

Abstract
Background
Lp(a) (lipoprotein (a)) is a risk factor for cardiovascular events, but the mechanism of increased risk is uncertain. This study evaluated the relationship between Lp(a) and coronary atheroma volume by intravascular ultrasound.
Methods and Results
This was a post hoc analysis of 6 randomized trials of coronary atheroma by intravascular ultrasound. The population was stratified into high (≥60 mg/dL) and low (<60 mg/dL) baseline serum Lp(a). The primary outcome was baseline coronary percent atheroma volume. A mixed model adjusted for baseline low density lipoprotein, ApoB (apoliporotein B100), non‐high density lipoprotein, sex, age, race, history of myocardial infarction, statin use, and intravascular ultrasound study was used to provide estimates of baseline plaque burden. Of 3943 patients, 17.3% (683) had Lp(a) ≥ 60 mg/dL and 82.7% (3260) had Lp(a) < 60 mg/dL. At baseline, uncorrected low density lipoprotein level (107.7 ± 32.0 versus 99.1 ± 31.5) and statin therapy (99.0% versus 97.0%) were higher in patients with high Lp(a) levels, but low density lipoprotein corrected for Lp(a) was lower (80.6 ± 32.0 versus 94.0 ± 31.4) in patients with high Lp(a) levels. Percent atheroma volume was significantly higher in the high Lp(a) group in unadjusted (38.2% [32.8, 43.6] versus 37.1% [31.4, 43.1], P=0.01) and risk‐adjusted analyses (38.7%±0.5 versus 37.5%±0.5, P<0.001). There was a significant association of increasing risk‐adjusted percent atheroma volume across quintiles of Lp(a) (Lp(a) quintiles 1‐5; 37.3 ± 0.5%, 37.2 ± 0.5%, 37.3 ± 0.5%, 38.0 ± 0.5%, 38.5 ± 0.5%, P=0.002).
Conclusions
Elevated Lp(a) is independently associated with increased percent atheroma volume. Further work is needed to clarify the relationship of Lp(a)‐lowering treatment with cardiovascular outcomes.
Keywords: atheroma, intravascular ultrasound, lipids, lipoprotein (a), prevention
Subject Categories: Lipids and Cholesterol, Ultrasound
Nonstandard Abbreviations and Acronyms
- Lp(a)
lipoprotein (a)
- PAV
percent atheroma volume
Clinical Perspective
What is New?
Lp(a) (lipoprotein (a)) has emerged as a strong risk factor for cardiovascular disease; however, the mechanism of risk is unknown.
This post hoc analysis of 6 clinical trials employing coronary intravascular ultrasound demonstrated that elevated Lp(a) is associated with increased coronary atheroma volume after adjusting for multiple covariates.
These data highlight an independent association between Lp(a) level and coronary atheroma volume, a finding that supports a proatherogenic mechanism of cardiovascular risk related to increased Lp(a).
What are the Clinical Implications?
Further work is needed to clarify whether specific therapies to reduce Lp(a) affect coronary atheroma volume over time and the effect of Lp(a) reduction on cardiovascular outcomes.
Cardiovascular disease remains the leading cause of death across the world, resulting in 18 million deaths annually. 1 Statin therapy is associated with significant reductions in cardiovascular events in primary and secondary prevention populations, and statin therapy remains the primary focus of current lipid guidelines. 2 Lp(a) (lipoprotein (a)) is a cholesterol particle consisting of ApoB (apolipoprotein B100) covalently bound to the glycoprotein ApoA (apoplipoprotein A), and a growing body of evidence links elevated Lp(a) levels to increased cardiovascular risk, even in patients treated with statin therapy. The mechanism of cardiovascular risk related to Lp(a) is not well understood. Prior reports have suggested multiple factors including increased atherogenesis, increased inflammation, abnormal endothelial function, and increased thrombosis. 3 , 4 , 5 , 6 , 7 However, data evaluating the mechanisms by which Lp(a) confers increased cardiovascular risk are sparse. We hypothesized that increased serum Lp(a) levels are associated with increased coronary atheroma burden. This was a post hoc analysis of 6 trials analyzed in a single core laboratory with standardized intravascular ultrasound (IVUS) measurements of coronary atheroma volume used to study the association between serum Lp(a) and coronary percent atheroma volume (PAV).
METHODS
Population
Between December 2002 and January 2015, 5393 patients were enrolled in the ACTIVATE (Adoptive Cell Therapy InVigorated to Augment Tumor Eradication), ASTEROID (A Study to Evaluate the Effect of Rosuvastatin on Intravascular Ultrasound‐Derived Coronary Atheroma Burden), ILLUSTRATE (Investigation of Lipid Level Management Using Coronary Ultrasound to Assess Reduction of Atherosclerosis by Cholesterylester Transfer Protein Inhibition and High‐Density Lipoprotein Elevation), STRADIVARIUS (Strategy to Reduce Atherosclerosis Development Involving Administration of Rimonabant—the Intravascular Ultrasound Study), SATURN (Study of Coronary Atheroma by Travascular Ultrasound: Effect of Rosuvastatin Versus Atorvastatin Trial), and GLAGOV (Global Assessment of Plaque Regression With a PCSK9 Antibody as Measured by Intravascular Ultrasound) trials. These were multicenter international trials with centralized IVUS measurement of coronary PAV and with Lp(a) available in study participants at baseline before treatment with study drugs. Our group performed all 6 trials, including IVUS core laboratory adjudication, and these trials had similar inclusion criteria and imaging methodology. The primary study methodology and results have been previously reported. 8 , 9 , 10 , 11 , 12 , 13 The institutional review boards of each participating center approved the trials, and each patient provided written informed consent. We performed an analysis of pooled patient level data. We excluded patients without available baseline Lp(a) data and those without paired IVUS data. We stratified the study population into patients with high (≥60 mg/dL) or low (<60 mg/dL) baseline levels of serum Lp(a) (Figure 1). An Lp(a) level < 30 mg/dL is generally accepted as desirable, but the specific cutoff level for abnormal Lp(a) is not well defined. Current US lipid guidelines use a cutoff value of 50 mg/dL, which corresponds to roughly 20% of the White American population. 2 A value of 60 mg/dL was chosen for this study to offer specificity in selecting a high‐risk population. Baseline demographics and comorbidities were recorded at the time of each trial. A central laboratory performed all biochemical determinations. Corrected low‐density lipoprotein cholesterol (LDL‐C) was calculated by subtracting 30% of the baseline Lp(a) value from the baseline serum LDL‐C value. 14
Figure 1. Study Population.
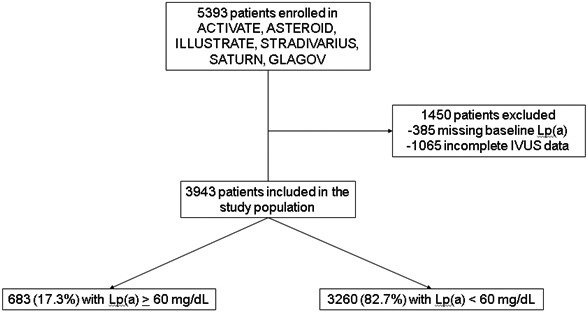
The selection of patients for the study population is shown. The population was stratified into those with elevated (≥60 mg/dL) and nonelevated (<60 mg/dL) levels of Lp(a). IVUS indicates intravascular ultrasound; and Lp(a), lipoprotein (a)
End Points
The methods used for IVUS assessment of coronary PAV have been previously described. 15 , 16 , 17 , 18 , 19 In short, each patient underwent baseline coronary IVUS by invasive catheterization before treatment with the study drug in each trial. The interventional operator at the time of catheterization identified a target vessel that did not have either prior revascularization or> 50% luminal narrowing throughout a target segment of ≥ 30 mm. A dose of 100 to 300 mcg of intracoronary nitroglycerin was administered, and an IVUS catheter was advanced into the target vessel with the ultrasound transducer positioned distal to a side branch. The transducer was withdrawn at a speed of 0.5 mm/sec with the use of a motor drive (“pullback”). Images were obtained at a rate of 30 frames/sec. The image quality of each recording was assessed at the Atherosclerosis Imaging Core Laboratory of the Cleveland Clinic Coordinating Center for Clinical Research, and only patients whose imaging met prespecified requirements for image quality were eligible to undergo randomization in the trials.
The primary end point of this study was baseline PAV by IVUS. PAV was calculated based on the following equation:
EEMarea is the cross‐sectional area of the external elastic membrane and Lumenarea is the cross‐sectional area of the lumen.
Statistical Analysis
Normally distributed continuous variables are presented as mean ± SD, and non‐normally distributed continuous variables are presented as median (25th percentile, 75th percentile). Categorical variables are presented with frequency (%). The primary end point, baseline PAV, was assessed using a mixed model with a random intercept term for trial and baseline Lp(a) (≥60 mg/dL vs < 60 mg/dL or quintile) as a fixed factor. Covariates included baseline serum LDL‐C level, ApoB level, non‐high‐density lipoprotein cholesterol level, sex, age, race, history of myocardial infarction, and baseline statin therapy use. Estimates of baseline PAV by Lp(a) level are presented as least square means with standard error. Trial was included as a random factor. Two risk‐adjusted models of the association between Lp(a) and PAV were performed: (1) stratified by high (≥60 mg/dL) versus low (<60 mg/dL) Lp(a) and (2) stratified by Lp(a) quintile. All reported P values are 2 sided with P<0.05 considered statistically significant. All statistical analyses were performed with SAS version 9.3 (SAS Institute, Cary, NC). The data underlying this article will not be shared publicly to protect the privacy of individuals who participated in the study.
This study complies with the Declaration of Helsinki, and the local institutional review board of each enrolling site approved the clinical trials from which these data were obtained. Informed consent was obtained from each subject or their legally authorized representative.
Results
Patients
Baseline Lp(a) values in the study population ranged from 0.1 to 305.6 mg/dL with a mean and median of 29.8 and 14.8 mg/dL respectively (Figure 2). Table 1 describes baseline demographics and comorbidities of the study groups. The studied population included 683 (17.3%) individuals with Lp(a) ≥60 mg/dL and 3260 (82.7%) with Lp(a) <60 mg/dL. Those with elevated Lp(a) values ≥ 60 mg/dL were more likely to be women (35.7% versus 27.4%,), less likely to be White (91.1% versus 94.9%), had a lower body mass index (28.9 [25.9, 32.7] versus 29.4 [26.6, 33.5]), and had higher rates of prior myocardial infarction (32.8% versus 27.9%), acute coronary syndromes (28.8% versus 24.6%), and family history of coronary artery disease (41.7% versus 33.6%). Baseline medical therapies were similar across both patient groups, aside from baseline statin therapy, which was more prevalent in those with elevated Lp(a) (99.0% versus 97.0%).
Figure 2. Lipoprotein (a) Values in the Study Population.
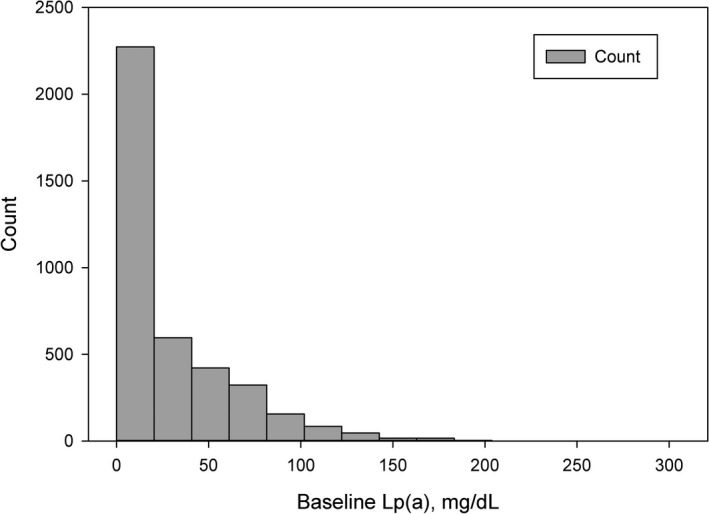
Lp(a) values in the study cohort are shown. Lp(a) ≥ 60 mg/dL was observed in 683 patients (17.4 %). Lp(a) indicates lipoprotein (a).
Table 1.
Baseline Characteristics
| Characteristic |
Lp(a)<60 mg/dL (N = 3260) |
Lp(a)≥60 mg/dL (N = 683) |
Absolute Standardized Difference (%) † |
|---|---|---|---|
| Baseline Demographics and Comorbidities | |||
| Age, median (25th, 75th percentile), years | 58.0 (52.0, 65.0) | 58.0 (51.0, 64.0) | 1.4 |
| Female, N (%) | 893 (27) | 244 (36) | 19.5 |
| White, N (%) | 3094 (95) | 622 (91) | 15.6 |
| Body mass index, median (25th, 75th percentile) | 29.4 (26.6, 33.5) | 28.9 (25.9, 32.7) | 9.0 |
| History of myocardial infarction, N (%) | 910 (28) | 224 (33) | 11.4 |
| History of percutaneous coronary revascularization, N (%) | 1054 (36) | 229 (36) | 0.3 |
| History of coronary artery bypass graft, N (%) | 57 (2) | 13 (2) | 1.4 |
| Acute coronary syndrome, N (%) | 802 (25) | 197 (29) | 7.4 |
| Angina, N (%) | 1291 (44) | 281 (44) | 1.4 |
| History of hypertension, N (%) | 2576 (79) | 523 (77) | 4.0 |
| Diabetes mellitus, N (%) | 754 (23) | 138 (20) | 6.9 |
| Family history of coronary artery disease, N (%) | 474 (34) | 164 (42) | 14.0 |
| Current smoker, N (%) | 734 (25) | 153 (24) | 1.1 |
| Concomitant Medical Therapy | |||
| Aspirin, N (%) | 3005 (92) | 628 (92) | 2.4 |
| Beta blockers, N (%) | 2472 (76) | 537 (79) | 7.0 |
| Angiotensin‐converting enzyme inhibitors, N (%) | 1803 (55) | 386 (57) | 2.0 |
| Angiotensin receptor blocker, N (%) | 768 (24) | 140 (21) | 5.1 |
| Nitrates, N (%) | 755 (23) | 139 (20) | 4.0 |
| Statin, N (%) | 3161 (97) | 676 (99) | 14.0 |
| Baseline Laboratory Values | |||
| LDL‐C, mean (SD), mg/dL | 99.1 ± 31.5 | 107.7 ± 32.0 | 25.3 |
| Corrected LDL‐C*, mean (SD), mg/dL | 94.0 ± 31.4 | 80.6 ± 32.0 | 43.8 |
| HDL‐C, mean (SD), mg/dL | 43.7 ± 11.7 | 46.2 ± 12.6 | 21.1 |
| Non‐HDL‐C, mean (SD), mg/dL | 128.6 ± 36.0 | 135.9 ± 36.6 | 18.9 |
| Triglyceride, median (25th, 75th percentile), mg/dL | 131.0 (94.0, 181.4) | 123.0 (92.0, 175.0) | 9.1 |
| Apolipoprotein A1, mean (SD), mg/dL | 131.1 ± 26.7 | 133.0 ± 27.4 | 9.2 |
| Apolipoprotein B‐100, mean (SD), md/dL | 90.4 ± 27.4 | 98.2 ± 28.7 | 27.0 |
| High sensitivity C‐reactive protein, median (25th, 75th percentile), mg/L | 1.9 (0.9, 4.4) | 2.0 (1.0, 4.0) | 1.0 |
HDL‐C indicates high density lipoprotein cholesterol; LDL‐C, low density lipoprotein cholesterol; and Lp(a), lipoprotein (a).
Corrected LDL‐C was calculated by subtracting 30% of the baseline LP(a) value from the baseline serum LDL‐C value.
Imbalance is defined as an absolute standardized difference> 20%.
Serum Lipid Levels
Baseline LDL‐C was higher in the elevated Lp(a) group (107.7 ± 32.0 versus 99.1 ± 31.5), but the LDL‐C level corrected for baseline Lp(a) level was lower in the elevated Lp(a) group (80.6 ± 32.0 versus 94.0 ± 31.4). high‐density lipoprotein cholesterol (46.2 ± 12.6 versus 43.7 ± 11.7) and non‐high‐density lipoprotein cholesterol (135.9 ± 36.6 versus 128.6 ± 36.0) were both higher in the elevated Lp(a) group, whereas triglyceride levels (123.0 [92.0, 175.0] versus 131.0 [94.0, 181.4]) were lower in the elevated Lp(a) group. ApoB levels were higher in the elevated Lp(a) group (98.2 ± 28.7 versus 90.4 ± 27.4).
Percent Atheroma Volume
Table 2 describes the association of Lp(a) with baseline PAV in unadjusted and risk‐adjusted analyses. Baseline unadjusted PAV (38.2% [32.8, 43.6] versus 37.1% [31.4, 43.1], P=0.01) and risk‐adjusted PAV (38.7% ± 0.5 versus 37.5% ± 0.5, P<0.001) were both significantly higher in patients with elevated Lp(a). We observed a significant association with increasing risk‐adjusted PAV across quintiles of baseline Lp(a) level (Figure 3). Risk‐adjusted PAV was 37.3% ± 0.5 in the lowest quintile versus 38.5% ±0.5 in the highest quintile of baseline Lp(a) (P=0.002 across quintiles).
Table 2.
Association Between Lipoprotein (a) and Percent Atheroma Volume
| Parameter |
Lp(a)<60 mg/dL (N = 3260) |
Lp(a)≥60 mg/dL (N = 683) |
P |
|---|---|---|---|
| Baseline unadjusted percent atheroma volume, median (25th, 75th percentile), % | 37.1 (31.4, 43.1) | 38.2 (32.8, 43.6) | 0.01 |
| Baseline risk‐adjusted percent atheroma volume, mean (SD), %* | 37.5 ± 0.5 | 38.7 ± 0.5 | <0.001 |
Lp(a) indicates lipoprotein (a).
Adjusted for baseline low‐density lipoprotein cholesterol, apolipoprotein B, non‐high‐density lipoprotein cholesterol, sex, age, race, history of myocardial infarction, baseline statin therapy, and intravascular ultrasound study.
Figure 3. Association of Baseline Lipoprotein (a) and Percent Atheroma Volume.
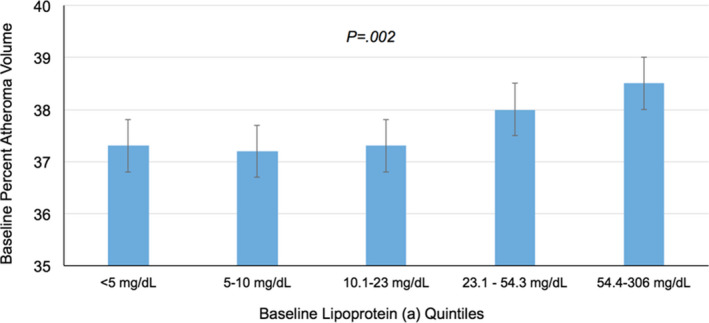
The population was stratified by quintiles of serum Lp(a) levels, and the risk‐adjusted coronary PAV by IVUS was assessed. PAV was similar between patients in the first three quintiles, and PAV was increased in patients in the fourth and fifth quintiles of Lp(a) levels with a significantly increasing trend. PAV is risk adjusted for potential confounders (baseline low‐density lipoprotein cholesterol, apolipoprotein B, non‐high‐density lipoprotein cholesterol, sex, age, race, history of myocardial infarction, baseline statin therapy, and IVUS study). IVUS indicates intravascular ultrasound; Lp(a), lipoprotein (a); and PAV, percent atheroma volume.
Discussion
Principal Findings
This post hoc analysis of 6 clinical trials employing coronary IVUS demonstrated that elevated Lp(a) is associated with increased coronary atheroma volume (PAV) after adjusting for multiple covariates (Summarizing Illustration). When considering the association between Lp(a) level and PAV, this study identified that PAV was similar in the first to third quintiles of Lp(a) but is associated with increasing PAV in the fourth and fifth Lp(a) quintiles. These data highlight an independent association between Lp(a) level and coronary atheroma volume, a finding that supports a proatherogenic mechanism of cardiovascular risk related to increased Lp(a).
Lp(a) as a Proatherogenic Cardiovascular Risk Factor
Prior reports have suggested a multitude of potential mechanisms of cardiovascular risk related to Lp(a) including increased atherogenesis, endothelial dysfunction, macrophage recruitment, oxidative stress, and prothrombotic factors. 3 , 4 , 5 , 6 , 7 Human atherosclerotic lesions express a VLDL (very low‐density lipoprotein) receptor that mediates endocytosis of circulating Lp(a), providing a plausible biologic mechanism to link Lp(a) and atheroma formation. 3 The findings of this study corroborate prior reports suggesting an association between Lp(a) and plaque burden on coronary imaging. A study of 269 Black participants without known cardiovascular disease demonstrated that Lp(a) was independently associated with total plaque volume by coronary computed tomographic angiography. 20 A study of 119 patients with nonculprit lesions evaluated with virtual histology IVUS demonstrated that Lp(a) was independently associated with necrotic core plaque progression after 8 months on statin therapy. 21 The findings of the present study contribute to a growing body of literature supporting the hypothesis that Lp(a) is associated with increased coronary atheroma volume.
However, the present study findings do not preclude the possibility that Lp(a) is also associated with separate mechanisms of cardiovascular risk independent of its association with atheroma burden. Lp(a) is a proinflammatory molecule related to its content of ApoB, ApoA, and oxidized phospholipids. 22 , 23 The inflammatory characteristics of Lp(a) may contribute not only to its atherogenicity but also to its prothrombotic potential as well. Further study is needed to determine if Lp(a) is independently associated with increased thrombotic risk or whether thrombotic events in patients with Lp(a) are mediated primarily by increased atheroma volume. 24 In a post hoc analysis of the SATURN trial, Lp(a) levels were not associated with plaque progression over time. However, cardiovascular event rates were higher at elevated Lp(a) levels with higher CRP (C‐reactive protein) > 2) suggesting a prothrombotic or inflammatory association. 24 , 25 , 26 , 27
Future Directions
Currently, there are no results of large clinical outcome trials testing therapies dedicated to lowering or inhibiting Lp(a). Mendelian randomization studies have suggested that large absolute reductions in plasma Lp(a) levels are likely to be more beneficial than the modest reductions seen with currently available therapies. 28 , 29 , 30 Antisense oligonucleotides have shown promise in this regard with up to an 80% reduction of Lp(a) levels. 31 Further research is needed to see if these large reductions translate to lower cardiovascular events. Future research is also needed to see if atheroma volume continues to progress over time in patients with elevated Lp(a) and whether Lp(a) lowering therapies promote atheroma regression and/or stabilization.
Limitations
First, this was a post hoc analysis of 6 separate clinical trials, and the population included in this study may be subject to selection bias inherent to clinical trial populations. Additionally, the Lp(a) assays used varied between the trials, which may have affected inclusion of patients in the high Lp(a) group. However, trial was included in the risk adjustment. Second, the population of this study was predominantly male and White, and the findings warrant confirmation in varied populations to confirm their generalizability. Third, residual confounding may contribute to the observed results despite attempts to perform risk adjustment for potential confounders. Fourth, although this study identified an association between Lp(a) levels and PAV, the mechanisms of increased PAV related to Lp(a) cannot be elucidated from the present analysis. Further investigation is needed to understand mechanisms of atherogenesis related to Lp(a). Despite these limitations, this is the first study to examine the relationship between Lp(a) levels and coronary atheroma burden in a large population of patients with standardized IVUS protocols and core laboratory adjudication.
Conclusions
Elevated Lp(a) is associated with increased coronary atheroma volume. Further work is needed to clarify whether specific therapies to reduce Lp(a) impact coronary atheroma volume over time and the effect of Lp(a) reduction on cardiovascular outcomes.
Sources of Funding
The study was internally funded at the Cleveland Clinic, and no external organization had any role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; or the decision to submit the manuscript for publication.
Disclosures
Dr. Nicholls reports research support from AstraZeneca, Amgen, Anthera, CSL Behring, Cerenis, Eli Lilly, Esperion, Resverlogix, Novartis, InfraReDx and Sanofi‐Regeneron and is a consultant for Amgen, Akcea, AstraZeneca, Boehringer Ingelheim, CSL Behring, Eli Lilly, Esperion, Kowa, Merck, Takeda, Pfizer, Sanofi‐Regeneron, and Novo Nordisk. Dr. Nissen reports that the Cleveland Clinic Center for Clinical Research has received funding to perform clinical trials from Abbvie, AstraZeneca, Amgen, Cerenis, Eli Lilly, Esperion, Medtronic, MyoKardia, Novartis, Pfizer, The Medicines Company, Silence Therapeutics, Takeda, and Orexigen. Dr. Nissen is involved in these clinical trials, but receives no personal remuneration for his participation. Dr. Nissen consults for many pharmaceutical companies, but requires them to donate all honoraria or consulting fees directly to charity so that he receives neither income nor a tax deduction. LC reports consulting for Amgen, Novartis, AstraZeneca, research Amgen, Novartis and Esperion. The remaining authors have no disclosures to report.
(J Am Heart Assoc. 2020;9:e018023 DOI: 10.1161/JAHA.120.018023.)
For Sources of Funding and Disclosures, see page 7.
REFERENCES
- 1. Roth GA, Johnson C, Abajobir A, Abd‐Allah F, Abera SF, Abyu G, Ahmed M, Aksut B, Alam T, Alam K, et al. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J Am Coll Cardiol. 2017;70:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Grundy SM, Stone NJ, Bailey AL, Beam C, Birtcher KK, Blumenthal RS, Braun LT, de Ferranti S, Faiella‐Tommasino J, Forman DE, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APHA/ASPC/NLA/PCNA Guideline on the management of blood cholesterol: A report of the american college of cardiology/american heart association task force on clinical practice guidelines. Circ. 2019;139:e1082–e1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Argraves KM, Kozarsky KF, Fallon JT, Harpel PC, Strickland DK. The atherogenic lipoprotein lp(a) is internalized and degraded in a process mediated by the vldl receptor. J Clin Invest. 1997;100:2170–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dangas G, Mehran R, Harpel PC, Sharma SK, Marcovina SM, Dube G, Ambrose JA, Fallon JT. Lipoprotein(a) and inflammation in human coronary atheroma: Association with the severity of clinical presentation. J Am Coll Cardiol. 1998;32:2035–2042. [DOI] [PubMed] [Google Scholar]
- 5. Schachinger V, Halle M, Minners J, Berg A, Zeiher AM. Lipoprotein(a) selectively impairs receptor‐mediated endothelial vasodilator function of the human coronary circulation. J Am Coll Cardiol. 1997;30:927–934. [DOI] [PubMed] [Google Scholar]
- 6. Takami S, Yamashita S, Kihara S, Ishigami M, Takemura K, Kume N, Kita T, Matsuzawa Y. Lipoprotein(a) enhances the expression of intercellular adhesion molecule‐1 in cultured human umbilical vein endothelial cells. Circulation. 1998;97:721–728. [DOI] [PubMed] [Google Scholar]
- 7. Tsimikas S, Brilakis ES, Miller ER, McConnell JP, Lennon RJ, Kornman KS, Witztum JL, Berger PB. Oxidized phospholipids, lp(a) lipoprotein, and coronary artery disease. N Engl J Med. 2005;353:46–57. [DOI] [PubMed] [Google Scholar]
- 8. Nicholls SJ, Ballantyne CM, Barter PJ, Chapman MJ, Erbel RM, Libby P, Raichlen JS, Uno K, Borgman M, Wolski K, et al. Effect of two intensive statin regimens on progression of coronary disease. N Engl J Med. 2011;365:2078–2087. [DOI] [PubMed] [Google Scholar]
- 9. Nicholls SJ, Puri R, Anderson T, Ballantyne CM, Cho L, Kastelein JJ, Koenig W, Somaratne R, Kassahun H, Yang J, et al. Effect of evolocumab on progression of coronary disease in statin‐treated patients: The glagov randomized clinical trial. JAMA. 2016;316:2373–2384. [DOI] [PubMed] [Google Scholar]
- 10. Nissen SE, Nicholls SJ, Sipahi I, Libby P, Raichlen JS, Ballantyne CM, Davignon J, Erbel R, Fruchart JC, Tardif JC, et al. Effect of very high‐intensity statin therapy on regression of coronary atherosclerosis: The asteroid trial. JAMA. 2006;295:1556–1565. [DOI] [PubMed] [Google Scholar]
- 11. Nissen SE, Nicholls SJ, Wolski K, Rodes‐Cabau J, Cannon CP, Deanfield JE, Despres JP, Kastelein JJ, Steinhubl SR, Kapadia S, et al. Effect of rimonabant on progression of atherosclerosis in patients with abdominal obesity and coronary artery disease: The stradivarius randomized controlled trial. JAMA. 2008;299:1547–1560. [DOI] [PubMed] [Google Scholar]
- 12. Nissen SE, Tardif JC, Nicholls SJ, Revkin JH, Shear CL, Duggan WT, Ruzyllo W, Bachinsky WB, Lasala GP, Tuzcu EM. Effect of torcetrapib on the progression of coronary atherosclerosis. N Engl J Med. 2007;356:1304–1316. [DOI] [PubMed] [Google Scholar]
- 13. Nissen SE, Tuzcu EM, Brewer HB, Sipahi I, Nicholls SJ, Ganz P, Schoenhagen P, Waters DD, Pepine CJ, Crowe TD, et al. Effect of acat inhibition on the progression of coronary atherosclerosis. N Engl J Med. 2006;354:1253–1263. [DOI] [PubMed] [Google Scholar]
- 14. Yeang C, Witztum JL, Tsimikas S. 'Ldl‐c' = ldl‐c + lp(a)‐c: Implications of achieved ultra‐low ldl‐c levels in the proprotein convertase subtilisin/kexin type 9 era of potent ldl‐c lowering. Curr Opin Lipidol. 2015;26:169–178. [DOI] [PubMed] [Google Scholar]
- 15. Eisen HJ, Tuzcu EM, Dorent R, Kobashigawa J, Mancini D, Valantine‐von Kaeppler HA, Starling RC, Sorensen K, Hummel M, Lind JM, et al. Everolimus for the prevention of allograft rejection and vasculopathy in cardiac‐transplant recipients. N Engl J Med. 2003;349:847–858. [DOI] [PubMed] [Google Scholar]
- 16. Nissen SE, Tsunoda T, Tuzcu EM, Schoenhagen P, Cooper CJ, Yasin M, Eaton GM, Lauer MA, Sheldon WS, Grines CL, et al. Effect of recombinant apoa‐i milano on coronary atherosclerosis in patients with acute coronary syndromes: A randomized controlled trial. JAMA. 2003;290:2292–2300. [DOI] [PubMed] [Google Scholar]
- 17. Nissen SE, Tuzcu EM, Libby P, Thompson PD, Ghali M, Garza D, Berman L, Shi H, Buebendorf E, Topol EJ. Effect of antihypertensive agents on cardiovascular events in patients with coronary disease and normal blood pressure: The camelot study: A randomized controlled trial. JAMA. 2004;292:2217–2225. [DOI] [PubMed] [Google Scholar]
- 18. Nissen SE, Tuzcu EM, Schoenhagen P, Brown BG, Ganz P, Vogel RA, Crowe T, Howard G, Cooper CJ, Brodie B, et al. Effect of intensive compared with moderate lipid‐lowering therapy on progression of coronary atherosclerosis: A randomized controlled trial. JAMA. 2004;291:1071–1080. [DOI] [PubMed] [Google Scholar]
- 19. Nissen SE, Tuzcu EM, Schoenhagen P, Crowe T, Sasiela WJ, Tsai J, Orazem J, Magorien RD, O'Shaughnessy C, Ganz P. Statin therapy, ldl cholesterol, c‐reactive protein, and coronary artery disease. N Engl J Med. 2005;352:29–38. [DOI] [PubMed] [Google Scholar]
- 20. Kral BG, Kalyani RR, Yanek LR, Vaidya D, Fishman EK, Becker DM, Becker LC. Relation of plasma lipoprotein(a) to subclinical coronary plaque volumes, three‐vessel and left main coronary disease, and severe coronary stenoses in apparently healthy african‐americans with a family history of early‐onset coronary artery disease. Am J Cardiol. 2016;118:656–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nozue T, Yamamoto S, Tohyama S, Fukui K, Umezawa S, Onishi Y, Kunishima T, Sato A, Nozato T, Miyake S, et al. Lipoprotein(a) is associated with necrotic core progression of non‐culprit coronary lesions in statin‐treated patients with angina pectoris. Lipids Health Dis. 2014;13:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Steinberg D, Witztum JL. Oxidized low‐density lipoprotein and atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30:2311–2316. [DOI] [PubMed] [Google Scholar]
- 23. Spence JD, Koschinsky M. Mechanisms of lipoprotein(a) pathogenicity: Prothrombotic, proatherosclerotic, or both? Arterioscler Thromb Vasc Biol. 2012;32:1550–1551. [DOI] [PubMed] [Google Scholar]
- 24. Boffa MB, Koschinsky ML. Lipoprotein (a): Truly a direct prothrombotic factor in cardiovascular disease? J Lipid Res. 2016;57:745–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van der Valk FM, Bekkering S, Kroon J, Yeang C, Van den Bossche J, van Buul JD, Ravandi A, Nederveen AJ, Verberne HJ, Scipione C, et al. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation. 2016;134:611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Figueroa AL, Abdelbaky A, Truong QA, Corsini E, MacNabb MH, Lavender ZR, Lawler MA, Grinspoon SK, Brady TJ, Nasir K, et al. Measurement of arterial activity on routine fdg pet/ct images improves prediction of risk of future cv events. JACC Cardiovasc Imaging. 2013;6:1250–1259. [DOI] [PubMed] [Google Scholar]
- 27. Rominger A, Saam T, Wolpers S, Cyran CC, Schmidt M, Foerster S, Nikolaou K, Reiser MF, Bartenstein P, Hacker M. 18f‐fdg pet/ct identifies patients at risk for future vascular events in an otherwise asymptomatic cohort with neoplastic disease. J Nucl Med. 2009;50:1611–1620. [DOI] [PubMed] [Google Scholar]
- 28. Ference BA. Causal effect of lipids and lipoproteins on atherosclerosis: Lessons from genomic studies. Cardiol Clin. 2018;36:203–211. [DOI] [PubMed] [Google Scholar]
- 29. Burgess S, Ference BA, Staley JR, Freitag DF, Mason AM, Nielsen SF, Willeit P, Young R, Surendran P, Karthikeyan S, et al. Association of lpa variants with risk of coronary disease and the implications for lipoprotein(a)‐lowering therapies: A mendelian randomization analysis. JAMA Cardiol. 2018;3:619–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tsimikas S. Rna‐targeted therapeutics for lipid disorders. Curr Opin Lipidol. 2018;29:459–466. [DOI] [PubMed] [Google Scholar]
- 31. Viney NJ, van Capelleveen JC, Geary RS, Xia S, Tami JA, Yu RZ, Marcovina SM, Hughes SG, Graham MJ, Crooke RM, et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): Two randomised, double‐blind, placebo‐controlled, dose‐ranging trials. Lancet. 2016;388:2239–2253. [DOI] [PubMed] [Google Scholar]