

Abstract
Animals can sense the presence of microbes in their tissues and mobilize their own defenses by recognizing and responding to conserved microbial structures (often called microbe-associated molecular patterns (MAMPs)). Successful host defenses may kill the invaders, yet the host animal may fail to restore homeostasis if the stimulatory microbial structures are not silenced. Although mice have many mechanisms for limiting their responses to lipopolysaccharide (LPS), a major Gram-negative bacterial MAMP, a highly conserved host lipase is required to extinguish LPS sensing in tissues and restore homeostasis. We review recent progress in understanding how this enzyme, acyloxyacyl hydrolase (AOAH), transforms LPS from stimulus to inhibitor, reduces tissue injury and death from infection, prevents prolonged post-infection immunosuppression, and keeps stimulatory LPS from entering the bloodstream. We also discuss how AOAH may increase sensitivity to pulmonary allergens. Better appreciation of how host enzymes modify LPS and other MAMPs may help prevent tissue injury and hasten recovery from infection.
Keywords: lipid A, lipopolysaccharide (LPS), Toll-like receptor 4 (TLR4), inflammation, tolerance, macrophage, natural killer cells (NK cells), colitis, acyloxyacyl hydrolase, AOAH, CD14
Animals can recognize and respond to bacteria that invade their tissues by sensing the invaders' MAMPs. Pro- and anti-inflammatory cytokines and other mediators are produced, neutrophils and other leukocytes are recruited to the infected site, blood flow to the tissue increases, and increased vascular endothelial permeability allows influx of antibodies, complement, and other molecules from the bloodstream into the tissue. Both extracellular (e.g. complement and antibodies) and intracellular mechanisms contribute to bacterial killing. The inflammatory response may also produce tissue injury and lead to harmful immunosuppression.
Although these features of the antibacterial host response are well-understood, much less is known about how the inflammatory process resolves, tissue injury and prolonged immunosuppression are avoided, and normal MAMP responsiveness is restored. Recent studies have shown that lipid mediators can help resolve inflammation following trauma (1, 2), but the studies reviewed here provide evidence that, if infection is the inciting event, it is also necessary to eliminate or inhibit the stimulatory MAMP(s). Insight into this aspect of recovery from MAMP-induced inflammation has come from studying how mice silence lipopolysaccharide (LPS, endotoxin), a potent Gram-negative bacterial MAMP that has long been implicated in the pathogenesis of infection-induced inflammation, sepsis, and chronic diseases (3, 4).
As Lewis Thomas wrote in The Lives of a Cell (4), “The Gram-negative bacteria … display lipopolysaccharide molecules in their walls, and these macromolecules are read by our tissues as the very worst of bad news …. There is nothing intrinsically poisonous about endotoxin, but it must look awful, or feel awful, when sensed by cells.” In fact, many host molecules can prevent cells from sensing LPS: anti-LPS antibodies, proteins that bind and sequester LPS (bactericidal permeability-increasing protein (5), lactoferrin, cathelecidin, and plasma lipoproteins), an intracellular phosphatase (6), and others (7). Remarkably, none of these molecules—alone or together—can completely silence LPS in mice, and bacteria do not usually destroy their own molecules, especially large macromolecules (8). Instead, LPS silencing is accomplished by a highly conserved host lipase, acyloxyacyl hydrolase (AOAH). Named for its chemical sites of action (9), AOAH removes from LPS the fatty acyl chains that are essential for its immunostimulatory activity and transforms this potent agonist into an effective LPS antagonist.
Here we review how LPS is sensed by animal cells, the importance of LPS acylation in determining the agonistic potency of LPS, the structure and enzymatic properties of AOAH, and evidence that AOAH plays significant roles in both moderating and terminating host inflammatory responses to LPS and Gram-negative bacteria. Recent findings have pointed to important roles for AOAH in limiting inflammatory responses to Gram-negative bacteria, restoring normal immune responsiveness after LPS exposure, preventing stimulatory LPS from entering the bloodstream, and reducing the agonistic activity of intestinal LPS. We then describe human genetic evidence that AOAH may be an “essential” gene and studies that have identified a unique trans (cross-chromosomal) regulatory mechanism and potential associations with colitis and asthma.
Sensing LPS
The LPS produced by most Gram-negative bacteria has a polysaccharide chain of variable length that is anchored to the bacterial outer membrane by the lipid A moiety (10) (Fig. 1). Animal cells sense extracellular LPS through the receptor known as MD2-TLR4 (myeloid differentiation factor 2–Toll-like receptor 4). Other host proteins (mainly LPS-binding protein (LBP), phospholipid transfer protein (11, 12), soluble and/or membrane CD14 (sCD14 and/or mCD14), and albumin (13)) can extract individual LPS molecules from bacterial membranes and transfer them to cell surface (or endosomal) MD-2–TLR4. (Fig. 2A) Activation occurs most potently (i.e. at picomolar concentrations (14)) when LPS molecules with hexaacyl lipid A (Fig. 1) form ternary complexes with MD-2–TLR4. These ternary complexes form stable dimers that promote assembly of multiprotein intracellular signaling complexes and initiate intracellular signal/transduction (15). MyD88-dependent signaling is initiated by activated receptor complexes at the cell surface, whereas MyD88-independent (TRIF-dependent) signaling is triggered by activated LPS–MD-2–TLR4 receptor complexes within endosomes (16). Together, these signals initiate inflammatory cellular and humoral responses to kill the invading bacteria.
Figure 1.
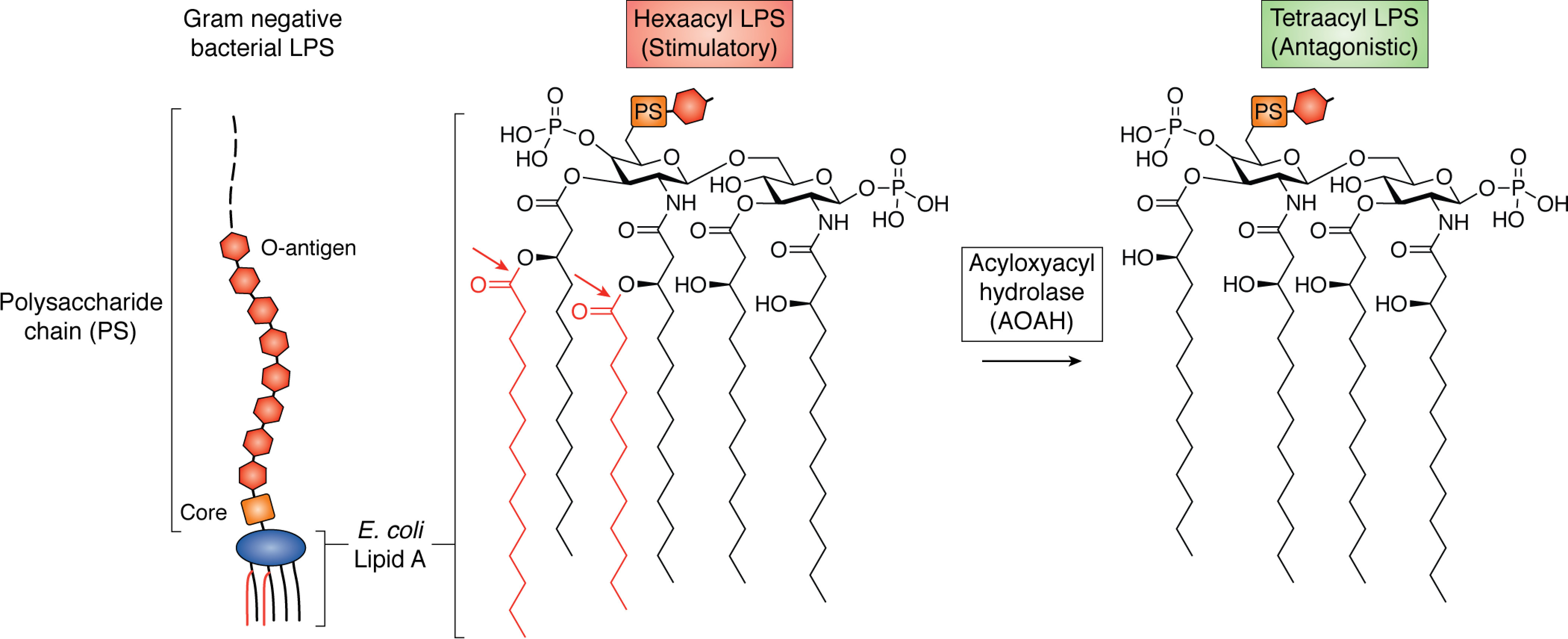
AOAH removes secondary acyl chains from LPS. E. coli LPS and lipid A are shown. The diglucosamine backbone is phosphorylated at 1 and 4′, and four primary 3-hydroxy fatty acyl chains are attached to the backbone in ester or amide linkage. Secondary acyl chains (red), usually myristate or laurate, are attached via acyloxyacyl linkage to two of the primary chains. Six acyl chains and both phosphates are required for optimal recognition by the MD-2–TLR4 receptor on animal cells. AOAH cleaves the acyloxyacyl linkages (arrows), converting stimulatory hexaacyl LPS into antagonistic tetraacyl LPS.
Figure 2.
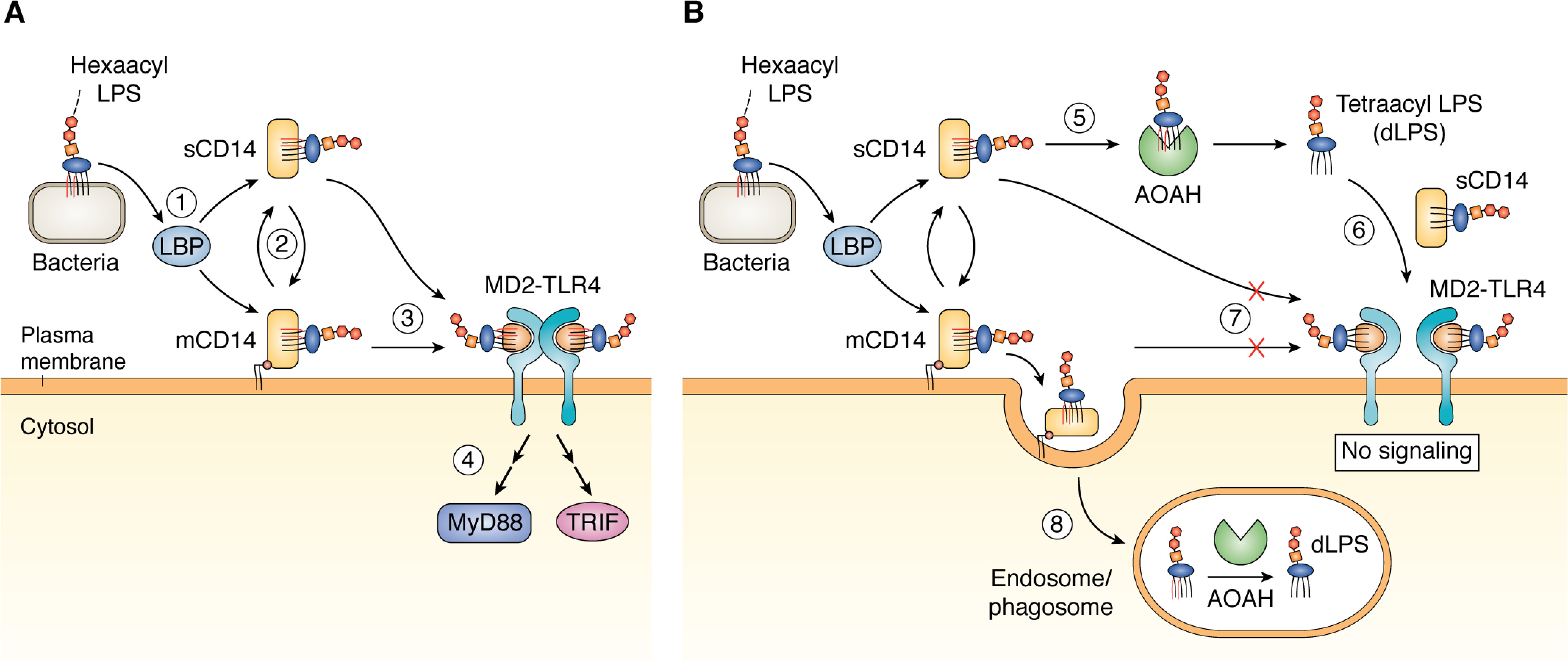
A, soluble cofactors enable LPS signaling. A single Gram-negative bacterium or outer membrane vesicle can contain >106 LPS molecules, all present in the outer leaflet of the outer membrane. 1, LBP binds to membrane particles containing hexaacyl (stimulatory) LPS and initiates the extraction and transfer of LPS monomers to sCD14 or mCD14. 2, LPS can be exchanged between sCD14 and mCD14. Either sCD14 or mCD14 can transfer the LPS monomer to MD2-TLR4 (3), which initiates TLR4 signaling following formation and dimerization of LPS-MD2-TLR4 ternary complexes (see text for more details) (4). B, AOAH deacylates LPS. 5, sCD14 can transfer LPS to mCD14 or to extracellular AOAH, which removes the two secondary acyl chains (red) to produce tetraacyl (≪stimulatory, antagonistic) dLPS. 6, dLPS is transferred back to sCD14 (no protein-free form of either hexaacyl or tetraacyl LPS is detected). sCD14 can transfer the tetraacyl dLPS to MD2-TLR4, but this ternary complex, unlike that of hexaacyl LPS-MD-2–TLR4 (Fig. 2A), does not form a stable dimerized ternary complex that can initiate signaling. 7, dLPS may also compete with hexaacyl LPS for binding MD2-TLR4. 8, mCD14 can promote uptake of LPS into an endosomal compartment, where it can be deacylated by AOAH.
From stimulus to inhibitor: the importance of LPS acylation
Most of the aerobic Gram-negative bacteria that inhabit mucosal surfaces and/or commonly cause disease in animals produce LPS that has hexaacyl lipid A (3). In Escherichia coli LPS, for example, myristoyl and lauroyl chains are attached via acyloxyacyl linkages to the hydroxyl functions of two of the four diglucosamine-linked 3-hydroxymyristoyl chains (Fig. 1). Some other Gram-negative aerobes and most Gram-negative anaerobes (e.g. Bacteroides species), produce pentaacyl LPS, a weaker MD-2/TLR4 agonist (17) that may inhibit hexaacyl LPS signaling (18). The fatty acid composition can vary both within and between species (19); in Vibrio cholerae and Pseudomonas aeruginosa, for example, secondary acyl chains can be hydroxylated (20, 21).
AOAH cleaves only the acyloxyacyl linkages, removing the secondary (or “piggyback”) acyl chains to produce pentaacyl or tetraacyl lipid A (Fig. 1). An early study found that human AOAH preferentially removed shorter (laurate > myristate), and nonhydroxylated (laurate > 2-OH–laurate) secondary chains (22). The resulting tetraacyl LPS still binds the MD-2–TLR4 receptor with comparable affinity, but the probability that the tetraacyl LPS–MD-2–TLR4 complex will form a stimulatory ternary complex is markedly reduced (23–25). This may reflect ligand acylation-dependent differences in the ability of the resulting ternary complexes to form the stable higher-order structures required for signal transduction (15).
The extraction and transfer of LPS monomers from LPS-rich surfaces (e.g. the bacterial outer membrane) to CD14 is necessary not only for efficient delivery of activating (hexaacyl) LPS to MD-2–TLR4 but also to present the LPS as a substrate for AOAH (26) (Fig. 2B). The apparent “Km” is 100-fold lower for transfer of the LPS monomer from sCD14 to MD-2–TLR4 than it is for deacylation by AOAH of LPS bound to sCD14 (26). However, the higher levels of sCD14 versus MD-2 often found in tissues suggest that monomeric LPS:sCD14 intermediates could accumulate, followed by AOAH action and delivery of much less stimulatory (“≪stimulatory”) AOAH-deacylated LPS (dLPS) to MD-2–TLR4 (Fig. 2B). Taken together, these properties seem most compatible with the ability of LPS at picomolar concentrations to activate potent immune responses acutely followed by a subsequent dLPS-assisted return to “resting” conditions.
Deacylation by AOAH reduces the stimulatory potency of hexaacyl LPS molecules by as much as 50–100-fold (27–30). Moreover, dLPS can competitively reduce binding of hexacyl LPS to MD-2–TLR4 and thereby inhibit formation of the hexaacyl LPS–MD-2–TLR4 ternary complexes needed to activate signaling (Fig. 2B). dLPS inhibition of LPS stimulation has been demonstrated in vitro using human umbilical vein endothelial cells (28, 31), human neutrophils (30), and murine macrophages and splenocytes (29). Even substoichiometric concentrations of dLPS may inhibit LPS signaling (32). In addition, dLPS can compete with LPS at each of the preceding steps involved in presenting LPS to MD-2–TLR4 (33) (i.e. by engaging LBP and/or CD14) (34). Teghanemt et al. (35) and Gioannini et al. (25, 26) demonstrated directly that dLPS interacts with MD-2 to form a complex with markedly reduced ability to activate TLR4, therefore acting as an MD-2–TLR4 antagonist. Activation of LPS-sensing cytosolic caspases by LPS also appears to depend upon the acylation state of LPS (36–38). An effect of AOAH-dependent LPS deacylation on regulating the activation of this cytosolic recognition/response system has not been reported.
Independent evidence for the importance of lipid A acylation in host–Gram-negative bacteria interactions has come from Gram-negative pathogens that can make hypoacylated (<6 acyl chains) LPS (39). Intracellular Shigella flexneri bacteria can avoid activating an inflammatory response by reducing LPS acylation (40), Salmonella typhimurium may produce a pentaacyl LPS with low stimulatory activity (41), Francisella spp. make tetraacyl LPS (42), and Yersinia pestis switches production from hexaacyl to tetraacyl LPS at mammalian body temperatures and prevents mobilization of TLR4-dependent host defenses (43, 44). For these intracellular bacteria, producing hypoacyl lipid A/LPS is an immune evasion mechanism.
As will be discussed below, AOAH-mediated transformation of LPS from hexaacyl to pentaacyl or tetraacyl can be anti-inflammatory (blunting LPS signaling via TLR4) as well as restorative (reestablishing normal host responsiveness to infection by inactivating stimulatory LPS). First, we summarize some of the enzyme's important features.
AOAH biosynthesis, structure
AOAH is a glycoprotein with Mr = 52,000–60,000 (45, 46). Its two potential subunits are joined by a single disulfide bond before the precursor peptide is cleaved to form the mature protein (46). The smaller subunit is a member of a protein family that includes several proteins that act at lipid-water or lipid-air interfaces or as cofactors for lysosomal hydrolases (47). The larger subunit (∼50,000 Da) is a GDSL lipase; the best-characterized member of this family is platelet-activating factor acetylhydrolase (48). When AOAH cDNA was expressed in cultured fibroblasts, much of the precursor protein was secreted, internalized by other cells, and cleaved within an acidic endosomal-lysosomal compartment to form the mature enzyme (49), which was more active toward LPS than was the precursor (48, 49).
Analysis of the crystal structure led Gorelik et al. (48) to conclude that “LPS binds to AOAH with its fatty acid tails covered by the hydrophobic pocket formed by the saposin and catalytic domains and a secondary (acyloxyacyl) chain buried in the hydrophobic tunnel at the active site.” A space-filling model resembles the “fingers in glove” appearance of lipid A inserted into MD-2 (50).
The DNA sequence that encodes AOAH has been found in all vertebrates studied except fish, which produce a TLR4 that is not activated by LPS (51), and also in many invertebrates (52). It is likely that AOAH was the LPS esterase originally described in the slime mold Dictyostelium discoideum (53), which also has lipid A–deacylating amidases and uses Gram-negative bacteria as a foodstuff (54). Although AOAH can act on other lipids, including phospholipids (55) and some bacterial lipoproteins (48), its activity toward lipid A/LPS is unique.
Cellular sources of AOAH
Although AOAH was intially found in neutrophils (9), cellular levels of AOAH are often lower in circulating and exudate neutrophils than in monocytes/macrophages, hepatic Kupffer cells (56), immature dendritic cells (57, 58), and NK cells. AOAH expression has also been found in mucosal ILC1 cells, which are closely related to NK cells but lack granulysin and perforin and have a genomic “superenhancer” region that includes part of the AOAH gene (59). Naive T cells just released from the thymus express AOAH mRNA along with mRNAs for complement receptors, IL-8, and TLR1 (60), consistent with a role in antimicrobial defense.
Macrophages internalize LPS into an endosomal compartment that contains AOAH (61); mCD14 can enable this uptake (62) (Fig. 2B). LPS deacylation occurs over several hours and can be inhibited by agents that prevent endosomal acidification (61), in keeping with the enzyme's low pH optimum for LPS deacylation in vitro. In general, cellular and secreted levels of AOAH increase for 1 or 2 days after the cells are stimulated with LPS or other agonists. Murine peritoneal macrophages greatly increased AOAH synthesis when they were stimulated in vitro with interferon-γ or LPS (63). Murine alveolar macrophages also constitutively express little AOAH, but 18 h following stimulation with LPS, either in vivo or ex vivo, they increased AOAH mRNA expression 40–100-fold (64). According to a recent comparison of mRNA abundance in murine and human brain cells, AOAH mRNA is much more highly expressed in human than in murine microglia (65). AOAH mRNA abundance increased in embryonic murine microglia, provided that the mother had received a standard (not germ-free) diet (66), pointing to a possible role for AOAH or translocated LPS (see below) in embryonic development.
Janelsins et al. (67) reported that resident colonic dendritic cells (DCs) express more AOAH than do DCs in other murine organs; AOAH mRNA abundance decreased when antibiotics were added to the drinking water and was barely detectable in colonic DCs from TLR4−/− mice. AOAH activity also increased when murine marrow–derived DCs were treated with LPS (57). Like peritoneal macrophages, the marrow-derived DCs deacylated the LPS in phagocytosed E. coli (57). Murray et al. (58) recently reported that circulating myeloid precursor DCs increased AOAH mRNA abundance 4-fold in people living with (latent) HIV.
There is an important exception to the generalization that AOAH is mainly produced by myeloid or lymphoid cells. When radiolabeled AOAH cDNA was used to perform a Northern blot analysis of mouse tissues, the greatest signal came from the kidney (68). In situ hybridization revealed that AOAH mRNA was localized to proximal tubule cells; further study found that mice and humans secrete mature AOAH into the urine. The AOAH could be taken up by bladder cells and deacylate LPS. A protective role for AOAH seems likely in the urinary tract, where E. coli and other aerobic Gram-negative bacteria are the most common pathogens.
AOAH can also deacylate LPS extracellularly (Fig. 2B). In ex vivo studies of a sterile inflammatory exudate induced in rabbits, Weinrauch et al. (69) showed the presence of AOAH in both inflammatory cells (macrophages > neutrophils) and in cell-free inflammatory fluid. Conversion of LPS to dLPS was demonstrable using either purified LPS or intact E. coli containing metabolically prelabeled LPS; it was greatest when macrophages were present (70).
In summary, although AOAH has been studied most often in phagocytes that respond to LPS and contribute to innate antibacterial defense—macrophages, DCs, and neutrophils—it is also produced by Kupffer cells, NK cells, ILC1 cells, recent thymic immigrant T cells, microglia, and renal proximal tubule cells. Cell stimulation is generally followed by gradual increases in AOAH abundance from low constitutive levels to maximal production within a day or two.
AOAH prevents, moderates, and terminates responses to LPS
Evidence that AOAH influences how animals respond to LPS and Gram-negative bacteria has come from studies in transgenic AOAH-producing (71) and Aoah−/− (57) mice. Significant roles have been found for preventing hexaacyl LPS from entering the bloodstream from a tissue site or the intestine, moderating and shortening the inflammatory response to Gram-negative bacteremia and pulmonary challenge, and terminating the immunosuppressive period of cellular reprogramming (“tolerance”) that follows exposure to LPS.
AOAH prevents stimulatory LPS from entering the bloodstream
The pathological consequences of Gram-negative bacteremia and endotoxemia have ranged from metabolic diseases to septic shock and death. Like Gram-negative bacteria, LPS moves via lymphatics from a subcutaneous injection site to draining lymph nodes and continues via lymphatics into the bloodstream. AOAH-dependent deacylation, largely carried out by macrophages, can inactivate most of the LPS before it reaches the circulation. When Aoah−/− and Aoah+/+ mice were compared, subcutaneously injected LPS induced more robust proliferation of B cells and plasmablasts in draining lymph nodes of Aoah−/− mice and increased their blood IgM and IgG3 levels for weeks (72, 73), confirming that AOAH participates in limiting responses to LPS even before the LPS molecules reach draining lymph nodes and the bloodstream.
Many investigators have studied the role of microbiota-derived LPS (endotoxemia) in the pathogenesis of diseases such as atherosclerosis, diabetes mellitus, metabolic syndrome, and others (74–77). There is evidence for translocation of stimulatory LPS into the bloodstream from the colon (78), where intestinal AOAH is most abundant (79). Qian et al. (79) obtained indirect evidence for AOAH-dependent LPS inactivation by measuring TLR4-stimulating activity using a TLR4 reporter cell line (80). In stool, mesenteric lymph nodes, plasma, and lung, TLR4 stimulation was greater in Aoah−/− mice than in Aoah+/+ littermate controls. The stimulatory activity decreased when polymyxin B was added to inhibit LPS or when antibiotics that kill aerobic Gram-negative bacteria were added to the drinking water to reduce hexaacyl LPS abundance in the intestine (79).
Protection from systemic stimulation by translocated intestinal LPS has often been attributed to the ability of intestinal alkaline phosphatase to inactivate LPS by removing one or both phosphates from lipid A (Fig. 1) (81, 82). With the recent discovery that certain primary (glucosamine-linked) acyl chains must be removed from lipid A before intestinal alkaline phosphatase can cleave either of the lipid A phosphates (Fig. 1) (83), AOAH became the most likely host mechanism for reducing the stimulatory potency of LPS molecules in the intestine by modifying lipid A structure. As noted above, its production by colonic DCs increases with hexaacyl LPS exposure.
As reported by d'Hennezel et al. (84), the net impact of the abundant Bacteroides (largely pentaacyl and monophosphoryl (85)) LPS in the human colon is to decrease TLR4 signaling by competing with TLR4-activating hexaacyl LPS (18, 86). Although Bacteroides genus tetraacyl lipid A was also found in the colon, the extent to which AOAH contributes to reducing levels of stimulatory intestinal LPS and generating increased levels of competing, weakly agonistic dLPS is not known. Colonic AOAH may help prevent excessive LPS-induced tolerance in DCs (see below), inflammation stimulated by excess hexaacyl LPS (as may accumulate during colitis (80)), and translocation of hexaacyl LPS into the bloodstream (79).
AOAH decreases LPS-induced inflammation in tissues
Although AOAH may be synthesized slowly after the onset of infection or LPS challenge, the enzyme can play a significant role in preventing potentially harmful consequences of the inflammatory response. For example, AOAH shortened the duration of LPS– or Klebsiella pneumoniae–induced pulmonary inflammation in models of experimental pneumonia (64). In the Aoah−/− animals, persistent LPS stimulated alveolar macrophages and epithelial cells to recruit more neutrophils to the lung, greatly delaying recovery and decreasing survival. AOAH has also improved survival from experimental Gram-negative bacteremia (71), hastened recovery in bacteremia survivors (87), and moderated LPS-induced hepatic inflammation (56, 71, 88).
In addition to the tissue injury that can be produced by excessive host responses to infection, tissue damage can be caused when host cells produce potentially toxic mediators, such as oxidized phospholipids and lysophospholipids. For example, dead cells can release oxidized phosphatidylcholine that mCD14 can deliver into DCs and activate an inflammatory response (89). Although AOAH can deacylate phospholipids and lysophospholipids in vitro (48, 55), a role for the enzyme in inactivating these or other lipids in vivo has not been established.
AOAH shortens endotoxin tolerance/immunosuppression
As if to prevent damage from “friendly fire,” the initial inflammatory response to LPS is typically followed by a state of tolerance (cellular reprogramming) (90–92) during which many of the host animal's pro-inflammatory responses to sensing LPS are diminished while some anti-inflammatory responses increase. Host defenses are typically weaker during this period of relative immunosuppression. Homeostasis is usually restored within a few days, and the animal's responses to subsequent LPS exposure (or microbial invasion) return to normal.
When Lu et al. gave a small intraperitoneal dose of LPS to Aoah−/− mice, however, the animals remained tolerant for many weeks and were more likely than Aoah+/+ mice to die from live E. coli challenge (93). Further study found that stimulatory LPS molecules persisted for many weeks in the peritoneal cavities of Aoah−/− mice and continued to tolerize macrophages there (94). AOAH was required to end the tolerant state (Fig. 3). It seems unlikely that long-term epigenetic changes prolonged tolerance because the tolerant peritoneal macrophages quickly regained responsiveness when they were transferred to a nontolerant, Aoah+/+ animal (94).
Figure 3.
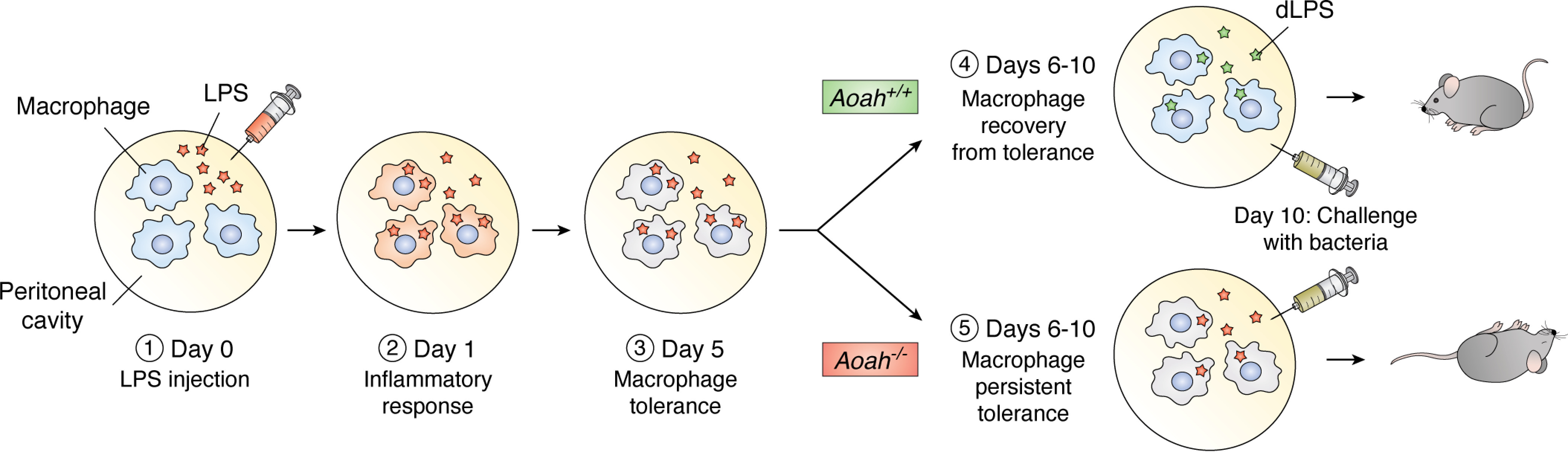
AOAH promotes recovery from tolerance. 1, LPS is injected into the peritoneal cavity of a naive Aoah+/+ or Aoah−/− mouse. 2, the resident and recruited peritoneal macrophages are stimulated by LPS and may internalize it. 3, after the initial inflammatory responses subside, the macrophages become tolerant; bioactive LPS (orange stars) remains. 4, in the Aoah+/+ mouse, AOAH transforms LPS to dLPS (green stars), and the macrophages recover from tolerance. 5, if AOAH is absent, LPS cannot be inactivated (orange stars). Bioactive LPS in extracellular fluid keeps stimulating macrophages locally, preventing them from recovering from tolerance. These mice are more likely to respond slowly and die quickly after they are challenged with live E. coli on day 10.
In keeping with these findings in living mice, Mages et al. (95) found large AOAH increases in LPS-tolerant marrow macrophages in vitro. In addition, stimulating cells with one TLR agonist may invoke tolerance to others (“cross-tolerance”), plausibly a general mechanism for reducing cell activation during recovery from infection. Lu et al. (57) found that AOAH activity in murine DCs increased when the cells were treated not only with LPS (TLR4) but also with TLR agonists CpG oligonucleotides (TLR9) and Micrococcus luteus (TLR2)—but not with inflammatory cytokines. AOAH production may increase during tolerance elicited by many different bacterial MAMPs.
Another role for AOAH in the regulation of LPS-induced tolerance was reported by Janelsins et al. (67), who found that CD103+CD11b+ALDH− colonic dendritic cells express much more AOAH than do DCs in other organs. Aoah−/− colonic DCs expressed more features of tolerance—less IL-6 production, less Th17 polarization, and greater Treg cell induction—than did colonic DCs from Aoah+/+ mice.
There is also evidence that intestinal AOAH can influence tolerance to pulmonary allergens (79). Previous studies had found that gut-derived or intratracheally administered hexaacyl LPS and other TLR agonists can decrease allergic TH2 responses linked to the development of asthma, whereas gut-derived pentaacyl LPS may permit them (96–99); in other studies, low LPS doses elicited TH2 allergic responses (e.g. IL-4, IL-5, and IL-10) when mice were challenged with house dust mite (HDM) extract, whereas high doses did not (100, 101). In keeping with these results, Qian et al. (79) found that LPS translocating from the intestine to the lungs reduced pulmonary epithelial cell TH2 responses to HDM, whereas translocating dLPS enhanced allergen sensitivity (Fig. 4).
Figure 4.

Intestinal AOAH modifies pulmonary immune responses. Colonic AOAH reduces translocation of bioactive LPS to the lungs, decreasing induction of tolerance in pulmonary epithelial cells to Th2 allergens, such as HDM.
Taken together, the in vivo studies have shown that AOAH may play important roles in terminating LPS- and Gram-negative bacteria-induced tolerance, restoring innate immune responsiveness, and influencing host susceptibility to pulmonary allergens.
An essential gene? Disease associations?
If AOAH is required to silence LPS in vivo in mice, does it play a role in human disease causation? Although intronic SNPs in the AOAH gene have been associated with chronic rhinosinusitis (102), asthma (103), and early onset Alzheimer's disease (104), how these mutations might alter AOAH abundance or function is not understood. Taking a different approach, Gorelik et al. determined the three-dimensional structure of AOAH, characterized its active site and catalytic mechanism, and used this information to derive a list of mutations that should alter the enzyme's function (48). A comparison of this list with the mutations listed in the human gnomAD browser (Broad Institute, 2020) did not identify a homozygote (missense or loss of function mutation) at any of the amino acid positions predicted to alter the enzyme's activity. This observation and the gene's conservation during evolution suggest that AOAH might be an “essential” gene. Although an essential role has not been apparent in AOAH-deficient C57Bl/6J or C3H/HeN laboratory mice, their ability to recover from natural infections and many other challenges has not been tested.
It is also possible that AOAH production differs from individual to individual, and an interesting regulatory mechanism has been suggested by human gene-disease association studies. Several research groups have found highly significant associations between polymorphisms in the same intron in the HLA-DR region on chromosome 6 (6p21) and AOAH expression on chromosome 7 (7p14.2). In one study, the polymorphism was also associated with reduced risk of asthma (105), in another study it was associated with lung cancer (106), and in several other studies (107–109) a highly significant association was found with colitis. The most detailed analysis was by Fairfax et al. (109), who found that the minor C allele at an intronic SNP (rs28366298) was specific to the HLA-DRB1*04, HLA-DRB1*07, and HLA-DRB1*09 alleles and that in each instance, AOAH mRNA abundance in peripheral blood monocytes was reduced and colitis risk was increased.
In both mice and humans, AOAH deficiency has been associated provisionally with less severe asthma and greater risk of colonic inflammation. Discovering how AOAH expression on chromosome 7 is regulated by an intronic sequence on chromosome 6 is a fascinating challenge. Studies to date have not found evidence for regulation by cis-mediation, noncoding RNA, or an intermediary gene product.
Conclusions
As Elsbach noted decades ago (7), “Effective host defense … requires that the inflammation-generating foreign materials be removed and the signals turned off.” We now know that many host molecules can transiently inhibit LPSs without permanently silencing them, whereas AOAH irreversibly transforms stimulatory LPS into a weak agonist that can competitively inhibit LPS signaling. Understanding how AOAH contributes to silencing LPS and possibly other lipid-containing MAMPs (such as bacterial outer membrane lipoprotein, a potent TLR2 agonist (48)) may yield insights applicable to MAMPs from other microbes, some of which may also require biochemical modification for sustained inactivation (110, 111). Just as “source control” (e.g. killing the bacteria, draining the abscess) is a critical step in managing infected patients who develop sepsis (112, 113), permanently silencing the patients' inciting MAMPs may be needed to prevent prolonged infection sequelae (114) and coordinate with other resolving mechanisms (1, 2) to clear the infection battlefield and restore homeostasis.
Acknowledgments
We dedicate this review to Emil C. Gotschlich, whose suggestion prompted the search for the LPS lipase; to Paul D. Rick, whose Salmonella mutant enabled its discovery and assay; and to the memory of Peter Elsbach (1924–2020), whose pioneering studies on the mechanisms, regulation, and possible roles of bacterial digestion by host enzymes set the stage for the studies described here. We also thank Wei Jiang and Luciana Giono for contributing to the figures.
Funding and additional information—This work was supported by National Natural Science Foundation of China Grants 31570910, 31770993, and 91742104 (to M. L.) (Fudan), National Institutes of Health Grants R01 AI18188 (to R. S. M.) and R01 AI059372 (to J. P. W.), the Division of Intramural Research, NIAID, National Institutes of Health (to R. S. M.), and Veterans Affairs Grant I01 BX000949/BX/BLRD (to J. P. W.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- MAMP
- microbe-associated molecular pattern
- LPS
- lipopolysaccharide
- AOAH
- acyloxyacyl hydrolase
- LBP
- LPS-binding protein
- sCD14
- soluble CD14
- mCD14
- membrane CD14
- dLPS
- AOAH-deacylated LPS
- NK
- natural killer
- DC
- dendritic cell
- IL
- interleukin
- HDM
- house dust mite.
References
- 1. Fullerton J. N., and Gilroy D. W. (2016) Resolution of inflammation: a new therapeutic frontier. Nat. Rev. Drug Discov. 15, 551–567 10.1038/nrd.2016.39 [DOI] [PubMed] [Google Scholar]
- 2. Serhan C. N., and Levy B. D. (2018) Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. J. Clin. Invest. 128, 2657–2669 10.1172/JCI97943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Munford R. S. (2008) Sensing Gram-negative bacterial lipopolysaccharides: a human disease determinant? Infect. Immun. 76, 454–465 10.1128/IAI.00939-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thomas L. (1974) The Lives of a Cell, Viking Press, New York [Google Scholar]
- 5. Elsbach P., Weiss J., Franson R. C., Beckerdite-Quagliata S., Schneider A., and Harris L. (1979) Separation and purification of a potent bactericidal/permeability-increasing protein and a closely associated phospholipase A2 from rabbit polymorphonuclear leukocytes: observations on their relationship. J. Biol. Chem. 254, 11000–11009 [PubMed] [Google Scholar]
- 6. Peterson A. A., and Munford R. S. (1987) Dephosphorylation of the lipid A moiety of Escherichia coli lipopolysaccharide by mouse macrophages. Infect. Immun. 55, 974–978 10.1128/IAI.55.4.974-978.1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Elsbach P. (2000) Mechanisms of disposal of bacterial lipopolysaccharides by animal hosts. Microbes Infect. 2, 1171–1180 10.1016/S1286-4579(00)01271-5 [DOI] [PubMed] [Google Scholar]
- 8. Elsbach P. (1980) Degradation of microorganisms by phagocytic cells. Rev. Infect. Dis. 2, 106–128 10.1093/clinids/2.1.106 [DOI] [PubMed] [Google Scholar]
- 9. Hall C. L., and Munford R. S. (1983) Enzymatic deacylation of the lipid A moiety of Salmonella typhimurium lipopolysaccharides by human neutrophils. Proc. Natl. Acad. Sci. U. S. A. 80, 6671–6675 10.1073/pnas.80.21.6671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Whitfield C., Williams D. M., and Kelly S. D. (2020) Lipopolysaccharide O-antigens—bacterial glycans made to measure. J. Biol. Chem. 295, 10593–10609 10.1074/jbc.REV120.009402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vesy C. J., Kitchens R. L., Wolfbauer G., Albers J. J., and Munford R. S. (2000) Lipopolysaccharide-binding protein and phospholipid transfer protein release lipopolysaccharides from Gram-negative bacterial membranes. Infect. Immun. 68, 2410–2417 10.1128/iai.68.5.2410-2417.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gautier T., Klein A., Deckert V., Desrumaux C., Ogier N., Sberna A. L., Paul C., Le Guern N., Athias A., Montange T., Monier S., Piard F., Jiang X. C., Masson D., and Lagrost L. (2008) Effect of plasma phospholipid transfer protein deficiency on lethal endotoxemia in mice. J. Biol. Chem. 283, 18702–18710 10.1074/jbc.M802802200 [DOI] [PubMed] [Google Scholar]
- 13. Esparza G. A., Teghanemt A., Zhang D., Gioannini T. L., and Weiss J. P. (2012) Endotoxin.albumin complexes transfer endotoxin monomers to MD-2 resulting in activation of TLR4. Innate Immun. 18, 478–491 10.1177/1753425911422723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gioannini T. L., Teghanemt A., Zhang D., Coussens N. P., Dockstader W., Ramaswamy S., and Weiss J. P. (2004) Isolation of an endotoxin-MD-2 complex that produces Toll-like receptor 4-dependent cell activation at picomolar concentrations. Proc. Natl. Acad. Sci. U. S. A. 101, 4186–4191 10.1073/pnas.0306906101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Latty S. L., Sakai J., Hopkins L., Verstak B., Paramo T., Berglund N. A., Cammarota E., Cicuta P., Gay N. J., Bond P. J., Klenerman D., and Bryant C. E. (2018) Activation of Toll-like receptors nucleates assembly of the MyDDosome signaling hub. eLife 7, e31377 10.7554/eLife.31377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kagan J. C., Su T., Horng T., Chow A., Akira S., and Medzhitov R. (2008) TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-β. Nat. Immunol. 9, 361–368 10.1038/ni1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Di Lorenzo F., De Castro C., Silipo A., and Molinaro A. (2019) Lipopolysaccharide structures of Gram-negative populations in the gut microbiota and effects on host interactions. FEMS Microbiol. Rev. 43, 257–272 10.1093/femsre/fuz002 [DOI] [PubMed] [Google Scholar]
- 18. Magnuson D. K., Weintraub A., Pohlman T. H., and Maier R. V. (1989) Human endothelial cell adhesiveness for neutrophils, induced by Escherichia coli lipopolysaccharide in vitro, is inhibited by Bacteroides fragilis lipopolysaccharide. J. Immunol. 143, 3025–3030 [PubMed] [Google Scholar]
- 19. Simpson B. W., and Trent M. S. (2019) Pushing the envelope: LPS modifications and their consequences. Nat. Rev. Microbiol. 17, 403–416 10.1038/s41579-019-0201-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hankins J. V., Madsen J. A., Giles D. K., Childers B. M., Klose K. E., Brodbelt J. S., and Trent M. S. (2011) Elucidation of a novel Vibrio cholerae lipid A secondary hydroxy-acyltransferase and its role in innate immune recognition. Mol. Microbiol. 81, 1313–1329 10.1111/j.1365-2958.2011.07765.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lo Sciuto A., Cervoni M., Stefanelli R., Spinnato M. C., Di Giamberardino A., Mancone C., and Imperi F. (2019) Genetic basis and physiological effects of lipid A hydroxylation in Pseudomonas aeruginosa PAO1. Pathogens 8, 291 10.3390/pathogens8040291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Erwin A. L., and Munford R. S. (1990) Deacylation of structurally diverse lipopolysaccharides by human acyloxyacyl hydrolase. J. Biol. Chem. 265, 16444–16449 [PubMed] [Google Scholar]
- 23. Ohto U., Fukase K., Miyake K., and Satow Y. (2007) Crystal structures of human MD-2 and its complex with antiendotoxic lipid IVa. Science 316, 1632–1634 10.1126/science.1139111 [DOI] [PubMed] [Google Scholar]
- 24. Kim H. M., Park B. S., Kim J. I., Kim S. E., Lee J., Oh S. C., Enkhbayar P., Matsushima N., Lee H., Yoo O. J., and Lee J. O. (2007) Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell 130, 906–917 10.1016/j.cell.2007.08.002 [DOI] [PubMed] [Google Scholar]
- 25. Gioannini T. L., Teghanemt A., Zhang D., Esparza G., Yu L., and Weiss J. (2014) Purified monomeric ligand.MD-2 complexes reveal molecular and structural requirements for activation and antagonism of TLR4 by Gram-negative bacterial endotoxins. Immunol. Res. 59, 3–11 10.1007/s12026-014-8543-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gioannini T. L., Teghanemt A., Zhang D., Prohinar P., Levis E. N., Munford R. S., and Weiss J. P. (2007) Endotoxin-binding proteins modulate the susceptibility of bacterial endotoxin to deacylation by acyloxyacyl hydrolase. J. Biol. Chem. 282, 7877–7884 10.1074/jbc.M605031200 [DOI] [PubMed] [Google Scholar]
- 27. Munford R. S., and Hall C. L. (1986) Detoxification of bacterial lipopolysaccharides (endotoxins) by a human neutrophil enzyme. Science 234, 203–205 10.1126/science.3529396 [DOI] [PubMed] [Google Scholar]
- 28. Riedo F. X., Munford R. S., Campbell W. B., Reisch J. S., Chien K. R., and Gerard R. D. (1990) Deacylated lipopolysaccharide inhibits plasminogen activator inhibitor-1, prostacyclin, and prostaglandin E2 induction by lipopolysaccharide but not by tumor necrosis factor-α. J. Immunol. 144, 3506–3512 [PubMed] [Google Scholar]
- 29. Erwin A. L., Mandrell R. E., and Munford R. S. (1991) Enzymatically deacylated Neisseria LPS inhibits murine splenocyte mitogenesis induced by LPS. Infect. Immun. 59, 1881–1887 10.1128/IAI.59.6.1881-1887.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nogare A. R., and Yarbrough W. C. Jr. (1990) A comparison of the effects of intact and deacylated lipopolysaccharide on human polymorphonuclear leukocytes. J. Immunol 144, 1404–1410 [PubMed] [Google Scholar]
- 31. Pohlman T. H., Munford R. S., and Harlan J. M. (1987) Deacylated lipopolysaccharide inhibits neutrophil adherence to endothelium induced by lipopolysaccharide in vitro. J. Exp. Med. 165, 1393–1402 10.1084/jem.165.5.1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kitchens R. L., Ulevitch R. J., and Munford R. S. (1992) Lipopolysaccharide (LPS) partial structures inhibit responses to LPS in a human macrophage cell line without inhibiting LPS uptake by a CD14-mediated pathway. J. Exp. Med. 176, 485–494 10.1084/jem.176.2.485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ryu J. K., Kim S. J., Rah S. H., Kang J. I., Jung H. E., Lee D., Lee H. K., Lee J. O., Park B. S., Yoon T. Y., and Kim H. M. (2017) Reconstruction of LPS transfer cascade reveals structural determinants within LBP, CD14, and TLR4-MD2 for efficient LPS recognition and transfer. Immunity 46, 38–50 10.1016/j.immuni.2016.11.007 [DOI] [PubMed] [Google Scholar]
- 34. Kitchens R. L., and Munford R. S. (1995) Enzymatically deacylated lipopolysaccharide (LPS) can antagonize LPS at multiple sites in the LPS recognition pathway. J. Biol. Chem. 270, 9904–9910 10.1074/jbc.270.17.9904 [DOI] [PubMed] [Google Scholar]
- 35. Teghanemt A., Zhang D., Levis E. N., Weiss J. P., and Gioannini T. L. (2005) Molecular basis of reduced potency of underacylated endotoxins. J. Immunol. 175, 4669–4676 10.4049/jimmunol.175.7.4669 [DOI] [PubMed] [Google Scholar]
- 36. Barker J. H., and Weiss J. P. (2019) Detecting lipopolysaccharide in the cytosol of mammalian cells: lessons from MD-2/TLR4. J. Leukoc. Biol. 106, 127–132 10.1002/JLB.3MIR1118-434R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lagrange B., Benaoudia S., Wallet P., Magnotti F., Provost A., Michal F., Martin A., Di Lorenzo F., Py B. F., Molinaro A., and Henry T. (2018) Human caspase-4 detects tetra-acylated LPS and cytosolic Francisella and functions differently from murine caspase-11. Nat. Commun. 9, 242 10.1038/s41467-017-02682-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shi J., Zhao Y., Wang Y., Gao W., Ding J., Li P., Hu L., and Shao F. (2014) Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192 10.1038/nature13683 [DOI] [PubMed] [Google Scholar]
- 39. Needham B. D., and Trent M. S. (2013) Fortifying the barrier: the impact of lipid A remodelling on bacterial pathogenesis. Nat. Rev. Microbiol. 11, 467–481 10.1038/nrmicro3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Paciello I., Silipo A., Lembo-Fazio L., Curcuru L., Zumsteg A., Noel G., Ciancarella V., Sturiale L., Molinaro A., and Bernardini M. L. (2013) Intracellular Shigella remodels its LPS to dampen the innate immune recognition and evade inflammasome activation. Proc. Natl. Acad. Sci. U. S. A. 110, E4345–E4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kawasaki K., Ernst R. K., and Miller S. I. (2004) 3-O-Deacylation of lipid A by PagL, a PhoP/PhoQ-regulated deacylase of Salmonella typhimurium, modulates signaling through Toll-like receptor 4. J. Biol. Chem. 279, 20044–20048 10.1074/jbc.M401275200 [DOI] [PubMed] [Google Scholar]
- 42. Gunn J. S., and Ernst R. K. (2007) The structure and function of Francisella lipopolysaccharide. Ann. N.Y. Acad. Sci. 1105, 202–218 10.1196/annals.1409.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Montminy S. W., Khan N., McGrath S., Walkowicz M. J., Sharp F., Conlon J. E., Fukase K., Kusumoto S., Sweet C., Miyake K., Akira S., Cotter R. J., Goguen J. D., and Lien E. (2006) Virulence factors of Yersinia pestis are overcome by a strong lipopolysaccharide response. Nat. Immunol. 7, 1066–1073 10.1038/ni1386 [DOI] [PubMed] [Google Scholar]
- 44. Chandler C. E., Harberts E. M., Pelletier M. R., Thaipisuttikul I., Jones J. W., Hajjar A. M., Sahl J. W., Goodlett D. R., Pride A. C., Rasko D. A., Trent M. S., Bishop R. E., and Ernst R. K. (2020) Early evolutionary loss of the lipid A modifying enzyme PagP resulting in innate immune evasion in Yersinia pestis. Proc. Natl. Acad. Sci. U. S. A. 117, 22984–22991 10.1073/pnas.1917504117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Munford R. S., and Hall C. L. (1989) Purification of acyloxyacyl hydrolase, a leukocyte enzyme that removes secondary acyl chains from bacterial lipopolysaccharides. J. Biol. Chem. 264, 15613–15619 [PubMed] [Google Scholar]
- 46. Hagen F. S., Grant F. J., Kuijper J. L., Slaughter C. A., Moomaw C. R., Orth K., O'Hara P. J., and Munford R. S. (1991) Expression and characterization of recombinant human acyloxyacyl hydrolase, a leukocyte enzyme that deacylates bacterial lipopolysaccharides. Biochemistry 30, 8415–8423 10.1021/bi00098a020 [DOI] [PubMed] [Google Scholar]
- 47. Munford R. S., Sheppard P. O., and O'Hara P. J. (1995) Saposin-like proteins (SAPLIP) carry out diverse functions on a common backbone structure. J. Lipid Res. 36, 1653–1663 [PubMed] [Google Scholar]
- 48. Gorelik A., Illes K., and Nagar B. (2018) Crystal structure of the mammalian lipopolysaccharide detoxifier. Proc. Natl. Acad. Sci. U. S. A. 115, E896–E905 10.1073/pnas.1719834115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Staab J. F., Ginkel D. L., Rosenberg G. B., and Munford R. S. (1994) A saposin-like domain influences the intracellular localization, stability, and catalytic activity of human acyloxyacyl hydrolase. J. Biol. Chem. 269, 23736–23742 [PubMed] [Google Scholar]
- 50. Park B. S., Song D. H., Kim H. M., Choi B. S., Lee H., and Lee J. O. (2009) The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature 458, 1191–1195 10.1038/nature07830 [DOI] [PubMed] [Google Scholar]
- 51. Sepulcre M. P., Alcaraz-Pérez F., López-Muñoz A., Roca F. J., Meseguer J., Cayuela M. L., and Mulero V. (2009) Evolution of lipopolysaccharide (LPS) recognition and signaling: fish TLR4 does not recognize LPS and negatively regulates NF-κB activation. J. Immunol. 182, 1836–1845 10.4049/jimmunol.0801755 [DOI] [PubMed] [Google Scholar]
- 52. Munford R. S., and Varley A. W. (2006) Shield as signal: lipopolysaccharides and the evolution of immunity to Gram-negative bacteria. PLoS Pathog. 2, e67 10.1371/journal.ppat.0020067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nigam V. N., Malchow D., Rietschel E. T., Lüderitz O., and Westphal O. (1970) Die enzymatische abspaltung langkettiger fettsäuren aus bakteriellen lipopolysacchariden mittels extrakten aus der amöbe von Dictyostelium discoideum. Hoppe-Seylers Z. Physiol. Chem. 351, 1123–1132 10.1515/bchm2.1970.351.2.1123 [DOI] [PubMed] [Google Scholar]
- 54. Verret C. R., Rosner M. R., and Khorana H. G. (1982) Fatty acyl amidases from Dictyostelium discoideum that act on lipopolysaccharide and derivatives. II. Aspects of substrate specificity. J. Biol. Chem. 257, 10228–10234 [PubMed] [Google Scholar]
- 55. Munford R. S., and Hunter J. P. (1992) Acyloxyacyl hydrolase, a leukocyte enzyme that deacylates bacterial lipopolysaccharides, has phospholipase, lysophospholipase, diacylglycerollipase, and acyltransferase activities in vitro. J. Biol. Chem. 267, 10116–10121 [PubMed] [Google Scholar]
- 56. Shao B., Lu M., Katz S. C., Varley A. W., Hardwick J., Rogers T. E., Ojogun N., Rockey D. C., DeMatteo R. P., and Munford R. S. (2007) A host lipase detoxifies bacterial lipopolysaccharides in the liver and spleen. J. Biol. Chem. 282, 13726–13735 10.1074/jbc.M609462200 [DOI] [PubMed] [Google Scholar]
- 57. Lu M., Zhang M., Kitchens R. L., Fosmire S., Takashima A., and Munford R. S. (2003) Stimulus-dependent deacylation of bacterial lipopolysaccharide by dendritic cells. J. Exp. Med. 197, 1745–1754 10.1084/jem.20030420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Murray S. M., Zhang Y., Douek D. C., and Sekaly R. P. (2020) Myeloid cells enriched for a dendritic cell population from people living with HIV have altered gene expression not restored by antiretroviral therapy. Front. Immunol. 11, 261 10.3389/fimmu.2020.00261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Koues O. I., Collins P. L., Cella M., Robinette M. L., Porter S. I., Pyfrom S. C., Payton J. E., Colonna M., and Oltz E. M. (2016) Distinct gene regulatory pathways for human innate versus adaptive lymphoid cells. Cell 165, 1134–1146 10.1016/j.cell.2016.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pekalski M. L., García A. R., Ferreira R. C., Rainbow D. B., Smyth D. J., Mashar M., Brady J., Savinykh N., Dopico X. C., Mahmood S., Duley S., Stevens H. E., Walker N. M., Cutler A. J., Waldron-Lynch F., et al. (2017) Neonatal and adult recent thymic emigrants produce IL-8 and express complement receptors CR1 and CR2. JCI Insight 2, e93739 10.1172/jci.insight.93739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Luchi M., and Munford R. S. (1993) Binding, internalization, and deacylation of bacterial lipopolysaccharides by human neutrophils. J. Immunol. 151, 959–969 [PubMed] [Google Scholar]
- 62. Poussin C., Foti M., Carpentier J. L., and Pugin J. (1998) CD14-dependent endotoxin internalization via a macropinocytic pathway. J. Biol. Chem. 273, 20285–20291 10.1074/jbc.273.32.20285 [DOI] [PubMed] [Google Scholar]
- 63. Cody M. J., Salkowski C. A., Henricson B. E., Detore G. R., Munford R. S., and Vogel S. N. (1997) Effect of inflammatory and antiinflammatory stimuli on acyloxyacyl hydrolase gene expression and enzymatic activity in murine macrophages. J. Endotoxin Res. 4, 371–379 10.1177/096805199700400509 [DOI] [Google Scholar]
- 64. Zou B., Jiang W., Han H., Li J., Mao W., Tang Z., Yang Q., Qian G., Qian J., Zeng W., Gu J., Chu T., Zhu N., Zhang W., Yan D., et al. (2017) Acyloxyacyl hydrolase promotes the resolution of lipopolysaccharide-induced acute lung injury. PLoS Pathog. 13, e1006436 10.1371/journal.ppat.1006436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gosselin D., Skola D., Coufal N. G., Holtman I. R., Schlachetzki J. C. M., Sajti E., Jaeger B. N., O'Connor C., Fitzpatrick C., Pasillas M. P., Pena M., Adair A., Gonda D. D., Levy M. L., Ransohoff R. M., et al. (2017) An environment-dependent transcriptional network specifies human microglia identity. Science 356, eaa3222 10.1126/science.aal3222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Thion M. S., Low D., Silvin A., Chen J., Grisel P., Schulte-Schrepping J., Blecher R., Ulas T., Squarzoni P., Hoeffel G., Coulpier F., Siopi E., David F. S., Scholz C., Shihui F., et al. (2018) Microbiome influences prenatal and adult microglia in a sex-specific manner. Cell 172, 500–516.e16 10.1016/j.cell.2017.11.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Janelsins B. M., Lu M., and Datta S. K. (2014) Altered inactivation of commensal LPS due to acyloxyacyl hydrolase deficiency in colonic dendritic cells impairs mucosal Th17 immunity. Proc. Natl. Acad. Sci. U. S. A. 111, 373–378 10.1073/pnas.1311987111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Feulner J. A., Lu M., Shelton J. M., Zhang M., Richardson J. A., and Munford R. S. (2004) Identification of acyloxyacyl hydrolase, a lipopolysaccharide-detoxifying enzyme, in the murine urinary tract. Infect. Immun. 72, 3171–3178 10.1128/IAI.72.6.3171-3178.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Weinrauch Y., Katz S. S., Munford R. S., Elsbach P., and Weiss J. (1999) Deacylation of purified lipopolysaccharides by cellular and extracellular components of a sterile rabbit peritoneal inflammatory exudate. Infect. Immun. 67, 3376–3382 10.1128/IAI.67.7.3376-3382.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Katz S. S., Weinrauch Y., Munford R. S., Elsbach P., and Weiss J. (1999) Deacylation of lipopolysaccharide in whole Escherichia coli during destruction by cellular and extracellular components of a rabbit inflammatory peritoneal exudate. J. Biol. Chem. 274, 36579–36584 10.1074/jbc.274.51.36579 [DOI] [PubMed] [Google Scholar]
- 71. Ojogun N., Kuang T. Y., Shao B., Greaves D. R., Munford R. S., and Varley A. W. (2009) Overproduction of acyloxyacyl hydrolase by macrophages and dendritic cells prevents prolonged reactions to bacterial lipopolysaccharide in vivo. J. Infect. Dis. 200, 1685–1693 10.1086/646616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lu M., Zhang M., Takashima A., Weiss J., Apicella M. A., Li X. H., Yuan D., and Munford R. S. (2005) Lipopolysaccharide deacylation by an endogenous lipase controls innate antibody responses to Gram-negative bacteria. Nat. Immunol. 6, 989–994 10.1038/ni1246 [DOI] [PubMed] [Google Scholar]
- 73. Lu M., and Munford R. S. (2011) The transport and inactivation kinetics of bacterial lipopolysaccharide influence its immunological potency in vivo. J. Immunol. 187, 3314–3320 10.4049/jimmunol.1004087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cani P. D., Bibiloni R., Knauf C., Waget A., Neyrinck A. M., Delzenne N. M., and Burcelin R. (2008) Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 57, 1470–1481 10.2337/db07-1403 [DOI] [PubMed] [Google Scholar]
- 75. Faraj T. A., McLaughlin C. L., and Erridge C. (2017) Host defenses against metabolic endotoxaemia and their impact on lipopolysaccharide detection. Int. Rev. Immunol. 36, 125–144 10.1080/08830185.2017.1280483 [DOI] [PubMed] [Google Scholar]
- 76. Munford R. S. (2016) Endotoxemia—menace, marker, or mistake? J. Leukoc. Biol. 100, 687–698 10.1189/jlb.3RU0316-151R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Guerville M., Leroy A., Sinquin A., Laugerette F., Michalski M. C., and Boudry G. (2017) Western-diet consumption induces alteration of barrier function mechanisms in the ileum that correlates with metabolic endotoxemia in rats. Am. J. Physiol. Endocrinol. Metab. 313, E107–E120 10.1152/ajpendo.00372.2016 [DOI] [PubMed] [Google Scholar]
- 78. Guerville M., and Boudry G. (2016) Gastrointestinal and hepatic mechanisms limiting entry and dissemination of lipopolysaccharide into the systemic circulation. Am. J. Physiol. Gastrointest. Liver Physiol. 311, G1–G15 10.1152/ajpgi.00098.2016 [DOI] [PubMed] [Google Scholar]
- 79. Qian G., Jiang W., Zou B., Feng J., Cheng X., Gu J., Chu T., Niu C., He R., Chu Y., and Lu M. (2018) LPS inactivation by a host lipase allows lung epithelial cell sensitization for allergic asthma. J. Exp. Med. 215, 2397–2412 10.1084/jem.20172225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Erridge C., Duncan S. H., Bereswill S., and Heimesaat M. M. (2010) The induction of colitis and ileitis in mice is associated with marked increases in intestinal concentrations of stimulants of TLRs 2, 4, and 5. PLoS One 5, e9125 10.1371/journal.pone.0009125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kaliannan K., Hamarneh S. R., Economopoulos K. P., Nasrin Alam S., Moaven O., Patel P., Malo N. S., Ray M., Abtahi S. M., Muhammad N., Raychowdhury A., Teshager A., Mohamed M. M., Moss A. K., Ahmed R., et al. (2013) Intestinal alkaline phosphatase prevents metabolic syndrome in mice. Proc. Natl. Acad. Sci. U. S. A. 110, 7003–7008 10.1073/pnas.1220180110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Rader B. A. (2017) Alkaline phosphatase, an unconventional immune protein. Front. Immunol. 8, 897 10.3389/fimmu.2017.00897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Komazin G., Maybin M., Woodard R. W., Scior T., Schwudke D., Schombel U., Gisch N., Mamat U., and Meredith T. C. (2019) Substrate structure-activity relationship reveals a limited lipopolysaccharide chemotype range for intestinal alkaline phosphatase. J. Biol. Chem. 294, 19405–19423 10.1074/jbc.RA119.010836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. d'Hennezel E., Abubucker S., Murphy L. O., and Cullen T. W. (2017) Total lipopolysaccharide from the human gut microbiome silences Toll-like receptor signaling. mSystems 2, e00046 10.1128/msystems.00046-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lindberg A. A., Weintraub A., Zähringer U., and Rietschel E. T. (1990) Structure-activity relationships in lipopolysaccharides of Bacteroides fragilis. Rev. Infect. Dis. 12, S133–S141 10.1093/clinids/12.Supplement_2.S133 [DOI] [PubMed] [Google Scholar]
- 86. Poxton I. R., and Edmond D. M. (1995) Biological activity of Bacteroides lipopolysaccharide—reappraisal. Clin. Infect. Dis. 20, S149–S153 10.1093/clinids/20.supplement_2.s149 [DOI] [PubMed] [Google Scholar]
- 87. Munford R., Lu M., Varley A. (2009) Chapter 2: Kill the bacteria … and also their messengers? Adv. Immunol. 103, 29–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Shao B., Kitchens R. L., Munford R. S., Rogers T. E., Rockey D. C., and Varley A. W. (2011) Prolonged hepatomegaly in mice that cannot inactivate bacterial endotoxin. Hepatology 54, 1051–1062 10.1002/hep.24488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zanoni I., Tan Y., Di Gioia M., Springstead J. R., and Kagan J. C. (2017) By capturing inflammatory lipids released from dying cells, the receptor CD14 induces inflammasome-dependent phagocyte hyperactivation. Immunity 47, 697–709.e3 10.1016/j.immuni.2017.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Henricson B. E., Benjamin W. R., and Vogel S. N. (1990) Differential cytokine induction by doses of lipopolysaccharide and monophosphoryl lipid A that result in equivalent early endotoxin tolerance. Infect. Immun. 58, 2429–2437 10.1128/IAI.58.8.2429-2437.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Medvedev A. E., Sabroe I., Hasday J. D., and Vogel S. N. (2006) Tolerance to microbial TLR ligands: molecular mechanisms and relevance to disease. J. Endotoxin Res. 12, 133–150 10.1179/096805106X102255 [DOI] [PubMed] [Google Scholar]
- 92. Collins P. E., and Carmody R. J. (2015) The regulation of endotoxin tolerance and its impact on macrophage activation. Crit. Rev. Immunol. 35, 293–323 10.1615/critrevimmunol.2015015495 [DOI] [PubMed] [Google Scholar]
- 93. Lu M., Varley A. W., Ohta S., Hardwick J., and Munford R. S. (2008) Host inactivation of bacterial lipopolysaccharide prevents prolonged tolerance following gram-negative bacterial infection. Cell Host Microbe 4, 293–302 10.1016/j.chom.2008.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lu M., Varley A. W., and Munford R. S. (2013) Persistently active microbial molecules prolong innate immune tolerance in vivo. PLoS Pathog. 9, e1003339 10.1371/journal.ppat.1003339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Mages J. R., Dietrich H., and Lang R. (2007) A genome-wide analysis of LPS tolerance in macrophages. Immunobiology 212, 723–737 10.1016/j.imbio.2007.09.015 [DOI] [PubMed] [Google Scholar]
- 96. Rodríguez D., Keller A. C., Faquim-Mauro E. L., De Macedo M. S., Cunha F. Q., Lefort J., Vargaftig B. B., and Russo M. (2003) Bacterial lipopolysaccharide signaling through Toll-like receptor 4 suppresses asthma-like responses via nitric oxide synthase 2 activity. J. Immunol. 171, 1001–1008 10.4049/jimmunol.171.2.1001 [DOI] [PubMed] [Google Scholar]
- 97. Velasco G., Campo M., Manrique O. J., Bellou A., He H., Arestides R. S., Schaub B., Perkins D. L., and Finn P. W. (2005) Toll-like receptor 4 or 2 agonists decrease allergic inflammation. Am. J. Respir. Cell Mol. Biol. 32, 218–224 10.1165/rcmb.2003-0435OC [DOI] [PubMed] [Google Scholar]
- 98. Schuijs M. J., Willart M. A., Vergote K., Gras D., Deswarte K., Ege M. J., Madeira F. B., Beyaert R., van Loo G., Bracher F., von Mutius E., Chanez P., Lambrecht B. N., and Hammad H. (2015) Farm dust and endotoxin protect against allergy through A20 induction in lung epithelial cells. Science 349, 1106–1110 10.1126/science.aac6623 [DOI] [PubMed] [Google Scholar]
- 99. Vatanen T., Kostic A. D., d'Hennezel E., Siljander H., Franzosa E. A., Yassour M., Kolde R., Vlamakis H., Arthur T. D., Hämäläinen A. M., Peet A., Tillmann V., Uibo R., Mokurov S., Dorshakova N., et al. (2016) Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell 165, 1551 10.1016/j.cell.2016.05.056 [DOI] [PubMed] [Google Scholar]
- 100. Daan de Boer J., Roelofs J. J., de Vos A. F., de Beer R., Schouten M., Hommes T. J., Hoogendijk A. J., de Boer O. J., Stroo I., van der Zee J. S., Veer C. V., and van der Poll T. (2013) Lipopolysaccharide inhibits Th2 lung inflammation induced by house dust mite allergens in mice. Am. J. Respir. Cell Mol. Biol. 48, 382–389 10.1165/rcmb.2012-0331OC [DOI] [PubMed] [Google Scholar]
- 101. Eisenbarth S. C., Piggott D. A., Huleatt J. W., Visintin I., Herrick C. A., and Bottomly K. (2002) Lipopolysaccharide-enhanced, Toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J. Exp. Med. 196, 1645–1651 10.1084/jem.20021340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zhang Y., Endam L. M., Filali-Mouhim A., Zhao L., Desrosiers M., Han D., and Zhang L. (2012) Polymorphisms in RYBP and AOAH genes are associated with chronic rhinosinusitis in a Chinese population: a replication study. PLoS One 7, e39247 10.1371/journal.pone.0039247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Barnes K. C., Grant A., Gao P., Baltadjieva D., Berg T., Chi P., Zhang S., Zambelli-Weiner A., Ehrlich E., Zardkoohi O., Brummet M. E., Stockton M., Watkins T., Gao L., Gittens M., et al. (2006) Polymorphisms in the novel gene acyloxyacyl hydroxylase (AOAH) are associated with asthma and associated phenotypes. J. Allergy Clin. Immunol. 118, 70–77 10.1016/j.jaci.2006.03.036 [DOI] [PubMed] [Google Scholar]
- 104. Velez J. I., Lopera F., Sepulveda-Falla D., Patel H. R., Johar A. S., Chuah A., Tobon C., Rivera D., Villegas A., Cai Y., Peng K., Arkell R., Castellanos F. X., Andrews S. J., Silva Lara M. F., et al. (2016) APOE*E2 allele delays age of onset in PSEN1 E280A Alzheimer's disease. Mol. Psychiatry 21, 916–924 10.1038/mp.2015.177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Ferreira M. A., Jansen R., Willemsen G., Penninx B., Bain L. M., Vicente C. T., Revez J. A., Matheson M. C., Hui J., Tung J. Y., Baltic S., Le Souëf P., Montgomery G. W., Martin N. G., Robertson C. F., et al. (2017) Gene-based analysis of regulatory variants identifies 4 putative novel asthma risk genes related to nucleotide synthesis and signaling. J. Allergy Clin. Immunol. 139, 1148–1157 10.1016/j.jaci.2016.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Lan Q., Hsiung C. A., Matsuo K., Hong Y. C., Seow A., Wang Z., Hosgood H. D. 3rd, Chen K., Wang J. C., Chatterjee N., Hu W., Wong M. P., Zheng W., Caporaso N., Park J. Y., et al. (2012) Genome-wide association analysis identifies new lung cancer susceptibility loci in never-smoking women in Asia. Nat. Genet. 44, 1330–1335 10.1038/ng.2456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Anderson C. A., Boucher G., Lees C. W., Franke A., D'Amato M., Taylor K. D., Lee J. C., Goyette P., Imielinski M., Latiano A., Lagacé C., Scott R., Amininejad L., Bumpstead S., Baidoo L., et al. (2011) Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 43, 246–252 10.1038/ng.764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Fehrmann R. S. N., Jansen R. C., Veldink J. H., Westra H. J., Arends D., Bonder M. J., Fu J., Deelen P., Groen H. J. M., Smolonska A., Weersma R. K., Hofstra R. M. W., Buurman W. A., Rensen S., Wolfs M. G. M., et al. (2011) Trans-eQTLs reveal that independent genetic variants associated with a complex phenotype converge on intermediate genes, with a major role for the HLA. PLoS Genet. 7, e1002197 10.1371/journal.pgen.1002197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Fairfax B. P., Makino S., Radhakrishnan J., Plant K., Leslie S., Dilthey A., Ellis P., Langford C., Vannberg F. O., and Knight J. C. (2012) Genetics of gene expression in primary immune cells identifies cell type-specific master regulators and roles of HLA alleles. Nat. Genet. 44, 502–510 10.1038/ng.2205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Jutras B. L., Lochhead R. B., Kloos Z. A., Biboy J., Strle K., Booth C. J., Govers S. K., Gray J., Schumann P., Vollmer W., Bockenstedt L. K., Steere A. C., and Jacobs-Wagner C. (2019) Borrelia burgdorferi peptidoglycan is a persistent antigen in patients with Lyme arthritis. Proc. Natl. Acad. Sci. U. S. A. 116, 13498–13507 10.1073/pnas.1904170116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Van Dyken S. J., and Locksley R. M. (2018) Chitins and chitinase activity in airway diseases. J. Allergy Clin. Immunol. 142, 364–369 10.1016/j.jaci.2018.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Jimenez M. F., and Marshall J. C., and International Sepsis Forum (2001) Source control in the management of sepsis. Intensive Care Med. 27, S49–S62 10.1007/pl00003797 [DOI] [PubMed] [Google Scholar]
- 113. Torgersen C., Moser P., Luckner G., Mayr V., Jochberger S., Hasibeder W. R., and Dunser M. W. (2009) Macroscopic postmortem findings in 235 surgical intensive care patients with sepsis. Anesth. Analg. 108, 1841–1847 10.1213/ane.0b013e318195e11d [DOI] [PubMed] [Google Scholar]
- 114. Cheng S. C., Scicluna B. P., Arts R. J., Gresnigt M. S., Lachmandas E., Giamarellos-Bourboulis E. J., Kox M., Manjeri G. R., Wagenaars J. A., Cremer O. L., Leentjens J., van der Meer A. J., van de Veerdonk F. L., Bonten M. J., Schultz M. J., et al. (2016) Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat. Immunol. 17, 406–413 10.1038/ni.3398 [DOI] [PubMed] [Google Scholar]