

Abstract
Uncovering additional long non-coding RNA (lncRNA)-disease associations has become increasingly important for developing treatments for complex human diseases. Identification of lncRNA biomarkers and lncRNA-disease associations is central to diagnoses and treatment. However, traditional experimental methods are expensive and time-consuming. Enormous amounts of data present in public biological databases are available for computational methods used to predict lncRNA-disease associations. In this study, we propose a novel computational method to predict lncRNA-disease associations. More specifically, a heterogeneous network is first constructed by integrating the associations among microRNA (miRNA), lncRNA, protein, drug, and disease, Second, high-order proximity preserved embedding (HOPE) was used to embed nodes into a network. Finally, the rotation forest classifier was adopted to train the prediction model. In the 5-fold cross-validation experiment, the area under the curve (AUC) of our method achieved 0.8328 ± 0.0236. We compare it with the other four classifiers, in which the proposed method remarkably outperformed other comparison methods. Otherwise, we constructed three case studies for three excess death rate cancers, respectively. The results show that 9 (lung cancer, gastric cancer, and hepatocellular carcinomas) out of the top 15 predicted disease-related lncRNAs were confirmed by our method. In conclusion, our method could predict the unknown lncRNA-disease associations effectively.
Keywords: lncRNA-disease associations, deep learning, heterogeneous information networks, rotation forest
Graphical Abstract
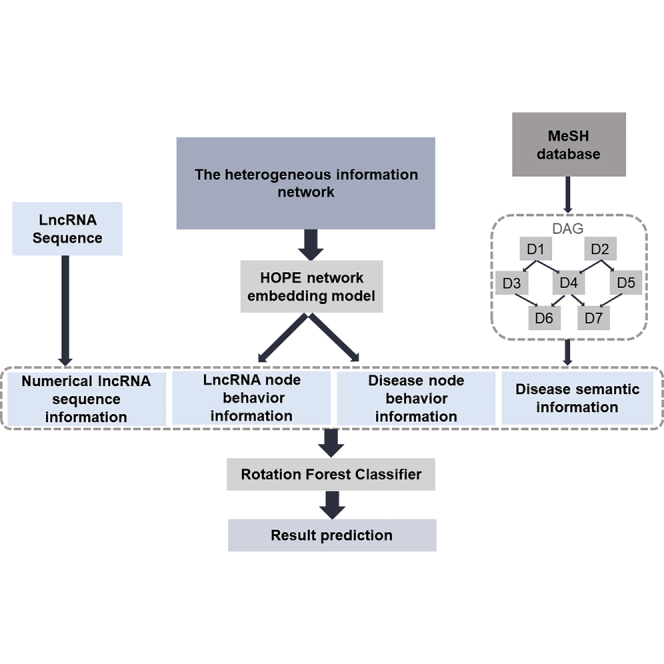
This article proposed a novel computational method to predict the potential associations between lncRNA and disease. This method extracts the information through the heterogeneous network, which is constructed by the information of serial biomolecular, drug, and disease. Then, the rotation forest was used as classifier to predict potential lncRNA-disease associations.
Introduction
RNA has been reported to play an intermediary role when the encoded protein is translated from DNA sequences.1 Only 2% of the Human genome encodes protein, and the remaining of 98% is known as non-coding RNAs (ncRNAs).2 Long non-coding RNA (lncRNA), which belongs to the heterogeneous class of ncRNAs, is an ncRNA with non-protein-coding transcripts longer than 200 nucleotides.3 Several previous works show that lncRNAs play a significant role in many biological processes, such as immune responses and chromosome, dynamic circuitry controlling pluripotency, and differentiation. Furthermore, lncRNA also play important role in many complex diseases, such as malignancies including lung cancer,4 hepatocellular cancer,5 and prostate cancer.6 For instance, the upregulation of DANCR is an essential factor in the development of lung cancer, especially in high-grade lung cancer tissues. The proliferation and colony formation of lung cancer is induced by ectopic DANCR expression. Thus, DANCR upregulation is an indicator of aggressive lung cancer. Another example is the HOTAIR, which is expressed about 100 to 2,000 times at the normal levels in breast cancer metastases.7 A similar association is found in other cancer types, including liver and gastric cancer. Therefore, finding an efficient way to predict additional associations among lncRNA and disease is a challenge for future progress.
In recent years, an increasing number of machine learning and data mining methods have been developed to take advantage of biological databases on large-scale lncRNA-disease associations. Blom et al.8 put forward a supervised machine learning for predicting gene-disease associations, in which a biased support vector machine is utilized. A random walk model with restart walking was proposed by Chen et al.9 to rank the microRNAs (miRNAs)-disease associations. However, these methods only recover a portion of all features contained in the databases. Currently, enormous datasets are populated using advanced technologies. A thorough understanding of the functions and mechanisms of lncRNAs will require a complete analysis of all this information.10 In addition, NONCODE provides a systematic platform including expression profiles and functions.11 Thus, constructing a network among different molecular nodes simultaneously and systemically is beneficial to capture the complicated relationships between ncRNA and diseases.
Xiong et al.12 constructed a heterogeneous biological network named HeteWalk, which includes miRNAs, genes, and diseases. You et al.13 developed a pathway method to construct the network through miRNA-miRNA, disease-disease similarities, and associations between miRNAs and diseases. Chen et al.14 proposed a network method, hyper-geometric distribution for lncRNA-disease association inference (HGLDA), in which the information of associations among miRNA and lncRNA, miRNA and disease is used to predict associations between lncRNA and miRNA. Sun et al.15 proposed a global network computational framework, RWRlncD, to identify possible associations between lncRNAs and diseases.
Previous efforts to capture attributes of proteins and ncRNAs and associations among these nodes form a substantive basis for additional work. The existing network graph method uses the associations among ncRNAs, proteins, and diseases. A more holistic and systematic heterogeneous network method would use a combination of known associations and attributes of all nodes in this existing network. There have several proposed methods focus on learning latent features from networks with multi-source features. He et al.16 proposed CCPMVFGC, which can discover clusters in graphs with multi-view vertex feature. For utilizing the constructed network, the network embedding method is introduced to our work to extract discriminative features from network topology. This method is popular for identifying potential relationships in social networks. This method allows for the preservation of vertex content, extra information, and topological structure, a low-dimensional feature space by mapping network data. In the low-dimensional vector space, each node in the network is mapped to a point, and connections between nodes are positively correlated to distances in the vector space.
The first challenge for constructing a heterogeneous network is choosing appropriate entities such as ncRNA and disease, along with other related items. Association among nodes can then be depicted concisely and clearly. A further challenge is identifying an efficient graph embedding algorithm. To address these issues, a heterogeneous network is constructed and analyzed by high-order proximity preserved embedding (HOPE) to identify potential associations among lncRNAs and diseases. First, a heterogeneous network is constructed by integrating the associations among miRNA, lncRNA, protein, disease, and drug. Second, each node in the network is represented as a vector by combining attribute feature of the node itself (e.g., sequences of ncRNAs and proteins, semantics of diseases and molecular fingerprints of drugs) and the behavior feature of the node in the complex network (associations with other nodes). Finally, the rotation forest model is chosen as the classifier for predicting new associations between lncRNA and disease. Furthermore, the proposed method was evaluated in lung, colorectal, and breast cancer. The experimental results demonstrate that the proposed method can quantitively identify the potential associations between lncRNA and disease.
Results
Performance evaluation measures
The k-fold cross-validation is a popular procedure in most algorithms designed for classifying two input categories of items or for comparing performance on a single dataset. The parameter k is set as 5 in our experiment. Specifically, a dataset is divided randomly into 5 disjointed folds of equal size. Each fold is then used in turns to test the model trained by the classifiers from the other 4 folds. The performance of classifiers is evaluated using the average of 5 values from 5-fold cross-validation. In practice, some nodes without association and nodes contribute simultaneously are isolated. Even though, the manual experiment of researchers can be simulated by this situation better. Manual assessment may address such a situation more efficiently.
For a comprehensive performance evaluation, a series of broader evaluation criteria are introduced to evaluate, including accuracy (Acc.), sensitivity (Sen.), specificity (Spec.), precision (Prec.), Matthews correlation coefficient (MCC), and area under the curve (AUC). The equations of accuracy (Acc.), sensitivity (Sen.), specificity (Spec.), precision (Prec.), and Matthews correlation coefficient (MCC) were listed as follows:
| (Equation 1) |
| (Equation 2) |
| (Equation 3) |
| (Equation 4) |
| (Equation 5) |
While in these equations, TP, TN, FP, FN, mean true positive, true negative, false positive, and false negative, respectively. In addition, the proposed model is evaluated using two visualization methods. First method uses the AUC, which is the area surrounded by the Receiver characteristic curve (ROC) in a coordinate system whose abscissa is the false positive rate (FPR) and the ordinate is the true positive rate (TPR). For another perspective, the area under the Precision-Recall curve is also applied. The abscissa for the graph is recall and the ordinate is precision. Average AUC and AUPR for performance of the proposed model are both positive correlations. The experimental results shown in Figure 1 and Figure 2 illustrate that our model is acceptable.
Figure 1.

The ROC curves of our methods
The AUC is the area under the receiver operating characteristic curves (ROC).
Figure 2.

The PR curves of our methods
The AUPR is the area under the precision-recall (PR) curves.
Comparison with different types of features
In the constructed network, each node can be represented by both attribute and behavior features. These two kinds of information can be combined. A comparison experiment is designed for assessing attribute and behavior information separately and in combination. In Table 1, the results of the average of Acc., Sen., Spec., Prec., MCC, and AUC is 78.69%, 75.06%, 82.32%, 80.93%, 57.54%, and 83.28%, respectively. These average values represent the ability of the proposed model to predict random associations in the constructed network.
Table 1.
5-fold cross-validation results of our method
| Fold | Acc. (%) | Sen. (%) | Spec. (%) | Prec. (%) | MCC (%) | AUC (%) |
|---|---|---|---|---|---|---|
| 0 | 78.87 | 76.49 | 81.25 | 80.31 | 57.8 | 84.86 |
| 1 | 75.15 | 70.83 | 79.46 | 77.52 | 50.49 | 79.63 |
| 2 | 81.55 | 77.68 | 85.42 | 84.19 | 63.28 | 85.78 |
| 3 | 78.12 | 74.40 | 81.85 | 80.39 | 56.41 | 82.84 |
| 4 | 79.76 | 75.89 | 83.63 | 82.26 | 59.70 | 83.28 |
| Average | 78.69 ± 2.36 | 75.06 ± 2.64 | 82.32 ± 2.28 | 80.93 ± 2.48 | 57.54 ± 4.71 | 83.28 ± 2.36 |
The experimental results are shown in Table 2 and Figures 3 and 4. Both AUPR and AUC performance is improved when attribute and behavior information are combined. The AUC is 71.92% and 81.88% when attribute and behavior information are used separately, but it improves to 83.28% combined.
Table 2.
Comparison of different features, respectively and simultaneously
| Feature | Acc. (%) | Sen. (%) | Spec. (%) | Prec. (%) | MCC (%) | AUC (%) |
|---|---|---|---|---|---|---|
| Attribute | 71.85 ± 1.55 | 66.79 ± 3.37 | 76.90 ± 1.63 | 74.31 ± 1.27 | 43.94 ± 2.97 | 71.92 ± 2.09 |
| Behavior | 79.16 ± 2.88 | 71.37 ± 3.11 | 86.96 ± 2.97 | 84.57 ± 3.41 | 59.06 ± 5.81 | 81.88 ± 2.61 |
| Both | 78.69 ± 2.36 | 75.06 ± 2.64 | 82.32 ± 2.28 | 80.93 ± 2.48 | 57.54 ± 4.71 | 83.28 ± 2.36 |
Figure 3.

Comparison of different features under 5-fold cross-validation, respectively
Figure 4.

Comparison of different features under 5-fold cross-validation simultaneously
Comparison with other methods
The rotation forest model is chosen as the classifier for predicting new lncRNA-disease associations. To show the outstanding performance of this classifier, several classifiers, including Random Forest, Decision Tree, Gradient Boost, and Naive Bayes were initially compared. Parameters were set as the default unless otherwise indicated. The experimental results are shown in Table 3. It can be observed that the prediction performance of rotation forest model is better than Random Forest due to its use of PCA. It can also see that Bagging is more suitable than boosting in this situation and an ensemble structure of “trees “outperforms a series of “weak” classifiers.
Table 3.
Comparison of different classifiers
| Classifier | Acc. (%) | Sen. (%) | Spec. (%) | Prec. (%) | MCC (%) | AUC (%) |
|---|---|---|---|---|---|---|
| Decision tree | 73.57 ± 1.74 | 66.07 ± 2.69 | 81.07 ± 2.39 | 77.77 ± 2.23 | 47.71 ± 3.51 | 73.55 ± 1.79 |
| GDBT | 78.69 ± 1.66 | 68.21 ± 2.42 | 89.17 ± 1.24 | 86.29 ± 1.68 | 58.69 ± 3.24 | 81.27 ± 1.30 |
| Naive bayes | 68.69 ± 2.66 | 48.16 ± 3.33 | 89.23 ± 2.25 | 81.67 ± 4.05 | 40.99 ± 5.64 | 79.02 ± 2.45 |
| Random forest | 78.75 ± 2.43 | 68.75 ± 2.68 | 88.75 ± 2.47 | 85.95 ± 3.05 | 58.69 ± 4.93 | 81.35 ± 1.85 |
| Rotation forest | 78.69 ± 2.36 | 75.06 ± 2.64 | 82.32 ± 2.28 | 80.93 ± 2.48 | 57.54 ± 4.71 | 83.28 ± 2.36 |
Case study
After running the proposed method through 5-fold cross-validation, a case study was introduced to evaluate the overall performance. Three types of cancer with high mortality rates including lung, breast, and colorectal cancer were chosen for the test. This case study was designed as follows. Known associations were removed before datasets were embedded using HOPE. Before ranking results, rest lncRNA-disease associations were trained. As a result, 9 of the top 15 lung cancer associations, 9 of the top 15 gastric cancer associations, and 9 of the top 15 hepatocellular carcinoma associations were validated in lnc2cancer v2.0.
The three cancer types were chosen for their high mortality and incidence rates. More than 1.3 million patients are estimated to die of lung cancer annually,17 with high incidence in China.18 Even in Europe, the 5-year survival rate is only approximately 10%. The high rate of treatment failure correlates with metastatic disease at diagnosis. Earlier diagnosis during the stage where surgery is a viable treatment increases the survival rate to more than 70%. Several lncRNAs show obviously higher expression in tumor tissues in patients with small-cell lung cancer (SCLC) and non-small-cell lung cancer (NSCLC). The expression of lncRNA is both upregulation and downregulation in lung cancer cells. For example, the higher expression of some lncRNA is considered as an indicator for NSCLC. To be more specific, HOTAIR was indicated that there is an upregulated association between this lncRNA and NSCLC. Further, loss of imprinting of H19 can cause lung cancer. Expression of KCNQ1OT1, demonstrated by the knockdown experiment, is correlated to a series of behaviors observed for lung adenocarcinomas, such as positive lymphatic metastasis and larger tumor size. In the NSCLC cases, XIST is recognized as an oncogenic lncRNA in driving tumorigenesis is overexpressed in these cells.
Even though there still exists much geographical variation, especially in East Asia and China, gastric cancer is the second most frequent cause of cancer-related deaths worldwide. Most patients with gastric cancer are diagnosed at a late stage of the disease, and mortality can be reduced by early detection. Some specific risk profiles, including the presence of precursor lesions and gene polymorphisms, should be considered while deciding on the diagnosis and treatment. Overexpression of H19 is known to promote proliferation, invasion, and other features of gastric cancer.19 The expression of ANRIL, measured by qPCR, is correlated with a higher TNM stage and larger tumor size. These features are independent predictors for overall survival.20 Downregulation associated with MEG3 is correlated with maternal expression of gene 3 and gastric cancer. Under-expression of MEG3 in gastric tissues is correlated with poor prognosis.21 Compared to patients with lower levels of NEAT1, patients with higher levels show poor survival. Associations between NEAT1 and gastric cancer include upregulation and overexpression of this lncRNA as an independent prognostic factor in these gastric cases.22
Hepatocellular carcinoma has become the fifth most common cancer and the third most common cause of cancer mortality worldwide in recent years. Mortality from this cancer increased by 25% between 2002 and 2012 and reached an incidence of 3.1 per 100,000 for men. In Asia, the incidence of liver cancer is expected to decrease. In Japan, the incidence was predicted to decrease to 5.4 per 10,000 for men in 2020. Except in East Asia, younger individuals from most regions show a more encouraging trend in the incidence of liver cancer than middle-aged individuals. It was also common that the mortality among women is lower than for men by 3-fold to 5-fold. H19 and IGF2 are regulated in parallel in the hepatocellular carcinomas. A common feature of this disease is the disruption of IGF2 prompter regulation.23 The tumor suppressor candidate 7 (TUSC7) suppresses epithelial-to-mesenchymal transformation through TUSC7-miR-10a-EphA4 axis, which is probably a potential target for treatment in hepatocellular carcinoma.24 The correct prediction of case study was shown in Table 4.
Table 4.
Marked lncRNAs associations between lung cancer, gastric cancer, and hepatocellular carcinomas
| lncRNA | Disease | Rank |
|---|---|---|
| HOTAIR | lung cancer | 3.10 |
| H19 | lung cancer | 6.7 |
| KCNQ1OT1 | lung cancer | 8 |
| MEG3 | lung cancer | 12 |
| UCA1 | lung cancer | 13 |
| XIST | lung cancer | 14 |
| linc-ROR | lung cancer | 15 |
| H19 | gastric cancer | 1 |
| MEG3 | gastric cancer | 3 |
| HOTAIR | gastric cancer | 6 |
| NEAT1 | gastric cancer | 7 |
| XIST | gastric cancer | 8 |
| UCA1 | gastric cancer | 9 |
| ANRIL | gastric cancer | 10 |
| CASC2 | gastric cancer | 12 |
| link-ROR | gastric cancer | 13 |
| H19 | hepatocellular carcinomas | 1.7 |
| HOTAIR | hepatocellular carcinomas | 2.3 |
| ANRIL | hepatocellular carcinomas | 5 |
| IGF2-AS | hepatocellular carcinomas | 9 |
| MEG3 | hepatocellular carcinomas | 12 |
| TUSC7 | hepatocellular carcinomas | 13 |
| NEAT1 | hepatocellular carcinomas | 15 |
Discussion
Recently, experimental technologies have proved plenty of known associations between lncRNA and disease. These associations play a crucial role in the prevention and treatment in severe disease, especially neoplasm. Due to the expensive and inefficient traditional experimental methods, we proposed this computational method based on known dataset and data mining method, for which the potential associations can be discovered.
This method has several advantages as listed. (1) The full use of biology information. The node representation was combined by not only the information of molecular function such as, lncRNA and miRNA molecular function, protein sequence, and semantic of disease, but also, the associations and interactions among the miRNA, lncRNA, protein, disease, and drug. (2) The HOPE was used to embed the network and present the node behavior. This method can preserve the feature information in graph network. (3) The rotation forest was used to train the model and predict the potential associations. The reason we chose these classifiers was for the following purposes: (1) for random forest classifier, principal-component analysis (PCA) was applied to each feature set to preserve variability information. The same number of “trees” was set at 45 for comparisons with rotation forest model. (2) Gradient boosting is a member of a family of classifiers; boosting uses an ensemble structure to boost some “weak” classifiers. (3) Decision tree model is a “weak” classifier chosen in the rotation forest because of its sensitivity to rotation of feature axes. (4) Naive Bayes is a popular “weak” classifier used when features are independent. The results of comparison with other classifiers shows that the process of PCA bring an outstanding performance.
Materials and methods
Construction of the molecular association network
Several databases that include known associations among miRNA, lncRNA, protein, disease, and drugs were used to construct the systematic and holistic molecular association network. The data used in the final model were selected by unifying identifiers, simplifying, eliminating redundancy, and deleting the extraneous items, which is shown in the Table 5. Divergent nodes were allocated separately from the Table 6 and concise amounts are provided in Table 6. The construction of the network is shown in Figure 5.
Table 5.
Nine associations involved in the heterogeneous network
| Relationship type | Database | Number of associations |
|---|---|---|
| miRNA-lncRNA | lncRNASNP225 | 8,374 |
| miRNA-disease | HMDD26 | 16,427 |
| miRNA-protein | miRTarBase27 | 4,944 |
| lncRNA-disease | lncRNAdisease28 | 1,680 |
| lncRNASNP225 | ||
| lnc2Cancer10 | ||
| lncRNA-protein | lncRNA2Target29 | 690 |
| protein-disease | DisGeNET30 | 25,087 |
| drug-protein | DrugBank31 | 11,107 |
| drug-disease | CTD32 | 18,416 |
| protein-protein | STRING33 | 19,237 |
| total | N/A | 105,963 |
Table 6.
The number of kinds of nodes in the heterogeneous network
| Node | Number of nodes |
|---|---|
| disease | 2,062 |
| lncRNA | 769 |
| miRNA | 1,023 |
| protein | 1,649 |
| drug | 1,025 |
| total | 6,528 |
Figure 5.

The network constructed by the multiple associations among different biomolecules
Representing sequences of lncRNA
To obtain the discriminative attribute feature, we downloaded sequences of lncRNA from Database: NONCODE.11 For simplifying our experiment, the sequences of lncRNA were encoded into a 64 (4 × 4 × 4) dimensional vector, where each dimension represents the normalized frequency of the corresponding 3-mer that was used in the lncRNA sequence (e.g., GAC, CUG, UGA).
MeSH for disease descriptions and directed acyclic graph
Medical Subject Headings (MeSH) are developed by the National Library of Medicine.34 An effective “tree structure” was introduced to automatically retrieve these descriptions. The MeSH tree structures are poly-hierarchical, that is, one main heading can be found in more than one subcategory. For instance, for the heading, Gallbladder Neoplasms, Neoplasms is a disease listed under Neoplasms and under Digestive System Disease. Within this structure, the most search is accurate and objective. The tree can be expressed in another form as a directed acyclic graph (DAG). The method for describing the disease in the DAG is as follows:
| (Equation 6) |
where N(d) means the points in the set that include all the diseases in the DAG(d). E(d) denotes the edges in the set that include all connections between nodes in the DAG(d). Figure 6 illustrates the description of the disease Astrocytoma.
Figure 6.

The directed acyclic graph of a type of digestive system disease, gastrointestinal neoplasms
For defining semantic similarity among diseases, we used an approach that calculates means of DAG. The following formula reveals how disease in any ancestral disease contributes to disease .
| (Equation 7) |
where, , a factor that contributes semantically, is set at 0.5 according to previous works. The weight of disease for itself is 1. The contribution of other nodes to this disease is attenuated and assessed to yield . From Equation (1), the sum of contributions of all ancestral nodes in the graph to is estimated as follows:
| (Equation 8) |
Moreover, the semantic similarity between the two diseases is calculated using the Jaccard similarity coefficient:
| (Equation 9) |
In this equation, p and q are two diseases, which would be calculated to find the similarity. would calculate all same father node of these two diseases and would calculate the mean of these two diseases.
Stacked auto-encoder
To avoid the labor-intensive and handcraft feature design, we used Sparse Auto-Encoder (SAE) in our method, which is an unsupervised feature leaning method. The hidden layer is acted as a feature extraction of the input layer. The output and input are equal in SAE finally. More specifically, SAE is an unsupervised feedforward neural network. is set as unsupervised examples. Encoder function,, maps the input layer n to hidden layer m, while decoder function, , is used to recover y from m. W is the connected parameter between two layers, b is a bias, and is a non-linear mapping. SAE can train a model that can approximate m with y, and the hidden layer is a new representation of data.
The encoder function and decoder are shown as below. The encoder function maps the input layer n to hidden layer m, while decoder function recover n from m. W is the connected parameters between two layers, p and q are bias. and denote non-linear mapping. SAE is for training a model that approximates x with y. The hidden layer can be considered as feature extraction of the input layer.
| (Equation 10) |
| (Equation 11) |
Additional constraints are added for avoiding training a trivial identity mapping, which may lead to a compressive representation. It will also add noises to data, in which SAE would recover the data from a corrupted version, and the representation would also be robust. Here, the ReLU is chosen as the function for activation.
| (Equation 12) |
Node representation
Two categories of information are used for representing nodes in the network. First, intrinsic attributes of nodes including sequences of miRNA, lncRNA, and protein, fingerprints of drugs, and semantic disease descriptions are used. Second, associations between nodes are defined as the behavior of nodes in the molecular network. Node behavior was represented using HOPE on the entire network.35
Recently, undirected graphs have been targeted by graph embedding methods. However, the method used for undirected graphs cannot be used for the directed graphs because of asymmetric transitivity, which is a fundamental characteristic of these graphs. HOPE preserves asymmetric transitivity in directed graphs during embedding learning. In this study, the loss function is minimized as follows:
| (Equation 13) |
In this formula, asymmetric transitivity can be affected by several high-order proximities. However, the approximation of proximities is facilitated by the general formula as follows:
| (Equation 14) |
Further, and are both polynomial matrices. A series of proximity measurements can be part of deduction in this equation, and all measurements can be classified into two categories. The first category is global proximity. Specifically, global asymmetric transitivity can be preserved with the Katz index and rooted PageRank, because these two measures are derived from a recurrent formula. The second category is local asymmetric transitivity. For instance, asymmetric transitivity only can be preserved in a local structure, because no recurrent structure exists in common neighbors and Adamic-Adar. Thus, an optimal rank-K approximation of the proximity matrix S can be determined by singular value decomposition (SVD). Embedding are constructed as follows:
| (Equation 15) |
where are singular values sorted in decreasing order; and are corresponding singular vectors of . Optimal embedding vectors can be expressed as follows:
| (Equation 16) |
| (Equation 17) |
To avoid calculating this matrix inversion in time complexity , it is necessary to use a previously proposed method to calculate by the equation below:
| (Equation 18) |
Finally, error bounds of this algorithm are produced as follows:
| (Equation 19) |
Conclusions
An increasing number of associations between lncRNA and disease have been detected by advanced biological techniques. A computational method was are increasingly important for identifying potential associations because detection such associations using biological experiments is expensive. In this study, a novel computational method was developed by constructing and embedding a heterogeneous network using HOPE. The experimental results demonstrated that the proposed method achieved an outstanding performance for predicting lncRNA-disease associations. The proposed method was further validated using three case studies of high mortality cancer types (lung, breast, and colorectal cancer) to demonstrate its predictive power and reliability.
Acknowledgments
This work is supported by the Science and Technology Innovation 2030-New Generation Artificial Intelligence Major Project (No. 2018AAA0100100), and in part by the NSFC Excellent Young Scholars Program under grant 61722212.
Author contributions
J.-R.Z. and Z.-H.Y. conceived the algorithm, carried out analyses, prepared the data sets, carried out experiments, and wrote the manuscript. Other authors designed, performed, and analyzed experiments and wrote the manuscript. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
References
- 1.Crick F.H.C., Barnett L., Brenner S., Watts-Tobin R.J. General nature of the genetic code for proteins. Nature. 1961;192:1227–1232. doi: 10.1038/1921227a0. [DOI] [PubMed] [Google Scholar]
- 2.Goldman N., Bertone P., Chen S., Dessimoz C., LeProust E.M., Sipos B., Birney E. Towards practical, high-capacity, low-maintenance information storage in synthesized DNA. Nature. 2013;494:77–80. doi: 10.1038/nature11875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kapranov P., Cheng J., Dike S., Nix D.A., Duttagupta R., Willingham A.T., Stadler P.F., Hertel J., Hackermüller J., Hofacker I.L. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316:1484–1488. doi: 10.1126/science.1138341. [DOI] [PubMed] [Google Scholar]
- 4.Loewen G., Jayawickramarajah J., Zhuo Y., Shan B. Functions of lncRNA HOTAIR in lung cancer. J. Hematol. Oncol. 2014;7:90. doi: 10.1186/s13045-014-0090-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Panzitt K., Tschernatsch M.M.O., Guelly C., Moustafa T., Stradner M., Strohmaier H.M., Buck C.R., Denk H., Schroeder R., Trauner M. Characterization of HULC, a Novel Gene With Striking Up-Regulation in Hepatocellular Carcinoma. as Noncoding RNA. 2007;132:330–342. doi: 10.1053/j.gastro.2006.08.026. [DOI] [PubMed] [Google Scholar]
- 6.Pickard M.R., Mourtada-Maarabouni M., Williams G.T. Long non-coding RNA GAS5 regulates apoptosis in prostate cancer cell lines. Biochim. Biophys. Acta. 2013;1832:1613–1623. doi: 10.1016/j.bbadis.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 7.Gupta R.A., Shah N., Wang K.C., Kim J., Horlings H.M., Wong D.J., Tsai M.-C., Hung T., Argani P., Rinn J.L. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–1076. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh-Blom U.M., Natarajan N., Tewari A., Woods J.O., Dhillon I.S., Marcotte E.M. Prediction and validation of gene-disease associations using methods inspired by social network analyses. PLoS ONE. 2013;8:e58977. doi: 10.1371/journal.pone.0058977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen H., Zhang Z. Prediction of associations between OMIM diseases and microRNAs by random walk on OMIM disease similarity network. ScientificWorldJournal. 2013;2013:204658. doi: 10.1155/2013/204658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ning S., Zhang J., Peng W., Hui Z., Wang J., Yue L., Yue G., Guo M., Ming Y., Wang L. Lnc2Cancer: a manually curated database of experimentally supported lncRNAs associated with various human cancers. Nucleic Acids Res. 2015;44:980–985. doi: 10.1093/nar/gkv1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bu D., Yu K., Sun S., Xie C., Geir S., Miao R., Hui X., Qi L., Luo H., Zhao G. NONCODE v3.0: integrative annotation of long noncoding RNAs. Nucleic Acids Res. 2011;D1:D1. doi: 10.1093/nar/gkr1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiong Y., Ruan L., Guo M., Tang C., Wang W. BIBM; 2018. Predicting Disease-Related Associations by Heterogeneous Network Embedding; pp. 548–555. (IEEE International Conference on Bioinformatics and Biomedicine). [Google Scholar]
- 13.You Z.H., Huang Z.A., Zhu Z., Yan G.Y., Li Z.W., Wen Z., Chen X. PBMDA: A novel and effective path-based computational model for miRNA-disease association prediction. PLoS Comput. Biol. 2017;13:e1005455. doi: 10.1371/journal.pcbi.1005455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen X. Predicting lncRNA-disease associations and constructing lncRNA functional similarity network based on the information of miRNA. Sci. Rep. 2015;5:13186. doi: 10.1038/srep13186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun J., Shi H., Wang Z., Zhang C., Liu L., Wang L., He W., Hao D., Liu S., Zhou M. Inferring novel lncRNA-disease associations based on a random walk model of a lncRNA functional similarity network. Mol. Biosyst. 2014;10:2074–2081. doi: 10.1039/c3mb70608g. [DOI] [PubMed] [Google Scholar]
- 16.Perozzi B., Al-Rfou R., Skiena S. Vol. 2014. 2014. DeepWalk: Online Learning of Social Representations; pp. 701–710. (Proceedings of the 20th ACM SIGKDD international conference). [Google Scholar]
- 17.Travis W.D. Lung Cancer. 2015;75:191–202. doi: 10.1002/1097-0142(19950101)75:1+<191::aid-cncr2820751307>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 18.Zheng R., Zeng H., Zuo T., Zhang S., Qiao Y., Zhou Q., Chen W. Lung cancer incidence and mortality in China, 2011. Thorac. Cancer. 2016;7:94–99. doi: 10.1111/1759-7714.12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H., Yu B., Li J., Su L., Yan M., Zhu Z., Liu B. Overexpression of lncRNA H19 enhances carcinogenesis and metastasis of gastric cancer. Oncotarget. 2014;5:2318–2329. doi: 10.18632/oncotarget.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang E.B., Kong R., Yin D.D., You L.H., Sun M., Han L., Xu T.P., Xia R., Yang J.S., De W., Chen J.f. Long noncoding RNA ANRIL indicates a poor prognosis of gastric cancer and promotes tumor growth by epigenetically silencing of miR-99a/miR-449a. Oncotarget. 2014;5:2276–2292. doi: 10.18632/oncotarget.1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun M., Xia R., Jin F., Xu T., Liu Z., De W., Liu X. Downregulated long noncoding RNA MEG3 is associated with poor prognosis and promotes cell proliferation in gastric cancer. Tumour Biol. 2014;35:1065–1073. doi: 10.1007/s13277-013-1142-z. [DOI] [PubMed] [Google Scholar]
- 22.Fu J.W., Kong Y., Sun X. Long noncoding RNA NEAT1 is an unfavorable prognostic factor and regulates migration and invasion in gastric cancer. J. Cancer Res. Clin. Oncol. 2016;142:1571–1579. doi: 10.1007/s00432-016-2152-1. [DOI] [PubMed] [Google Scholar]
- 23.Li X., Nong Z., Ekström C., Larsson E., Nordlinder H., Hofmann W.J., Trautwein C., Odenthal M., Dienes H.P., Ekström T.J., Schirmacher P. Disrupted IGF2 promoter control by silencing of promoter P1 in human hepatocellular carcinoma. Cancer Res. 1997;57:2048–2054. [PubMed] [Google Scholar]
- 24.Wang Y., Liu Z., Yao B., Dou C., Xu M., Xue Y., Ding L., Jia Y., Zhang H., Li Q. Long non-coding RNA TUSC7 acts a molecular sponge for miR-10a and suppresses EMT in hepatocellular carcinoma. Tumour Biol. 2016;37:11429–11441. doi: 10.1007/s13277-016-4892-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miao Y.-R., Liu W., Zhang Q., Guo A.-Y. lncRNASNP2: an updated database of functional SNPs and mutations in human and mouse lncRNAs. Nucleic Acids Res. 2018;46(D1):D276–D280. doi: 10.1093/nar/gkx1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang Z., Shi J., Gao Y., Cui C., Zhang S., Li J., Zhou Y., Cui Q. HMDD v3.0: a database for experimentally supported human microRNA-disease associations. Nucleic Acids Res. 2019;47(D1):D1013–D1017. doi: 10.1093/nar/gky1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chou C.-H., Shrestha S., Yang C.-D., Chang N.-W., Lin Y.-L., Liao K.-W., Huang W.-C., Sun T.-H., Tu S.-J., Lee W.-H. miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018;46(D1):D296–D302. doi: 10.1093/nar/gkx1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen G., Wang Z., Wang D., Qiu C., Liu M., Chen X., Zhang Q., Yan G., Cui Q. LncRNADisease: a database for long-non-coding RNA-associated diseases. Nucleic Acids Res. 2013;41(Database issue, D1):D983–D986. doi: 10.1093/nar/gks1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheng L., Wang P., Tian R., Wang S., Guo Q., Luo M., Zhou W., Liu G., Jiang H., Jiang Q. LncRNA2Target v2.0: a comprehensive database for target genes of lncRNAs in human and mouse. Nucleic Acids Res. 2019;47(D1):D140–D144. doi: 10.1093/nar/gky1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Piñero J., Bravo À., Queralt-Rosinach N., Gutiérrez-Sacristán A., Deu-Pons J., Centeno E., García-García J., Sanz F., Furlong L.I. DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017;45(D1):D833–D839. doi: 10.1093/nar/gkw943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wishart D.S., Feunang Y.D., Guo A.C., Lo E.J., Marcu A., Grant J.R., Sajed T., Johnson D., Li C., Sayeeda Z. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46(D1):D1074–D1082. doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davis A.P., Grondin C.J., Johnson R.J., Sciaky D., McMorran R., Wiegers J., Wiegers T.C., Mattingly C.J. The Comparative Toxicogenomics Database: update 2019. Nucleic Acids Res. 2019;47(D1):D948–D954. doi: 10.1093/nar/gky868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Szklarczyk D., Morris J.H., Cook H., Kuhn M., Wyder S., Simonovic M., Santos A., Doncheva N.T., Roth A., Bork P. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–D368. doi: 10.1093/nar/gkw937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lowe H.J., Barnett G.O. Understanding and using the medical subject headings (MeSH vocabulary to perform literature. JAMA. 1994;271:1103–1108. [PubMed] [Google Scholar]
- 35.Ou M., Peng C., Jian P., Zhang Z., Zhu W. Vol. 2016. 2016. Asymmetric Transitivity Preserving Graph Embedding; pp. 1105–1114. (Proceedings of the 22nd ACM SIGKDD International Conference). [Google Scholar]