

Abstract
Small‐molecule stabilization of protein–protein interactions (PPIs) is a promising concept in drug discovery, however the question how to identify or design chemical starting points in a “bottom‐up” approach is largely unanswered. We report a novel concept for identifying initial chemical matter for PPI stabilization based on imine‐forming fragments. The imine bond offers a covalent anchor for site‐directed fragment targeting, whereas its transient nature enables efficient analysis of structure–activity relationships. This bond enables fragment identification and optimisation using protein crystallography. We report novel fragments that bind specifically to a lysine at the PPI interface of the p65‐subunit‐derived peptide of NF‐κB with the adapter protein 14‐3‐3. Those fragments that subsequently establish contacts with the p65‐derived peptide, rather than with 14‐3‐3, efficiently stabilize the 14‐3‐3/p65 complex and offer novel starting points for molecular glues.
Keywords: 14-3-3 proteins, cooperative effects, fragment-based drug discovery, imine chemistry, protein–protein interactions
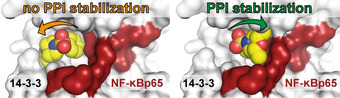
A novel concept for optimizing orthosteric protein–protein interaction (PPI) stabilization is reported. Increasing interactions with the protein partner that contributes less to the composite binding pocket of the stabilizer (NF‐κB, red surface) results in increased stabilization, whereas further enhancing the interaction with the dominant partner protein (14‐3‐3, white surface) does not contribute to the stabilizing effect.
Small‐molecule modulation of protein‐protein interactions (PPIs) is one of the most exciting and promising conceptual strategies in drug discovery and chemical biology, facilitating modulation of clinically relevant targets not previously druggable using conventional approaches.[ 1 , 2 , 3 ] The field of targeted PPI inhibition has matured into a successful drug discovery approach,[ 4 , 5 ] while the opposite strategy of PPI stabilization has been largely a domain of serendipity and retrospective elucidation of modes‐of‐action.[ 1 , 6 ] Examples of the immunomodulatory drug (IMiD) Lenalidomide [7] (Revlimid®) and the immunesuppressant Rapamycin [8] (Rapamune®) illustrate the tremendous potential of PPI stabilization. One of the biggest challenges that PPI stabilization is facing, is the systematic identification of chemical matter for compound development. In recent years, the drug discovery field has shifted to the implementation of fragment‐based drug discovery (FBDD)[ 9 , 10 , 11 ] as a “bottom‐up” strategy for drug development. Alternative to the traditional FBDD, site‐directed fragment “tethering” approaches enable localization of a fragment to a specific site within a protein due to covalent bond formation. Common to all site‐directed fragment libraries is an electrophilic chemical handle (such as a disulfide, acryl or haloketone moiety) which enables conjugation to the protein.[ 12 , 13 , 14 ] By mutational insertion of a cysteine at the PPI interface, control of localization can be achieved as previously shown by our research group, providing the first fragment‐based stabilization of the PPI complex of 14‐3‐3 and the estrogen receptor α. [15]
Lysines constitute a large percentage of the proteinogenic amino acids, with concomitant covalent and dynamic covalent drug targeting approaches developed for this amino acid.[ 16 , 17 , 18 ] Aldehydes forming aldimine bonds provide attractive entries for targeting lysine sidechains, but have typically only been successful when the imine bond was intrinsically stabilized by flanking chemical functionalities which trap the imine bond via an intramolecular hydrogen bond, such as the drug Voxelotor used to treat sickle cell disease (Figure S1).[ 16 , 18 , 19 ] Nevertheless, we reasoned the reversible nature of imine bonds to be of high potential for tethered FBDD of PPI complexes. The formation of non‐trapped aldimines would potentially aid in identifying fragments with beneficial contacts to the target pocket, as templating effects would facilitate aldimine bond formation. Here we show for the first time the use of dynamic covalent fragments which stabilize a protein complex, using imine chemistry as covalent anchor. Illustrated using the 14‐3‐3/NF‐κB interaction, a high value drug target,[ 20 , 21 ] we reveal how a composite PPI binding pocket featuring the hydrophobically buried Lys122 provides entry to selective PPI stabilizers (Figure 1). Notably, hit compounds are specific Lys122 binders, affording exquisite control over localization. Additionally, our study reveals that only those fragments that feature enhanced contacts with the NF‐κB element, rather than with 14‐3‐3 alone, provide the best starting points as molecular glues. These results further support previous observation using semisynthetic analogues of fusicoccin and disulfide trapping experiments.[ 15 , 21 ]
Figure 1.
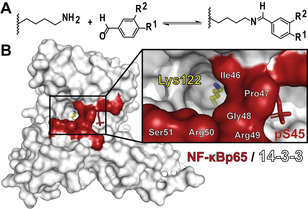
Concept of imine tethering. A) Lysine residues can be targeted with aldehydes to form an aldimine bond. B) Lysine 122 of 14‐13‐3 is located in a deep composite binding pocket created by the NF‐κB/14‐3‐3 complex (surface representation of 14‐3‐3 in white and the p65 subunit of NF‐κB in red).
14‐3‐3σ, exemplary as one of the seven 14‐3‐3 isoforms,[ 22 , 23 ] features 18 mostly solvent exposed lysine residues (Figure S2). In silico analysis of the local pK a values (Table S1) with the Rosetta webtool[ 24 , 25 ] showed that Lys122 and Lys159 feature the lowest predicted pK a’s both around 10, suggesting these two residues to be most amenable to imine bond formation. Lys122 is of particular interest, as the amino acid is located within a predominately hydrophobic region of the 14‐3‐3 phosphopeptide binding groove (Figure 1 and S2). Lys122 is part of the so‐called “Fusicocin binding pocket” — a preferred drug targeting pocket for 14‐3‐3 PPI stabilization (Figure S2)[ 23 , 26 ] — and thus ideally positioned to explore for fragment‐based PPI stabilization via imine‐based tethering. The crystal structure of 14‐3‐3 in complex with the p65 subunit of NF‐κB [21] offers an excellent opportunity for fragment crystal soaking, also because the hydrophobic microenvironment around Lys122 is further extended by three hydrophobic residues of p65, Ile46, Pro47 and Gly48 (Figure 1).
Initially, we tested a small collection of 10 aldehyde‐bearing fragments (Figure S3) and soaked these fragments individually into crystals of the binary p65/14‐3‐3 complex. [21] These fragments were part of an “in‐house” fragment cocktail library (≈160 cocktails, 5 fragments/cocktail) which consisted of covalent and non‐covalent fragments. [27] Following X‐ray diffraction data analysis, additional density was observed for three fragments binding to Lys122: 1 (TCF521), 2 (TCF569) and 3 (TCF789) (Figure 2 A). Fragments 2 and 3 showed partial electron density coverage and at least three other lysine residues elicited extra electron density, indicating non‐specific reactivity of these compounds (Figure S3).
Figure 2.
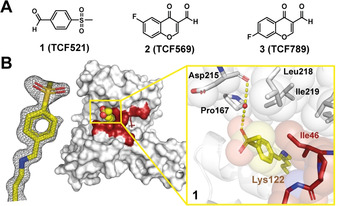
Initial hit fragments that form a covalent bond with Lys122 of 14‐3‐3. A) Overview of compounds binding to Lys122. For details on 2 and 3 see Figure S3. B) The electron density map (grey mesh contoured at 1σ) for 1 (yellow sticks, spheres) binding at the interface of p65 (red) and 14‐3‐3 (white). The p65/14‐3‐3 complex is represented as either a van der Waals surface (middle) or sticks and spheres (close‐up). Water molecule: red sphere; hydrogens bonds: yellow dashed.
In contrast, 1 was completely covered by the electron density map allowing the unambiguous elucidation of the molecular orientation (Figure 2 B). Gratifyingly, no secondary binding site was detected for 1, testifying to its potential for selective targeting of the Lys122. The balanced reactivity and specificity of 1 for Lys122 likely relates to the aldehyde moiety being activated by the electron‐withdrawing sulfonyl moiety in combination with templating effects based on hydrophobic contacts of the benzyl ring with the side chain of Ile46 (Figure 2 B). Replacement of the aldehyde moiety with related functional groups like acid, alcohol, amine, ketone and methyl impeded binding in the crystal structure, highlighting the essential contribution of the imine bond formation (Figure S4).
An extended fragment library consisting of 34 commercially available aldehydes with various ring substitutions on the benzaldehyde core was assembled and subsequently tested in the crystal screening setup (Figure S5). Interestingly, only those fragments featuring an electron withdrawing group, activating the aldehyde for imine formation, showed electron density in the crystal structures (Figure S5, S6, Table S3). Importantly though, all compounds again specifically bound to Lys122 and a conserved water mediated hydrogen bond was observed between the sulfone group and the back‐bone carbonyl of Asp215. These results further support the importance of a balanced activation of the aldehyde for effective but specific imine formation with Lys122. We also soaked the ortho‐hydroxy variant of 1, featuring a hydrogen bond donor group typically used for imine bond trapping. [18] However, of all investigated aldehydes this was the only one inducing crystal cracking potentially caused by pan‐labeling of the majority of the lysine residues.
Given the well‐defined binding mechanism of 1 and its chemical tractability, we sought to grow this compound into a stabilizer of the p65/14‐3‐3 interaction. To this end, we designed a focused library of extended fragments, of which derivatives 4 (TCF521‐123) and 5 (TCF521‐129), showed highly interesting binding characteristics. Briefly, 4 and 5 were accessed via a sulfonyl amide coupling of 4‐formylbenzenesulfonyl chloride with 4‐acetylpiperazin‐1‐yl (4) or 2,6‐dimethyl‐morpholine (5), respectively (Scheme 1).
Scheme 1.
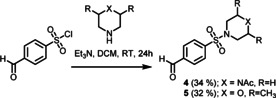
The synthesis of 4‐morpholino‐ (4; TCF521‐123) and 4‐acetylpipererazinyl‐ (5; TCF521‐129) benzenesufonamides analogues.
Structural data on the binding of both compounds was acquired using crystal soaking experiments. The additional electron density was again specifically confined to Lys122 and both compounds were completely covered by the electron density map (Figure 3, Table S4). The aldehyde functionalities of 4 and 5 account for a continuous electron density with the Lys122 side chain, verifying the covalent imide bond formation. The coupling of a single compound to 14‐3‐3 was also verified with mass spectroscopy after reductive amination of the imine bond (Figure S7). The aromatic element of the benzaldehyde ring of both compounds engages in hydrophobic contacts with Ile46 of p65 (Figure 3 A,B). The sulfonamide groups of both molecules make additional water mediated contacts with 14‐3‐3 via Asn42 and the backbone of Asp215. These interactions are analogous to those found for the starting fragment 1 and clarify the basal binding affinity of these fragments to the 14‐3‐3 scaffold.
Figure 3.
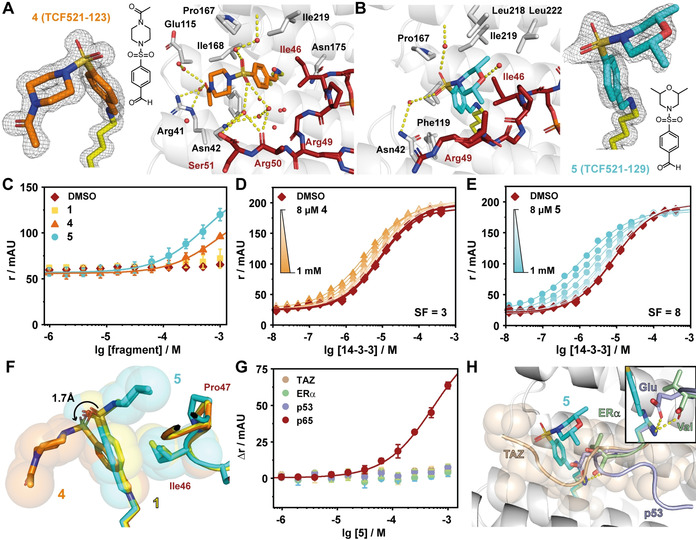
Structure and activity of extended fragments derived from the initial hit TCF521. A) Crystal structure of 4 (TCF521‐123; orange sticks) binding to the complex of 14‐3‐3σ (white sticks & cartoon) and a peptide derived from p65 (red sticks). The final 2 F o−F c electron density for the fragment is shown as grey mesh (contoured at 1σ), polar contacts are indicated by yellow dashes and water molecules are shown as red spheres. B) Crystal structure of 5 (TCF521‐129; cyan sticks) binding to 14‐3‐3 and p65. Details as described in (A). C) Fluorescence anisotropy (FA: r in mAU) assay shows binding of 100 nM of FITC‐labelled monovalent p65 to 14‐3‐3 in the presence of increasing concentrations of the indicated fragments. Shown are mean+/‐SD (n=3). D) 2D Titrations of 4 binding to the p65/14‐3‐3 complex in FA assay. 14‐3‐3 Protein was titrated to 100 nM of FITC‐labelled bivalent p65 in presence of various constant concentrations of fragment. The highest concentration of 4 was 1 mM which was titrated stepwise in a 1:1 dilution series down to 8 μM (n=1, DMSO control n=3). The stabilization factor (SF) describes the fragment induced shift in binding affinities, comparing the apparent K D of the DMSO control and the highest concentration of fragment. E) 2D Titrations of 5 binding to the p65/14–3‐3 complex in FA assay. Details as described in (D) (n=1, DMSO control n=3). F) Overlay of the crystal structures of TCF521 (yellow sticks & spheres), 4 (orange sticks & spheres) and 5 (cyan sticks & spheres). Additional to the compounds, an overlay of the Ile46 and Pro47 of p65 are shown, colouring follows the stick colours of the compounds, respectively. The tilted conformation of 4 increases the distance of the benzaldehyde core, potentially reducing the hydrophobic contact with Ile46 (indicated by sphere representation). G) Fragment 5 is not a pan‐stabilizer of 14‐3‐3 interactions. The fragment was titrated to constant concentrations of 14‐3‐3 and either FITC‐TAZ, FITC‐ERα, TAMRA‐p53 or FITC‐p65. H) Overlay of the 5/p65/14‐3‐3 crystal structure with TAZ (beige cartoon and spheres), ERα (green cartoon), and p53 (violet cartoon). TAZ occupies the whole binding groove and the close‐up shows how the glutamate of p53 and the C‐terminal carboxy of ERα engage Lys122 in polar interactions (for more details see Figure S8).
A significant difference between both fragments was found regarding the orientation of their sulfonyl amide head groups. These newly inserted functionalities, as compared to 1, adopt opposite conformations within the PPI interface. The substituted morpholino ring system of 5 actively engages with elements of the p65 peptide, while the piperazine functionality of 4 adopts an opposite orientation and points away from the p65 element. The sulfonamide oxygens in 4 are engaged in a complex water network with additional water‐mediated contacts to Arg41 of 14‐3‐3 and the Arg50 and Ser51 main‐chain carbonyls of the p65 peptide (Figure 3 A). 5 is engaged in a less pronounced water network and its morpholino group bends off to p65. In addition, one of the methyl groups of 5 makes additionally contacts with Pro47 and Gly48 of p65. The other methyl group is engaging in hydrophobic contacts with the “roof” of the 14‐3‐3 groove comprised of residues Leu218, Ile219, and Leu222 (Figure 3 B).
As ultimate proof of the potential stabilizing capacity of these Lys122‐specific imines forming compounds, biochemical PPI stabilization studies were performed. Compound titrations of 4 and 5 with a fluorescently‐labeled monovalent p65 peptide and 14‐3‐3 protein induced a concentration dependent increase in fluorescence anisotropy (FA), indicative for compound driven complex stabilization. Whereas both compounds induce an increase in anisotropy, 5 is active at lower concentrations and shows a stronger increase in anisotropy (Figure 3 C). The stabilizing effect of the compounds was quantified by titrating 14‐3‐3 to a bivalent p65 peptide and multiple constant concentrations of compound (Figure 3 D,E). Decreasing apparent dissociation constants (K D) due to increasing compound concentrations imply complex stabilization. Comparing the K D of the DMSO control (K D=8.6±0.3 μM) and of the highest compound concentration reveals a stabilization factor (SF) of SF=3 for 4 (K D,app=3.4 μM) and SF=8 for 5 (K D,app=1.1 μM).
The observed stabilizing effect of 4 is probably solely caused by the hydrophobic contact between the benzaldehyde ring and Ile46 of p65 (Figure 3 F). The tilted conformation of 4 has the overall effect of an increased distance between the benzaldehyde ring and Ile46 of the peptide, potentially weakening this hydrophobic contact. The orientation of 5 overlays more precisely with that of the initial hit 1, reaching the full potential of this hydrophobic contact.
The combined structural and biochemical data reveal that the additional contacts made by the morpholino ring of 5 with both the 14‐3‐3 protein and the p65 peptide are beneficial for the ternary small molecule‐stabilized complex. In contrast, the additional contacts of 4 by virtue of its piperazine functionality and the extensive contacts with the water network are exclusively engaged with 14‐3‐3. While such observations are highly valuable towards affinity optimization and selectivity considerations, here specifically these do not contribute to p65/14‐3‐3 stabilization.
As an adapter protein, 14‐3‐3 binds to multiple other interaction partners. Nevertheless, 4 and 5 are not able to stabilize the TAZ/14‐3‐3, ERα/14‐3‐3, nor the p53/14‐3‐3 interaction (Figure 3 G). These are three representative 14‐3‐3 client proteins covering a typical interaction partner binding in an elongated manner in the binding groove (TAZ), one with a phosphorylated C‐terminus (ERα) and a partner with a bent conformation alike the one p65 but with a bulky amino acid in +1 position of the phosphorylation site (p53) (Figure 3 H, Figure S8). The transient nature of the imine bond prevents binding competition of compound and TAZ peptide binding, while both ERα and p53 engage Lys122 in polar bonds, hence prevent imine formation. The specific molecular nature of p65, making a sharp turn out of the 14‐3‐3 binding pocket, governed by Ile46, Pro47, and Gly48 of p65, provides access to the Lys122 in a uniquely generated composite hydrophobic pocket. This reflects a principal feature of orthosteric PPI stabilization which is based on the direct, simultaneous physical interaction of the stabilizer with both protein partners.
In conclusion, we have developed the first small molecule compounds that stabilize the 14‐3‐3/p65 complex, using a site‐directed fragment screening approach. This screening approach is unique to other covalent aldehyde chemical probes, which are reliant on trapping moieties. The lack of trapping moieties enables us to exploit templating effects caused by the binding of the p65 subunit. The unique pK a profile of Lys122, in combination with templating effects of the partner peptide facilitates the specific aldimine bond formation with Lys122. Further, we demonstrate how initial fragments can be rapidly developed into extended stabilizing fragments which elicit promising activity. Further we show that the unique interface of 14‐3‐3/p65 enables the development of selective fragments. This concept provides valuable starting points for further PPI drug development.
Conflict of interest
L.B. and C.O. are scientific founders of AmbAgon Therapeutics.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The research was supported by funding from the European Union through the TASPPI project (H2020‐MSCA‐ITN‐2015, grant number 675179), through the Eurotech Postdoctoral Fellow program (Marie Skłodowska‐Curie Co‐funded, grant number 754462) and through The Netherlands Organization for Scientific Research (NWO) via VICI grant 016.150.366 and via Gravity Program 024.001.035.
M. Wolter, D. Valenti, P. J. Cossar, L. M. Levy, S. Hristeva, T. Genski, T. Hoffmann, L. Brunsveld, D. Tzalis, C. Ottmann, Angew. Chem. Int. Ed. 2020, 59, 21520.
Contributor Information
Dr. Dimitrios Tzalis, Email: DTzalis@taros.de.
Prof. Dr. Christian Ottmann, Email: C.Ottmann@tue.nl.
References
- 1. Milroy L.-G., Grossmann T. N., Hennig S., Brunsveld L., Ottmann C., Chem. Rev. 2014, 114, 4695–4748. [DOI] [PubMed] [Google Scholar]
- 2. Scott D. E., Bayly A. R., Abell C., Skidmore J., Nat. Rev. Drug Discovery 2016, 15, 533–550. [DOI] [PubMed] [Google Scholar]
- 3. Schapira M., Calabrese M. F., Bullock A. N., Crews C. M., Nat. Rev. Drug Discovery 2019, 18, 949–963. [DOI] [PubMed] [Google Scholar]
- 4. Arkin M. R., Tang Y., Wells J. A., Chem. Biol. 2014, 21, 1102–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Valeur E., Guéret S. M., Adihou H., Gopalakrishnan R., Lemurell M., Waldmann H., Grossmann T. N., Plowright A. T., Angew. Chem. Int. Ed. 2017, 56, 10294–10323; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10428–10459. [Google Scholar]
- 6. Andrei S. A., Sijbesma E., Hann M., Davis J., O'Mahony G., Perry M. W. D., Karawajczyk A., Eickhoff J., Brunsveld L., Doveston R. G., Milroy L.-G., Ottmann C., Expert Opin. Drug Discovery 2017, 12, 925–940. [DOI] [PubMed] [Google Scholar]
- 7. Morabito F., Skafi M., Recchia A. G., Kashkeesh A., Hindiyeh M., Sabatleen A., Morabito L., Alijanazreh H., Hamamreh Y., Gentile M., Expert Opin. Pharmacother. 2019, 20, 487–494. [DOI] [PubMed] [Google Scholar]
- 8. Li J., Kim S. G., Blenis J., Cell Metab. 2014, 19, 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Price A. J., Howard S., Cons B. D., Essays Biochem. 2017, 61, 475–484. [DOI] [PubMed] [Google Scholar]
- 10. Draxler S. W., Bauer M., Eickmeier C., Nadal S., Nar H., Rangel Rojas D., Seeliger D., Zeeb M., Fiegen D., J. Med. Chem. 2020, 63, 5856—5864. [DOI] [PubMed] [Google Scholar]
- 11. Hassaan E., Eriksson P.-O., Geschwindner S., Heine A., Klebe G., ChemMedChem 2020, 15, 324–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Erlanson D. A., Braisted A. C., Raphael D. R., Randal M., Stroud R. M., Gordon E. M., Wells J. A., Proc. Natl. Acad. Sci. USA 2000, 97, 9367–9372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Erlanson D. A., Davis B. J., Jahnke W., Cell Chem. Biol. 2019, 26, 9–15. [DOI] [PubMed] [Google Scholar]
- 14. Dalton S. E., Campos S., ChemBioChem 2020, 21, 1080–1100. [DOI] [PubMed] [Google Scholar]
- 15. Sijbesma E., Hallenbeck K. K., Leysen S., de Vink P. J., Skóra L., Jahnke W., Brunsveld L., Arkin M. R., Ottmann C., J. Am. Chem. Soc. 2019, 141, 3524–3531. [DOI] [PubMed] [Google Scholar]
- 16. Pettinger J., Jones K., Cheeseman M. D., Angew. Chem. Int. Ed. 2017, 56, 15200–15209; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15398–15408. [Google Scholar]
- 17. Hacker S. M., Backus K. M., Lazear M. R., Forli S., Correia B. E., Cravatt B. F., Nat. Chem. 2017, 9, 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cuesta A., Taunton J., Annu. Rev. Biochem. 2019, 88, 365–381. [DOI] [PubMed] [Google Scholar]
- 19. Oksenberg D., Dufu K., Patel M. P., Chuang C., Li Z., Xu Q., Silva-Garcia A., Zhou C., Hutchaleelaha A., Patskovska L., Patskovsky Y., Almo S. C., Sinha U., Metcalf B. W., Archer D. R., Br. J. Haematol. 2016, 175, 141–153. [DOI] [PubMed] [Google Scholar]
- 20. Taniguchi K., Karin M., Nat. Rev. Immunol. 2018, 18, 309–324. [DOI] [PubMed] [Google Scholar]
- 21. Wolter M., de Vink P., Neves J. F., Srdanovic S., Higuchi Y., Kato N., Wilson A. J., Landrieu I., Brunsveld L., Ottmann C., J. Am. Chem. Soc. 2020, 142, 11772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Obsil T., Obsilova V., Semin. Cell Dev. Biol. 2011, 22, 663–672. [DOI] [PubMed] [Google Scholar]
- 23. Stevers L. M., Sijbesma E., Botta M., MacKintosh C., Obsil T., Landrieu I., Cau Y., Wilson A. J., Karawajczyk A., Eickhoff J., Davis J., Hann M., O'Mahony G., Doveston R. G., Brunsveld L., Ottmann C., J. Med. Chem. 2018, 61, 3755–3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kilambi K. P., Gray J. J., Biophys. J. 2012, 103, 587–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lyskov S., Chou F.-C., Conchúir S. Ó., Der B. S., Drew K., Kuroda D., Xu J., Weitzner B. D., Renfrew P. D., Sripakdeevong P., Borgo B., Havranek J. J., Kuhlman B., Kortemme T., Bonneau R., Gray J. J., Das R., PLoS ONE 2013, 8, e63906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Andrei S. A., de Vink P., Sijbesma E., Han L., Brunsveld L., Kato N., Ottmann C., Higuchi Y., Angew. Chem. Int. Ed. 2018, 57, 13470–13474; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 13658–13662. [Google Scholar]
- 27. Valenti D., Neves J. F., Cantrelle F.-X., Hristeva S., Lentini Santo D., Obšil T., Hanoulle X., Levy L. M., Tzalis D., Landrieu I., Ottmann C., MedChemComm 2019, 10, 1796–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary