
Fig 3 |. Bioinformatics workflow.
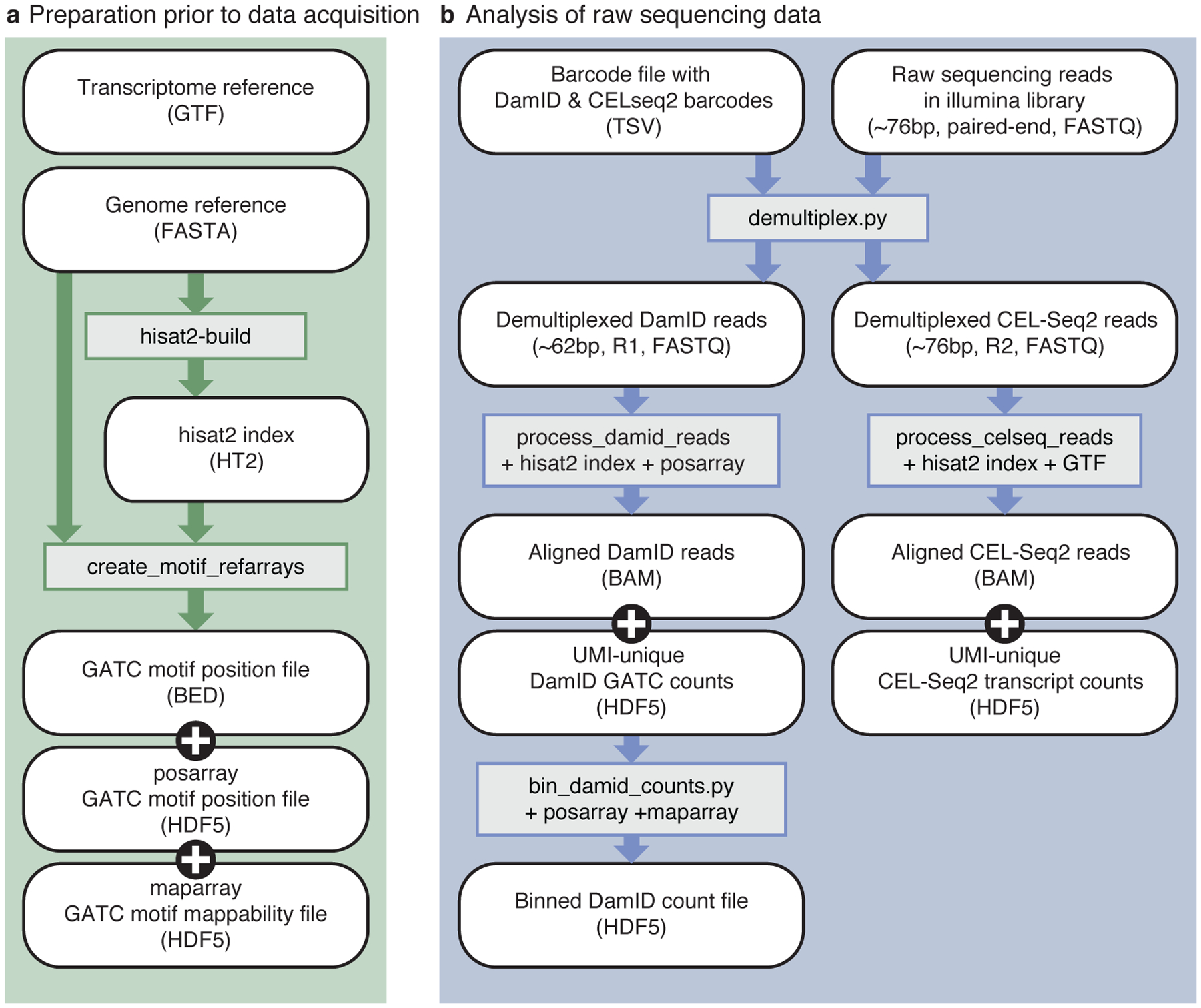
a) Preparation of reference files, which only needs to be performed once per reference genome. The genome reference (FASTA) file is used as input to generate the HISAT2 index, as well as the motif arrays. b) Processing of raw sequencing data to tables of unique DamID and CEL-Seq2 counts. White, rounded boxes show (intermediate) files; grey, rectangular boxes show programs and necessary reference files. Arrows indicate which files are used as input for subsequent programs.