−SnPh3]+ is now endothermic (ΔE ≠ TS1=14.1, ΔE=9.5 kcal mol−1; Figure 2, in blue) and the subsequent deprotonation faces a sizeable barrier (ΔE ≠ TS2=30.9, ΔE=−0.2 kcal mol−1). The near thermoneutral reaction profile and high barrier accounts for the slow and modest formation of [tBu3PH][HB(C6F5)3] and explains why the [tBu3P‐SnPh3][HB(C6F5)3] intermediate can be isolated after short reaction times and immediate work‐up. [2c]
To support the intermediacy of the bridging [Ph3Sn−H−SnPh3]+ cation, we combined [Ph3C][B(C6F5)4] with 2 equiv of Ph3SnH in C6H5Cl at −35 °C in order to access this species by hydride abstraction (Scheme 4). Indeed, after 1 hour, the characteristic yellow color of the trityl cation disappeared, and an expected downfield shift of the aromatic 1H nuclei of the Ph3Sn species in combination with a broadening of the hydride peak at 6.91 ppm was observed by 1H NMR spectroscopy (Supporting Information, Figures S20, S21). [15] In addition, the spectrum evidenced formation of triphenylmethane (δ1H=5.55). [18] As predicted by DFT, subsequent addition of PMes3 led to formation of [Mes3PH][B(C6F5)4] by deprotonation, as observed by 1H‐ and 31P‐NMR spectroscopy (Supporting Information, Figure S23, S24), and the formation of Ph3Sn−SnPh3, evidenced by 119Sn NMR spectroscopy (Supporting Information, Figure S25). Addition of PtBu3 instead of PMes3 afforded both [tBu3PH][B(C6F5)4] and [tBu3P‐SnPh3][B(C6F5)4] according to 31P‐NMR spectroscopy (approx. 4:5 ratio; Supporting Information, Figure S26), since both the forward and reverse pathways (in blue, Figure 2) have accessible barriers (21.4 and 4.6 kcal mol−1, respectively); this yields the thermodynamically controlled product distribution. These findings demonstrate that after heterolytic Sn−H bond cleavage to form [R3P‐SnPh3][HB(C6F5)3], subsequent bond metathesis leads to formation of [R3PH][HB(C6F5)3] via a bridging hydride intermediate and highlights that the complete reaction of PMes3/B(C6F5)3 and PtBu3/B(C6F5)3 with Ph3SnH is accessible via heterolytic polar pathways.
Scheme 4.

Hydride abstraction from Ph3SnH using [Ph3C][B(C6F5)4] and subsequent reaction with PMes3.
Next, we set out to analyze the reaction of PMes3/B(C6F5)3 with tetrachloro‐1,4‐benzoquinone (TCQ) for which Stephan et al. detected radical formation (PMes3 .+) by EPR spectroscopy. [2c] They postulated that this proceeds via SET from PMes3 to B(C6F5)3 to form the corresponding radical ion pair [PMes3 .+, B(C6F5)3 .−], after which 2 equiv of B(C6F5)3 .− react with the quinone to form dianion 7, while 1 equiv of B(C6F5)3 .− affords the neutral adduct Mes3P‐TCQ‐B(C6F5)3 4 (Scheme 5). [2c] We performed this reaction in the absence of light and found that the reaction still proceeds rapidly, forming a deep purple solution for which EPR spectroscopy confirmed the formation of PMes3 .+ (two‐line signal simulated with g iso=2.0050, A iso=670.00 MHz),[ 2c , 19 ] but also showed for the first time a featureless signal (g iso=2.0058) that we attribute to a TCQ centered radical anion, most likely TCQ‐B(C6F5)3 .− (Figure 3). Furthermore, we noted an unknown smaller third signal, which was also reported by Müller and Klare et al. when combining PMes3 and the strongly accepting silyl and trityl cations.[ 20 , 21 ]
Scheme 5.
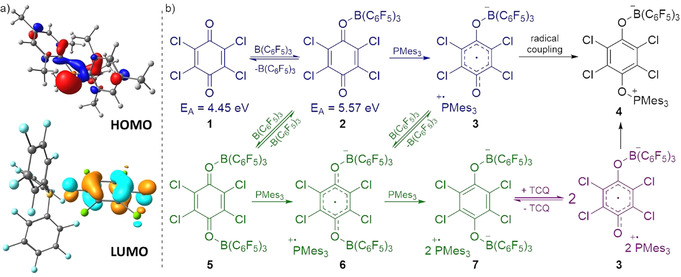
a) Orbitals involved in the SET between PMes3 and the TCQ‐B(C6F5)3 adduct. b) Reactivity, featuring all possible pathways for the reaction of TCQ, B(C6F5)3, and PMes3.
Figure 3.

Experimental EPR spectrum (bottom) for reaction of PMes3, B(C6F5)3 and TCQ (2:2:1) and simulated spectra for PMes3 .+, TCQ‐B(C6F5)3 .− and the third smaller signal. See the Supporting Information for experimental and simulation parameters. HFI=hyperfine interaction.
So, how is it possible that radicals are formed in the dark? Clearly, a strong electron acceptor is required to oxidise PMes3 (IED=5.25 eV; SCRF [15] /ωB97X‐D/6–311+G(d,p), solvent=chlorobenzene) and neither B(C6F5)3 nor TCQ are suitable (E A=3.31 and 4.45 eV, respectively) [15] to accommodate the needed thermal SET. Yet, B(C6F5)3 can coordinate to one of the carbonyl moieties of TCQ, affording the corresponding Lewis adduct TCQ‐B(C6F5)3, which has an increased electron affinity (E A=5.57 eV) and therefore should be capable of oxidizing PMes3. [11] Note that such interactions between a Lewis acid and a carbonyl moiety are typically exploited in photo‐redox catalysis to facilitate SET events. [22] As the carbonyl moieties of TCQ are electron poor, the interaction with B(C6F5)3 is weak (ΔE=−4.6, ΔG°298K=10.7 kcal mol−1) leading to an equilibrium featuring low concentrations of the TCQ‐B(C6F5)3 adduct, which supports the notion of Stephan et al. that no interaction between B(C6F5)3 and TCQ is observable by NMR spectroscopy. [2c] However, in presence of PMes3, the transient TCQ‐B(C6F5)3 adduct will undergo SET from the PMes3 HOMO to the TCQ‐B(C6F5)3 LUMO (Scheme 5, left) generating the radical ion pair [PMes3 .+, TCQ‐B(C6F5)3 .−] 3, which drives the equilibrium towards the TCQ‐B(C6F5)3 adduct (Scheme 5 b, blue). Subsequent radical coupling of PMes3 .+ and TCQ‐B(C6F5)3 .− via a computed 8 kcal mol−1 (ΔG°298 K) barrier leads to the formation of Mes3P‐TCQ‐B(C6F5)3 4 as observed experimentally by Stephan et al. (Scheme 5 b, black). [2c] This mechanism highlights that, rather than directly participating in SET, B(C6F5)3 is facilitating the process by increasing the electron affinity of the quinone acceptor.
As TCQ features two carbonyl moieties, coordination of two B(C6F5)3 molecules can also occur prior to SET (2→5 ΔE=−16.0, ΔG°298 K=−2.9 kcal mol−1; coordination after SET is unlikely: 3→6 ΔE=2.7, ΔG°298 K=18.8 kcal mol−1), yielding radical ion pair [PMes3 .+, (C6F5)3B‐TCQ‐B(C6F5)3 .−] 6 (Scheme 5 b, green). The radical anion of 6 has a high electron affinity (E A=6.11 eV), which allows another SET from a second equiv of PMes3 to generate dianion 7. [2c] To complete the picture, dianion 7 is in equilibrium with TCQ‐B(C6F5)3 .− radical anion 3 (ΔE=5.2, ΔG°298 K=2.3 kcal mol−1, Scheme 5 b, purple) that can, as noted earlier, undergo radical coupling with PMes3 .+ to form 4 (ΔE=−48.9, ΔG°298 K=−20.4 kcal mol−1; Scheme 5 b, black).
Changing the phosphine to PtBu3 was shown by Stephan et al. to only yield the tBu3P−TCQ−B(C6F5)3 adduct, akin to 4, without detectable radicals or dianion 7, which could indicate a different mechanism. The ionization energy of tBu3P (IED=5.54 eV), however, suggests that SET from the phosphine to the TCQ‐B(C6F5)3 adduct (E A=5.57 eV) is still feasible. In this case, though, the subsequent radical coupling is barrierless, [23] which leads to the immediate formation of tBu3P‐TCQ‐B(C6F5)3 (ΔE=−56.5, ΔG°298 K=−20.4 kcal mol−1) and prevents detection of radical species or subsequent reactivity to form dianion 7. This shows that, similar to our findings for Ph3SnH, changing from PMes3 to PtBu3 does not alter the mechanism, but merely the energy levels along the reaction path leading to observation of radical intermediates for PMes3, but not in case of PtBu3.
Since the groups of Melen and Ooi recently reported FLP type reactions featuring radical formation when using B(C6F5)3 and carbonyl containing substrates,[ 6 , 7 ] we postulated that also in these cases Lewis acid coordination to the substrate could increase its electron acceptor capacity and promote SET (Scheme 6). Indeed, for methylvinylketone (MVK), the substrate utilized by Ooi et al. (Scheme 1 d), [7] we found that B(C6F5)3 forms an adduct (ΔE=−16.3, ΔG°298 K=0.9 kcal mol−1; SCRF/ωB97X‐D/6–311+G(d,p), solvent=dichloroethane) and increases its electron affinity from 1.43 to 2.73 eV (Scheme 7, left). This decreases the energy gap between the ground state amine donor (IED=5.11 eV; R=Me; Scheme 1 d) and methylvinylketone acceptor pair to the corresponding radical ion pair [amine.+, MVK‐B(C6F5)3 .−] from 3.68 to 2.38 eV, which results in visible light induced (1.5–3.1 eV, 400–800 nm) SET becoming feasible. Indeed, Ooi et al. used 400 nm light to promote this reaction. In case of substrate 9, used by Melen et al. (Scheme 1 c), [6] we found a similar result. Binding of B(C6F5)3 (ΔE=−19.1, ΔG°298 K=−0.2 kcal mol−1; SCRF/ωB97X‐D/6–311+G(d,p), solvent=THF) increases the electron affinity from 2.31 to 3.56 eV (Scheme 7, right) bringing the energy required for SET (with PMes3 as donor) down from 2.89 to 1.74 eV. This reduced energy gap (40.0 kcal mol−1) is still sizeable and suggests that, also in this case, the SET is photo‐induced and thus performing this reaction in broad daylight (or using a high‐power LED) will be beneficial. These results, in combination with the transient nature of the highly reactive B(C6F5)3 .− species in solution,[ 3a , 24 ] make it highly plausible that also for these systems, B(C6F5)3 is facilitating SET through binding to the substrate and increasing its electron affinity, instead of participating directly in SET.
Scheme 6.
Lewis acid coordination to a carbonyl moiety facilitating SET. LB=Lewis base.
Scheme 7.
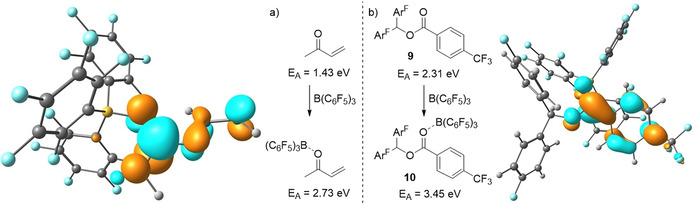
Change in electron affinity when B(C6F5)3 coordinates and the resulting LUMO for two different B(C6F5)3‐coordinated substrates.
Conclusion
Although the archetypal PMes3/B(C6F5)3 and PtBu3/B(C6F5)3 FLP systems can form high energy radical ion pairs via photo‐induced single‐electron transfer, we found that this pathway is not a major contributor in the reaction with H2 or Ph3SnH, and that in both cases the reactions occur via polar, heterolytic mechanisms. Furthermore, we discovered that the SET reactivity observed for FLP systems with substrates featuring carbonyl moieties is not the result of SET from the Lewis base directly to the borane Lewis acid. Instead, adduct formation between the Lewis acid and substrate activates the substrate for SET, after which electron donor‐acceptor complex formation with the Lewis base provides the corresponding radical ion pair, via either thermal or photoinduced SET, depending on the energy required. To promote radical reactivity in cases based on photoinduced SET, it is thus important to locate the CT‐band arising after Lewis acid coordination to determine the optimal wavelength for irradiation of reaction mixtures. These important mechanistic insights are of fundamental importance for both efficient usage of current radical FLP systems as well as the design of novel radical FLP systems and new examples of main‐group redox catalysis, [25] which we are currently exploring in our laboratories.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Council for Chemical Sciences of The Netherlands Organization for Scientific Research (NWO/CW) by a VIDI grant (J.C.S.), NWA Idea Generator grant (J.C.S.) and a VENI grant (A.R.J.).
F. Holtrop, A. R. Jupp, B. J. Kooij, N. P. van Leest, B. de Bruin, J. C. Slootweg, Angew. Chem. Int. Ed. 2020, 59, 22210.
References
- 1.
- 1a. Welch G. C., San Juan R. R., Masuda J. D., Stephan D. W., Science 2006, 314, 1124–1126; [DOI] [PubMed] [Google Scholar]
- 1b. Paradies J., Coord. Chem. Rev. 2019, 380, 170–183; [Google Scholar]
- 1c. Skara G., De Vleeschouwer F., Geerlings P., De Proft F., Pinter B., Sci. Rep. 2017, 7, 16024; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Stephan D. W., Science 2016, 354, aaf7229;27940818 [Google Scholar]
- 1e. Stephan D. W., J. Am. Chem. Soc. 2015, 137, 10018–10032; [DOI] [PubMed] [Google Scholar]
- 1f. Stephan D. W., Erker G., Angew. Chem. Int. Ed. 2015, 54, 6400–6441; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6498–6541; [Google Scholar]
- 1g. Jupp A. R., Stephan D. W., Trends Chem. 2019, 1, 35–48. [Google Scholar]
- 2.
- 2a. Paradies J., Eur. J. Org. Chem. 2019, 283–294; [Google Scholar]
- 2b. Liu L., Cao L. L., Zhu D., Zhou J., Stephan D. W., Chem. Commun. 2018, 54, 7431–7434; [DOI] [PubMed] [Google Scholar]
- 2c. Liu L., Cao L. L., Shao Y., Ménard G., Stephan D. W., Chem 2017, 3, 259–267; [Google Scholar]
- 2d. Rocchigiani L., Ciancaleoni G., Zuccaccia C., Macchioni A., J. Am. Chem. Soc. 2014, 136, 112–115; [DOI] [PubMed] [Google Scholar]
- 2e. Rokob T. A., Bakó I., Stirling A., Hamza A., Pápai I., J. Am. Chem. Soc. 2013, 135, 4425–4437; [DOI] [PubMed] [Google Scholar]
- 2f. Grimme S., Kruse H., Goerigk L., Erker G., Angew. Chem. Int. Ed. 2010, 49, 1402–1405; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 1444–1447; [Google Scholar]
- 2g. Daru J., Bakó I., Stirling A., Pápai I., ACS Catal. 2019, 9, 6049–6057. [Google Scholar]
- 3.
- 3a. Lawrence E. J., Oganesyan V. S., Wildgoose G. G., Ashley A. E., Dalton Trans. 2013, 42, 782–789; [DOI] [PubMed] [Google Scholar]
- 3b. Piers W. E., Marwitz A. J. V., Mercier L. G., Inorg. Chem. 2011, 50, 12252–12262; [DOI] [PubMed] [Google Scholar]
- 3c. Dong Z., Cramer H. H., Schmidtmann M., Paul L. A., Siewert I., Müller T., J. Am. Chem. Soc. 2018, 140, 15419–15424; [DOI] [PubMed] [Google Scholar]
- 3d. Liu L. L., Stephan D. W., Chem. Soc. Rev. 2019, 48, 3454–3463; [DOI] [PubMed] [Google Scholar]
- 3e. Dong Z., Pezzato C., Sienkiewicz A., Scopelliti R., Fadaei-Tirani F., Severin K., Chem. Sci. 2020, 11, 7615–7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bentley J. N., Pradhan E., Zeng T., Caputo C. B., Dalton Trans. 2020, 10.1039/d0dt00506a. [DOI] [PubMed] [Google Scholar]
- 5.Note triphenylphosphine forms an adduct with TCQ; see: Ramirez F., Dershowitz S., J. Am. Chem. Soc. 1956, 78, 5614–5622. [Google Scholar]
- 6. Soltani Y., Dasgupta A., Gazis T. A., Ould D. M. C., Richards E., Slater B., Stefkova K., Vladimirov V. Y., Wilkins L. C., Wilcox D., Melen R. L., Cell Rep. Phys. Chem. 2020, 1, 10016. [Google Scholar]
- 7. Aramaki Y., Imaizumi N., Hotta M., Kumagai J., Ooi T., Chem. Sci. 2020, 11, 4305–4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Mulliken R. S., J. Am. Chem. Soc. 1952, 74, 811–824; [Google Scholar]
- 8b. Foster R., J. Phys. Chem. 1980, 84, 2135–2141; [Google Scholar]
- 8c. Rosokha S. V., Kochi J. K., Acc. Chem. Res. 2008, 41, 641–653; [DOI] [PubMed] [Google Scholar]
- 8d.for an ethylene/I2 EDA complex, see: Kalume A., George L., Powell A. D., Dawes R., Reid S. A., J. Phys. Chem. A 2014, 118, 6838–6845. [DOI] [PubMed] [Google Scholar]
- 9.For selected examples of electron donor–acceptor complexes in photocatalysis, see:
- 9a. Arceo E., Jurberg I. D., Álvarez-Fernández A., Melchiorre P., Nat. Chem. 2013, 5, 750–756; [DOI] [PubMed] [Google Scholar]
- 9b. Zhu Y., Zhang L., Luo S., J. Am. Chem. Soc. 2014, 136, 14642–14645; [DOI] [PubMed] [Google Scholar]
- 9c. Nappi M., Bergonzini G., Melchiorre P., Angew. Chem. Int. Ed. 2014, 53, 4921–4925; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 5021–5025; [Google Scholar]
- 9d. Woźniak Ł., Murphy J. J., Melchiorre P., J. Am. Chem. Soc. 2015, 137, 5678–5681; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9e. Kandukuri S. R., Bahamonde A., Chatterjee I., Jurberg I. D., Escudero-Adan E. C., Melchiorre P., Angew. Chem. Int. Ed. 2015, 54, 1485–1489; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1505–1509; [Google Scholar]
- 9f. Sandfort F., Strieth-Kalthoff F., Klauck F. J. R., James M. J., Glorius F., Chem. Eur. J. 2018, 24, 17210–17214; [DOI] [PubMed] [Google Scholar]
- 9g. Wu J., Grant P. S., Li X., Noble A., Aggarwal V. K., Angew. Chem. Int. Ed. 2019, 58, 5697–5701; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 5753–5757; [Google Scholar]
- 9h.for a recent review, see: Crisenza G. E. M., Mazzarella D., Melchiorre P., J. Am. Chem. Soc. 2020, 142, 5461–5476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Holtrop F., Jupp A. R., Van Leest N. P., Paradiz Dominguez M., Williams R. M., Brouwer A. M., De Bruin B., Ehlers A. W., Slootweg J. C., Chem. Eur. J. 2020, 26, 9005–9011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.For increasing the electron-acceptor capacity of quinones by hydrogen-bond donors, see:
- 11a. Turek A. K., Hardee D. J., Ullman A. M., Nocera D. G., Jacobsen E. N., Angew. Chem. Int. Ed. 2016, 55, 539–544; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 549–554; for the B(C6F5)3 coordination to and thereby redox-activation of S-nitrosothiols, see: [Google Scholar]
- 11b. Hosseininasab V., McQuilken A. C., Bakhoda A., Bertke J. A., Timerghazin Q. K., Warren T. H., Angew. Chem. Int. Ed. 2020, 59, 10854–10858; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 10946–10950; for the reduction of disulfides by ferrocene enabled by Lewis acids like B(C6F5)3, see: [Google Scholar]
- 11c. Liu L. L., Cao L. L., Shao Y., Stephan D. W., J. Am. Chem. Soc. 2017, 139, 10062–10071. [DOI] [PubMed] [Google Scholar]
- 12. Welch G. C., Stephan D. W., J. Am. Chem. Soc. 2007, 129, 1880–1881. [DOI] [PubMed] [Google Scholar]
- 13.Note that the conversion values are based on the relative integrals of the tBu3P and [tBu3PH]+ resonances in each case. These values do not accurately correspond to conversion because the spectra were not obtained in a quantitative manner; however, the key result is that the values are the same regardless of whether the samples were irradiated or not.
- 14.
- 14a. Houghton A. Y., Hurmalainen J., Mansikkamäki A., Piers W. E., Tuononen H. M., Nat. Chem. 2014, 6, 983–988; [DOI] [PubMed] [Google Scholar]
- 14b. Rendler S., Oestreich M., Angew. Chem. Int. Ed. 2008, 47, 5997–6000; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 6086–6089; [Google Scholar]
- 14c. Mewald M., Oestreich M., Chem. Eur. J. 2012, 18, 14079–14084. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Horton A. D., de With J., Organometallics 1997, 16, 5424–5436; [Google Scholar]
- 15b. Blackwell J. M., Piers W. E., Parvez M., Org. Lett. 2000, 2, 695–698; [DOI] [PubMed] [Google Scholar]
- 15c. Beringhelli T., Donghi D., Maggioni D., D'Alfonso G., Coord. Chem. Rev. 2008, 252, 2292–2313; [Google Scholar]
- 15d. Piers W. E., Adv. Organomet. Chem. 2005, 52, 1–76. [Google Scholar]
- 16.See the Supporting Information.
- 17. Neumann W. P., Schneider B., Sommer R., Justus Liebigs Ann. Chem. 1966, 692, 1–11. [Google Scholar]
- 18. Horn M., Schappele L. H., Lang-Wittkowski G., Mayr H., Ofial A. R., Chem. Eur. J. 2013, 19, 249–263. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Il'yasov A. V., Kargin Y. M., Nikitin E. V., Vafina A. A., Romanov G. V., Parakin O. V., Kazakova A. A., Pudovik A. N., Phosphorus Sulfur Relat. Elem. 1980, 8, 259–262; [Google Scholar]
- 19b. Culcasi M., Berchadsky Y., Gronchi G., Tordo P., J. Org. Chem. 1991, 56, 3537–3542. [Google Scholar]
- 20. Merk A., Großekappenberg H., Schmidtmann M., Luecke M.-P., Lorent C., Driess M., Oestreich M., Klare H. F. T., Müller T., Angew. Chem. Int. Ed. 2018, 57, 15267–15271; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 15487–15492. [Google Scholar]
- 21.The third signal corresponds to either a radical species which shows hyperfine coupling with two 31P nuclei, or a species featuring two uncoupled (that is, non-interacting) unpaired electrons, for which one shows hyperfine coupling to a 31P nucleus. One possible assignment would be Mes3P−PMes3 .+, although this species has no stable minimum on the potential energy surface; see also: Tojo S., Yasui S., Fujitsuka M., Majima T., J. Org. Chem. 2006, 71, 8227–8232.17025316 [Google Scholar]
- 22.
- 22a. Du J., Skubi K. L., Schultz D. M., Yoon T. P., Science 2014, 344, 392–396; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22b. Yoon T. P., Acc. Chem. Res. 2016, 49, 2307–2315; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22c. Lee K. N., Ngai M.-Y., Chem. Commun. 2017, 53, 13093–13112; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22d. Speckmeier E., Fuchs P. J. W., Zeitler K., Chem. Sci. 2018, 9, 7096–7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.The computed barrier (ΔG°298 K=−0.4 kcal mol−1) is less than the error margin and can thus be considered approximately equal to zero.
- 24. Kwaan R. J., Harlan C. J., Norton J. R., Organometallics 2001, 20, 3818–3820. [Google Scholar]
- 25.
- 25a. McAtee R. C., McClain E. J., Stephenson C. R. J., Trends Chem. 2019, 1, 111–125; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25b. Romero N. A., Nicewicz D. A., Chem. Rev. 2016, 116, 10075–10166; [DOI] [PubMed] [Google Scholar]
- 25c. Fu M.-C., Shang R., Zhao B., Wang B., Fu Y., Science 2019, 363, 1429–1434; [DOI] [PubMed] [Google Scholar]
- 25d. Deng Z., Lin J.-H., Xiao J.-C., Nat. Commun. 2016, 7, 10337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary