

Abstract
FMS‐like tyrosine kinase 3 (FLT3) plays a key role in hematopoiesis. However, the oncogenic role of FLT3 amplification in patients with metastatic colorectal cancer (mCRC) remains unclear. Here, we aimed to evaluate the characteristics, prognosis, and treatment efficacy of an FLT3 inhibitor (regorafenib) in patients with mCRC with FLT3 amplifications. Tumor tissue samples from 2329 patients were sequenced using NGS in the Nationwide Cancer Genome Screening Project in Japan. The effects of clinicopathological features, co‐altered genes, prognosis, and efficacy of regorafenib were investigated. Between April 2015 and June 2018, 85 patients with mCRC with FLT3 amplification were observed. There were no differences in baseline characteristics between patients with or without FLT3 amplification. The frequency of RAS or other gene co‐alterations was inversely correlated with the copy number status. Median survival time in patients with FLT3 amplification was significantly shorter compared with those with non‐FLT3 amplification. Further investigations of FLT3 amplification as a potential treatment target in mCRC are warranted.
Keywords: colorectal cancer, copy number status, FLT3 amplification, next‐generation sequencing, prognosis
High FLT3 amplification may function not only as a prognostic factor but a promising treatment target in metastatic colorectal cancer.

Abbreviations
- CLIA
clinical laboratory improvement amendments
- EGFR
epidermal growth factor receptor
- FLT3
FMS‐like tyrosine kinase 3
- mCRC
metastatic colorectal cancer
- NGS
next‐generation sequencing
- OCA
OncomineTM Comprehensive Assay
- OCP
Oncomine™ Cancer Research Panel
- OS
Overall survival
- PDC
patient‐derived tumor cells
- PDGFR
platelet‐derived growth factor receptor
- PFS
progression‐free survival
- RTK
receptor tyrosine kinase
- STAT
signal transducer and activator transcription factor
- VEGFR
vascular endothelial growth factor receptor
1. INTRODUCTION
FMS‐like tyrosine kinase 3 (FLT3) is a member of the class III receptor tyrosine kinase family, which includes PDGF‐R, KIT, and FMS. 1 The FLT3 protein is normally expressed in hematopoietic progenitor stem cells and plays a key role in controlling the proliferation and differentiation of hematopoietic precursor cells. 2 FLT3‐activating mutations have been identified in approximately 30% of patients with acute myeloid leukemia. 3 The overexpression of wild‐type FLT3 is observed mainly in MLL‐rearranged acute lymphoblastic leukemia. 4 Overexpression and activating mutations of FLT3 are important targets for molecular therapy and are unfavorable prognostic factors. FLT3 tyrosine kinase inhibitors have been shown to be effective in hematopoietic cancer cells with FLT3 gene alterations. 5 More recently, comprehensive genomic analyses have indicated that the majority of FLT3 alterations are somatic mutations, followed by amplification, and FLT3 alterations have also been observed in solid tumors. The frequency of FLT3 amplifications was reported to be 1.0% in breast cancer, 6 3.9% in metastatic colorectal cancer (mCRC), 7 1.7% in gastric cancer, 8 and 0.9% in lung adenocarcinoma. 9
Two case reports of patients with mCRC with FLT3 amplifications, which were identified by targeted genomic profiling, demonstrated the clinical benefit of a multikinase inhibitor with inhibitory activity against FLT3 such as regorafenib or sorafenib. 10 , 11 Regorafenib is an orally available multikinase inhibitor that showed survival improvement in salvage line treatment in mCRC. 12 It emerged from the process of optimizing sorafenib by modulating its molecular structure, 13 therefore, like sorafenib, regorafenib blocks similar kinases such as Raf serine/threonine kinases (Raf‐1, wild‐type B‐Raf and B‐Raf V600E), vascular endothelial growth factor receptor (VEGFR) 1‐3, platelet‐derived growth factor receptor (PDGFR)‐β and FLT3, c‐Kit, and RET. 14 In a previous study, Lim et al evaluated the efficacy of regorafenib in mCRC with FLT3 amplification preclinically and clinically. 11 In their clinical investigation, although, FLT3 expression was slightly reduced following treatment with regorafenib or sorafenib, using PDC from an FLT3‐amplified colorectal cancer patient, patients with high copy numbers of FLT3 achieved partial response. 11 Therefore, the association between copy number of FLT3 and the efficacy of multikinase inhibitors still remains unclear.
To date, the clinical impact and the status of genetic heterogeneity such as co‐alteration genes of FLT3 amplifications in patients with mCRC is yet to be fully evaluated. In this study, we evaluated the characteristics, prognosis, genetic heterogeneity, and treatment efficacy of an FLT3 inhibitor (regorafenib) in patients with FLT3‐amplified mCRC.
2. MATERIALS AND METHODS
2.1. Study design and patients
This was an observational, retrospective, multicenter study on patients with mCRC. Tumor tissue samples from 2329 patients with mCRC were sequenced using the Oncomine Comprehensive Assay, an NGS‐based assay, in the Nationwide Cancer Genome Screening Project in Japan (SCRUM‐Japan GI‐SCREEN). The included patients: (i) had a pathologically confirmed colorectal adenocarcinoma, (ii) received systemic chemotherapy, (iii) had a RAS mutational status identified with a PCR‐based assay, (iv) had an Eastern Cooperative Oncology Group performance status (ECOG PS) of 0‐1, (v) had adequate bone marrow, renal, and hepatic function at the initiation of chemotherapy, (vi) had no other severe medical conditions, and (vii) provided written informed consent. The ethical, medical, and scientific aspects of the study were reviewed and approved by the Institutional Review Board. This trial was registered in the University Hospital Medical Information Network Clinical Trials Registry (UMIN000016343). The study was conducted in accordance with the 1975 Declaration of Helsinki, revised in 2000.
2.2. Targeted sequencing
Patients’ biopsies or archived surgically resected samples were sent to a CLIA‐certified clinical laboratory in the United States. In the CLIA laboratory, tumor DNA and RNA were extracted and used for NGS‐based amplicon sequencing with the Oncomine™ Cancer Research Panel (OCP) or Ion Torrent™ Oncomine™ Comprehensive Assay (OCA; Thermo‐Fisher Scientific, Waltham, MA). These assays covered 143 (v1, 2015‐2017) and 161 (v3, 2017 to current day) of the most relevant cancer‐related genes, respectively, and detected relevant single nucleotide variants, copy number variations, gene fusions, and indels in 1 streamlined workflow. The annotated genome variant call format files and the binary version of the sequence alignment/map files were stored at the SCRUM‐Japan Data Center.
2.3. Copy number status of FLT3 and other genes using tissue NGS
FLT3 gene copy number alteration was evaluated (Figure S1), and FLT3 amplification and other genes were defined as copy number ≥ 7.0 by OCA/OCP, which has been analytically and clinically validated. 15 , 16 FLT3 amplification status was divided into 2 categories using the median copy number. High amplification was defined as copy number ≥ 10.0, and low amplification was defined as 7 ≤ copy number < 10.0. A mutation was identified if the allele frequency was more than 5% and the depth of coverage was more than 250.
We evaluated the frequency of co‐alteration status of potential driver genes such as RAS/ BRAFV600E and other genes related to RTKs or those located on chromosome 13 in FLT3‐amplified mCRC patients. In addition, we evaluated the prognosis in patients with FLT3 amplifications who received at least second‐line or more chemotherapy, which is widely accepted as the standard treatment for mCRC.
2.4. Statistical analyses
OS and PFS were calculated using the Kaplan‐Meier method and compared with the log‐rank test. During systemic chemotherapy, which included treatment with regorafenib, each patient was assessed for objective response to treatment in accordance with the Response Evaluation Criteria in Solid Tumors (v.1.1) with computed tomographic scans performed every 2‐3 mo until disease progression. The disease control rates represented the percentage of patients with a complete response, partial response, and stable disease. Differences in proportion were evaluated with Pearson chi‐square test. The significance of differences in age was estimated with Kruskal‐Wallis test. Statistical analyses were conducted using SAS 9.3 software (SAS Institute, Cary, NC). All tests were 2‐sided, and a P‐value < .05 was considered to indicate statistical significance.
3. RESULTS
3.1. Patient characteristics
Between April 2015 and June 2018, 2078 patients who met the study inclusion criteria were recruited (Figure 1). Of these, 85 patients (3.6%) with mCRC and who had FLT3 amplifications were analyzed. There were no significant differences in baseline characteristics between patients with FLT3 amplifications (high and low) and those with non‐FLT3 amplifications, as shown in Table 1. The OCP or OCA results demonstrated that enrichment of TP53 mutations showed a trend for higher frequency in FLT3 amplification mCRC patients compared with in non‐FLT3 amplification patients, but the difference did not reach statistical significance (P = .16). The frequency of RAS mutations was similar in these 3 cohorts (P = .12). In contrast, there were activating alterations in both BRAFV600E (2.5% in low amplification vs 0% in high amplification vs 6.2%, P = .03) and there was a trend toward a lower frequency of PIK3CA mutations in patients with high FLT3 amplification compared with those with low or non‐FLT3 amplification (7.3% in non‐amplification and 10% in low amplification vs 2.2% in high amplification, P = .14).
FIGURE 1.
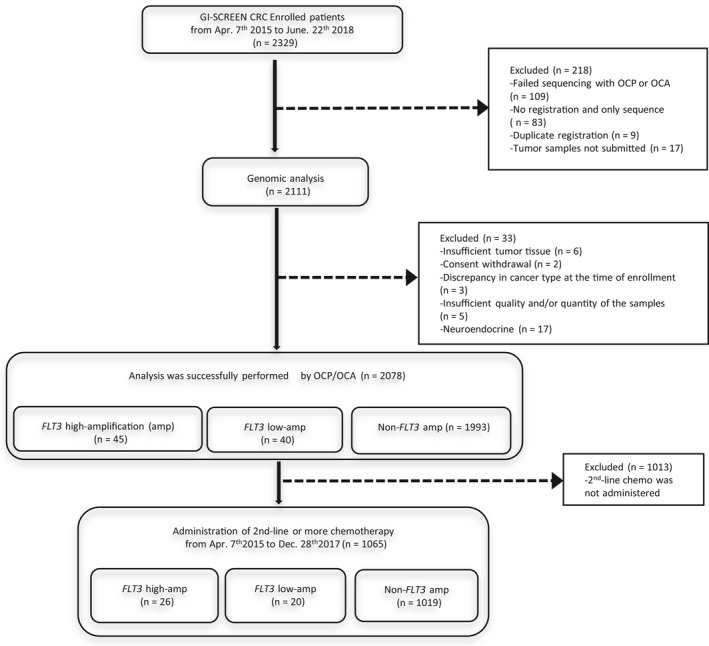
Flow diagram of this study. amp, amplification
TABLE 1.
Patients’ characteristics
| Non‐FLT3 amp (n = 1993) | FLT3 amp (n = 85) | P a | P b | ||
|---|---|---|---|---|---|
| Low amp (n = 40, 1.9%) n (%) | High amp (n = 45, 2.2%) n (%) | ||||
| Age | |||||
| Median, (range) (y) | 64 (21‐88) | 65 (35‐84) | 65 (32‐83) | .85 | .88 |
| Gender | |||||
| Male | 1103 (55.3) | 29 (72.5) | 21 (46.7) | .56 | .02 |
| Female | 90 (44.7) | 11 (28.5) | 24 (54.3) | ||
| Primary site | |||||
| Right colon | 619 (31.1) | 11 (27.5) | 13 (28.9) | <.01 | .06 |
| Left colon | 718 (36.0) | 15 (37.5) | 12 (71.1) | ||
| Rectum | 628 (31.5) | 10 (25.0) | 20 (44.4) | ||
| Unknown | 28 (1.4) | 4 (10.0) | 0 (0.0) | ||
| Histology (grade) | |||||
| Grade 1 | 543 (27.2) | 8 (20.0) | 9 (20.0) | .20 | .75 |
| Grade 2 | 1201 (60.3) | 26 (65.0) | 33 (73.3) | ||
| Grade 3 | 94 (4.7) | 3 (7.5) | 1 (2.2) | ||
| Others | 23 (5.2) | 2 (5.0) | 1 (2.2) | ||
| Unknown | 52 (2.6) | 1 (2.5) | 1 (2.2) | ||
| RAS | |||||
| Wild‐type | 1149 (57.2) | 16 (40.0) | 28 (63.2) | .12 | .04 |
| Mutant | 844 (42.8) | 24 (60.0) | 17 (37.8) | ||
| BRAF V600E | |||||
| Wild‐type | 1869 (93.8) | 39 (97.5) | 45 (100.0) | .03 | .29 |
| Mutant | 124 (6.2) | 1 (2.5) | 0 (0.0) | ||
| PIK3CA | |||||
| Wild‐type | 1763 (88.4) | 36 (90.0) | 44 (97.8) | .14 | |
| Mutant | 230 (7.3) | 4 (10.0) | 1 (2.2) | .13 | |
| ERBB2 | |||||
| Non‐Amp | 1938 (97.2) | 39 (97.5) | 45 (93.3) | .37 | .29 |
| Amp (CN ≥ 7.0) | 55 (2.8) | 1 (5.0) | 0 (0.0) | ||
| TP53 | |||||
| Wild‐type | 711 (35.7) | 11 (27.5) | 11 (24.5) | .16 | .75 |
| Mutant | 1282 (64.3) | 29 (72.5) | 34 (75.5) | ||
| BRCA1 | |||||
| Wild‐type | 1859 (93.2) | 31 (77.5) | 38 (80.0) | <.01 | .41 |
| Mutant | 134 (6.8) | 9 (22.5) | 7 (15.6) | ||
| BRCA2 | |||||
| Wild‐type | 1731 (86.9) | 30 (75.0) | 44 (97.8) | <.01 | <.01 |
| Mutant | 262 (13.1) | 0 (0.0) | 0 (0.0) | ||
| Amp (CN ≥ 7.0) | 0 (0.0) | 10 (25.0) | 1 (2.2) | ||
Abbreviations: amp, amplification; CN, copy number.
P‐values were calculated including 3 cohorts (Non‐FLT3 amp and FLT3 amp: Low amp and high amp) using Pearson chi‐square test.
P‐values were calculated between 2 cohorts (Low amp and high amp) by Pearson chi‐square test.
3.2. Correlation between FLT3 amplification status and clinicopathological features, including the status of other gene alterations
We evaluated clinicopathological variables according to FLT3 amplification status to investigate the clinical relevance of copy numbers of FLT3 amplifications. FLT3 amplification status was not associated with age or pathological grading, whereas the status of high FLT3 amplification was more frequently observed in females and in the rectum compared with that of low FLT3 amplification (Table 1). The frequency of RAS mutations was significantly lower in patients with high FLT3 amplification compared with in patients with low FLT3 amplification (37.8% vs 60.0%, P = .04), although the frequency of TP53 was similar regardless of FLT3 amplification status (Figure 2A,B). With regards to the co‐amplification status related to RTKs such as ERBB2, EGFR, FGFR, and MET, concurrent amplification was more common in low FLT3 amplification (Figure 2A). To assess the co‐amplification genes on the long arm of chromosome 13 at position 12 where FLT3 is located, we evaluated amplifications in BRCA2, which is also located on the long arm of chromosome 13 at position 13.1 and GAS6, which is located on the long arm of chromosome 13 at position 34, in addition to RAS, BRAF, and ERBB2, which are known oncogenic driver genes in mCRC patients (Figure 3A,B). More than half of the patients with low FLT3 amplification had co‐mutations in RAS and 40% of these patients had co‐amplification of BRCA2 or GAS6 on chromosome 13 (Figure 3A). In contrast, more than half of patients with high FLT3 amplification had no co‐occurring alterations (Figure 3B). The frequency of co‐alterations was significantly lower in patients with high FLT3 amplification compared with those with low FLT3 amplification (75% vs 38%, P < .001).
FIGURE 2.
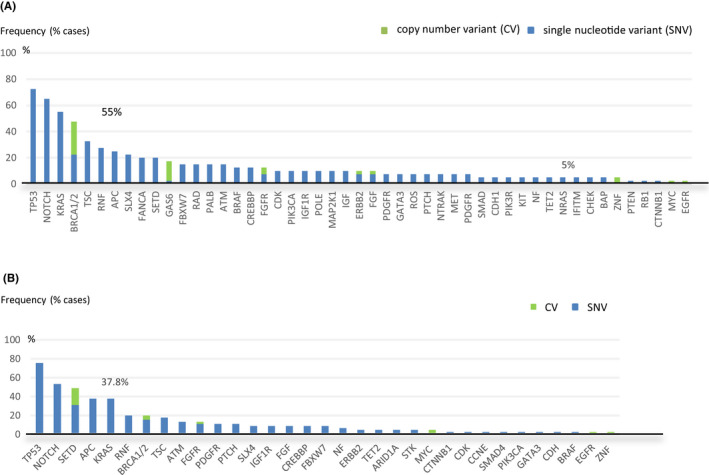
Long tail plots for FLT3 amplified metastatic colorectal cancer. A, Concurrent genomic alterations in FLT3 low‐amplified cases (n = 40). B, Concurrent genomic alterations in FLT3 high‐amplified cases (n = 45)
FIGURE 3.
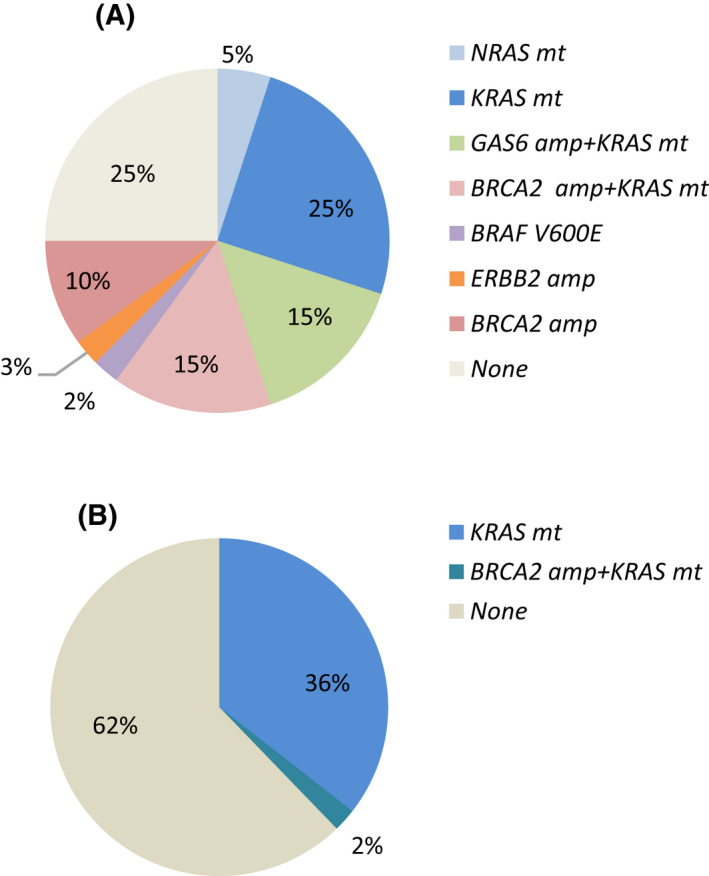
Overlapping alterations on chromosome 13 and other potential driver genes in FLT3 amplified metastatic colorectal cancer. A, Overlapping alterations in FLT3 low‐amplified cases (n = 40). B, Overlapping alterations in FLT3 high‐amplified cases (n = 45). amp, amplification; mt, mutation
3.3. Prognostic significance of FLT3 amplification status
To determine the prognostic significance of FLT3 amplification, we performed survival analyses for 1065 patients who received at least second‐line or more chemotherapy. The mean follow‐up time was 24 mo (range: 6‐127 mo), and 451 patients (45.3%) died. There were no significant differences in patient characteristics and treatment regimens between the cohorts (Table S1). The median OS from first‐line chemotherapy was significantly shorter in patients with low or high amplification compared with those without FLT3 amplification (29.5 mo in low amplification and 24.5 mo in high amplification vs 43.6 mo without FLT3 amplification, P = .003 and P = .002, respectively; Figure 4). In comparison between patients with high and low FLT3 amplification, the median OS in patients with low amplification tended to be better compared with that in patients with high amplification (29.5 mo in low amplification vs 24.5 mo in high amplification, P = .12). Furthermore, among patients with low FLT3 amplification, 13 of 20 (65.0%) patients had RAS mutation and 1 (5%) patient had ERBB2 amplification (Table S1), while the others (n = 6) did not.
FIGURE 4.
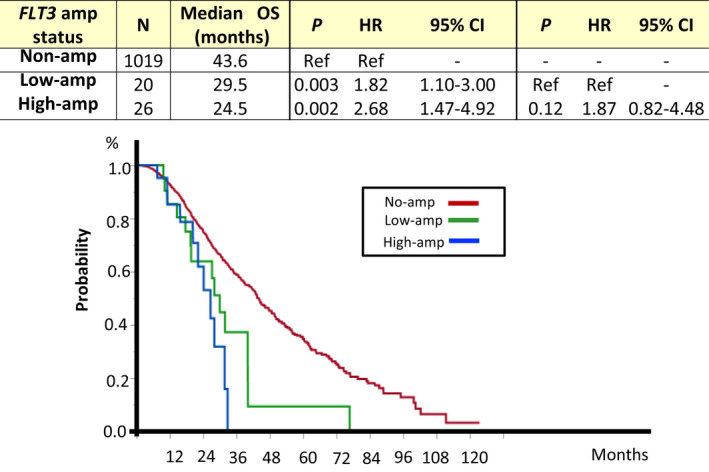
Overall survival according to FLT3 amplification status. Kaplan‐Meier curve for patients with high FLT3 amplification (n = 26, blue) and low FLT3 amplification (n = 20, green), and those without FLT3 amplification (n = 1019, red). amp, amplification; CI, confidence interval; HR, hazard ratio; OS, overall survival
3.4. Treatment efficacy of regorafenib according to FLT3 gene copy number status
The treatment efficacy of regorafenib was evaluated in 20 patients for whom the data of tumor responses to regorafenib were available among all patients whose gene analysis was successfully performed (n = 2078) (Table 2). The disease control rate was numerically but not statistically higher in patients with FLT3 amplification compared with in those with non‐FLT3 amplification, as shown in Table 2 (57.1% vs. 23.0%).
TABLE 2.
Treatment efficacy of regorafenib in mCRC patients
| FLT3 amp status | n | CR + PR | SD | PD | DCR |
|---|---|---|---|---|---|
| Non‐amplification | 13 | 0 | 3 | 10 | 23.0% |
| Amplification | 7 | 0 | 4 | 3 | 57.1% |
Abbreviations: amp, amplification; CR, complete remission; DCR, disease control rate; PD, progressive disease; PR, partial response; SD, stable disease.
4. DISCUSSION
This study aimed to identify the clinical significance and status of genetic heterogeneity in patients with FLT3‐amplified mCRC. Here, we observed FLT3 amplifications in approximately 4% of patients including cases with high copy number of FLT3. In the current study, FLT3 amplifications were identified more commonly in left side colon compared with in right side colon, and the amplification status was associated with co‐alteration gene status of other driver genes and other genes on chromosome 13. Furthermore, the copy number status was associated with prognosis, and the efficacy of regorafenib. To the best of our knowledge, this is the first study to evaluate the characteristics and status of genetic heterogeneity according to copy number status of FLT3 using NGS in a large‐scale study of FLT3‐amplified mCRC.
To date, the status of concurrent alterations in FLT3‐amplified mCRC remains unclear. In our study, c. 50% of the patients with FLT3 amplifications also harbored RAS mutations, which are common driver genes for mCRC, 17 although other driver genes, such as BRAF and PIK3CA, were rarely co‐mutated with the FLT3 gene. With regards to patients with FLT3 amplification status, the frequency of co‐occurring RAS mutation was higher in patients with low FLT3 amplification compared with in those with high FLT3 amplification. This frequency was inversely correlated with FLT3 gene copy number. These findings suggested that the status of high FLT3 amplifications may function as an oncogenic driver alteration that promotes the proliferation of cancer cells, similar to a RAS mutation. Regarding the co‐amplification gene status on chromosome 13 where the FLT3 gene is located and other co‐altered oncogenic driver genes, the frequency of co‐alteration genes was higher in patients with low FLT3 amplification compared with those with high FLT3 amplification. The co‐occurring alteration status of other genes was inversely associated with the copy number status of the FLT3 gene. In hematopoietic cells, FLT3 induces the activation of signal transduction networks mainly through PI3K and the RAS cascade, supporting the activation of AKT, signal transducer and activator transcription factor (STAT), and extracellular‐signal regulated kinases (ERK) 1 and 2. 18 Therefore, the presence of co‐amplified and co‐altered genes is implicated in innate and acquired resistance to receptor tyrosine kinase‐targeted therapies for FLT3.
Two types of amplification patterns have been reported in ERBB2 or MET genes, ie, focal and non‐focal amplifications, in which the gene copy number gain was due to chromosomal aneuploidy. Among patients with increased MET copy numbers, polysomy for chromosome 7 (the chromosome on which MET is located) was observed in c. 30% of patients with non–small‐cell lung cancer 19 and gastric cancer. 20 Furthermore, it was reported that non‐focal MET amplification did not act as an oncogenic driver. 20 Therefore, low copy numbers of the FLT3 gene, regarded as non‐focal amplification, may not function as an oncogenic driver, unlike high amplification of the FLT3 gene. Indeed, patients with high FLT3 amplification had less driver co‐alteration genes, while 14 of 20 (70.0%) patients with low FLT3 amplification had some driver co‐alteration genes such as RAS or ERBB2 (Table S1), associated with poor prognosis. The trend of shorter OS in patients with high FLT3 amplification, compared with those with low FLT3 amplification, suggested that high FLT3 amplification may be associated with the tumor biology and be a potential treatment target, similar to ERBB2 amplification in mCRC. 21
As for the efficacy of regorafenib in our study, disease control rate in patients with FLT3 amplification tended to be slightly better compared with that in patients with no amplification. In the current study and another previous report, 11 regorafenib, which is a mild inhibitor of FLT3 with an IC50 of 162 nmol/L, 14 showed moderate activity in patients with FLT3 amplifications. Moreover, previous case reports have reported that patients with metastatic colon cancer harboring FLT3 amplifications, determined by targeted sequencing, exhibited partial responses when heavily treated with regorafenib or sorafenib, 10 , 11 which partially blocked the activity of FLT3. Conversely, the recent TAPUR trial investigated the efficacy of sunitinib administration in patients with mCRC with FLT3 amplification. 22 However, monotherapy with sunitinib did not demonstrate sufficient activity in these patients. This may have been due to the inhibition of phosphorylation of wild‐type FLT3 by sunitinib with an IC50 of 250 nmol/L in vitro. 23 In contrast, a previous case report showed promising efficacy for sorafenib with an IC50 of 58 nmol/L in wild‐type FLT3. 10 , 24 Given the limited efficacy of regorafenib with a disease control rate of 40% in mCRC, 25 a therapeutic strategy with specific inhibition of FLT3 needs to be explored for FLT3‐amplified mCRC in the future.
Additionally, Siravegna et al, 26 reported that patients with FLT3 amplifications may be resistant to anti‐EGFR therapy. In their report, 2 of 10 patients with mCRC with primary resistance to anti‐EGFR therapy harbored FLT3 amplifications. In our study, it was difficult to evaluate the presence of primary resistance because there were few patients who had received anti‐EGFR therapy as first‐line treatment. Further evaluation of resistance to anti‐EGFR therapy is also needed for patients with FLT3 amplifications.
Our study has several limitations. We did not assess FLT3 expression immunohistochemically or conduct basic research to clarify the molecular mechanisms of the antitumor activity of the multikinase inhibitors of FLT3 amplification. As for the efficacy of regorafenib, in our study, it was difficult to evaluate accurately whether this drug specifically inhibited the activation of FLT3 in FLT3‐amplified mCRC in terms of spectrum of kinase inhibitory profile and in a small number of patients. Large‐scale studies and further basic research are required to validate our results.
In conclusion, 3.6% of patients with mCRC exhibited FLT3 amplifications. The copy number status was inversely related to the frequency of co‐existing alterations, and prognosis. High FLT3 amplification most commonly existed without other alterations, indicating focal amplification. The association between copy number status of FLT3 and clinical outcome requires further investigations to clarify whether it could be a promising target for future cancer treatment in mCRC.
CONFLICT OF INTEREST
Hiroya Taniguchi received research funding from Eli Lilly Japan Co., Ltd., Takeda, and Daiichi Sankyo. Yoshiaki Nakamura received research funding from Taiho Pharmaceutical. Satoshi Yuki received research funding from Chugai Pharmaceutical Co., Ltd. and Eli Lilly Japan Co., Ltd. Toshikazu Moriwaki received research honoraria from Taiho Pharmaceutical Co., Ltd and Chugai Pharmaceutical Co., Ltd. Eiji Oki received honoraria from Chugai Pharmaceutical Co., Ltd., Taiho Pharmaceutical Co., Ltd., and Eli Lilly Japan Co., Ltd. Akihito Tuji received honoraria from Taiho Pharmaceutical Co., Ltd. and Chugai Pharmaceutical Co., Ltd. Takayuki Yoshino received research funding from Novartis Pharma KK, Chugai Pharmaceutical Co., Ltd., and Daiichi Sankyo. All other authors have nothing to declare.
Supporting information
Fig S1
Table S1
ACKNOWLEDGMENTS
This work was supported by SCRUM‐Japan funds. We appreciate the participation of patients and contribution of the members that participated in this study. We would like to thank Editage for English language review.
Hasegawa H, Taniguchi H, Nakamura Y, et al. FMS‐like tyrosine kinase 3 (FLT3) amplification in patients with metastatic colorectal cancer. Cancer Sci. 2021;112:314–322. 10.1111/cas.14693
Clinical trial registration: The Nationwide Cancer Genome Screening Project for Gastrointestinal Cancer in Japan (SCRUM‐Japan GI‐SCREEN), University Hospital Medical Information Network Clinical Trials Registry (UMIN000016343).
REFERENCES
- 1. Blume‐Jensen P, Hunter T. Oncogenic kinase signaling. Nature. 2001;411:55‐365. [DOI] [PubMed] [Google Scholar]
- 2. Mackarehtschian K, Hardin JD, Moore KA, Boast S, Goff SP, Lemischka IR. Targeted disruption of the flk2/flt3 gene leads to deficiencies in primitive hematopoietic progenitors. Immunity. 1995;3:147‐161. [DOI] [PubMed] [Google Scholar]
- 3. Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100:1532‐1542. [DOI] [PubMed] [Google Scholar]
- 4. Armstrong SA, Kung AL, Mabon ME, et al. Inhibition of FLT3 in MLL. Validation of a therapeutic target identified by gene expression based classification. Cancer Cell. 2003;3:173‐183. [DOI] [PubMed] [Google Scholar]
- 5. Williams B, Atkins A, Zhang H, et al. Cell‐based selection of internalizing fully human antagonistic antibodies directed against FLT3 for suppression of leukemia cell growth. Leukemia. 2005;19:1432‐1438. [DOI] [PubMed] [Google Scholar]
- 6. Network CGA. Comprehensive molecular portraits of human breast tumors. Nature. 2012;490:61‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cancer Genome Atlas Network: Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cancer Genome Atlas Network: Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cancer Genome Atlas Network: Comprehensive molecular characterization of lung adenocarcinoma. Nature. 2014;511:543‐550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moreira RB, Peixoto RD, de Sousa Cruz MR. Clinical Response to Sorafenib in a patient with metastatic colorectal cancer and FLT3 amplification. Case Rep Oncol. 2015;8:83‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lim SH, Kim SY, Kim K, et al. The implication of FLT3 amplification for FLT targeted therapeutics in solid tumors. Oncotarget. 2017;10:3237‐3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grothey A, Van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo‐controlled, phase 3 trial. Lancet. 2013;381:303‐312. [DOI] [PubMed] [Google Scholar]
- 13. de la Fouchardière C. Regorafenib in the treatment of metastatic colorectal cancer. Future Oncol. 2018;14:2239‐2246. [DOI] [PubMed] [Google Scholar]
- 14. Wilhelm SM, Dumas J, Adnane L, et al. Regorafenib (BAY 73–4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer. 2011;129:245‐255. [DOI] [PubMed] [Google Scholar]
- 15. Yuki A, Chitwood J, Sidhu H, et al. Analytical validation of the oncomine comprehensive assay v3 with FFPE and cell line tumor specimens in a CAP‐accredited and CLIA‐certified clinical laboratory. J Mol Diagn. 2018;20:972, ST016. [Google Scholar]
- 16. Lih CJ, Harrington RD, Sims DJ, et al. Analytical validation of the next‐generation sequencing assay for a nationwide signal‐finding clinical trial: molecular analysis for therapy choice clinical trial. J Mol Diagn. 2017;19:313‐327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Janakiraman M, Vakiani E, Zeng Z, et al. Genomic and biological characterization of exon 4 KRAS mutations in human cancer. Cancer Res. 2010;70:5901‐5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Scholl C, Gilliland DG, Frohling S. Deregulation of signaling pathways in acute myeloid leukemia. Semin Oncol. 2008;35:336‐345. [DOI] [PubMed] [Google Scholar]
- 19. Go H, Jeon YK, Park HJ, Sung SW, Seo JW, Chung DH. High Met gene copy number leads to shorter survival in patients with non‐small cell lung cancer. J Thorac Oncol. 2010;5:305‐313. [DOI] [PubMed] [Google Scholar]
- 20. Janjigian YY, Tang LH, Coit DG, et al. Met expression and amplification in patients with localized gastric cancer. Cancer Epidemiol Biomarkers Prev. 2011;20:1021‐1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sawada K, Nakamura Y, Yamanaka T, et al. Prognostic and predictive value of HER2 amplification in patients with metastatic colorectal cancer. Clin Colorectal Cancer. 2018;17:198‐205. [DOI] [PubMed] [Google Scholar]
- 22. Alvarez RH, Garrett‐Mayer E, Halabi S, et al. Sunitinib (S) in patients (Pts) with metastatic colorectal cancer (mCRC) with FLT‐3 alterations: Results from the Targeted Agent and Profiling Utilization Registry (TAPUR) Study. Proc Am Assoc Cancer Res Annu Meet USA. 2019;79(13):CT146. [Google Scholar]
- 23. O'Farrell AM, Abrams TJ, Yuen HA, et al. SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood. 2003;101:3597‐3605. [DOI] [PubMed] [Google Scholar]
- 24. Wilhelm SM, Carter C, Tang L, et al. BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099‐7109. [DOI] [PubMed] [Google Scholar]
- 25. Grothey A, Van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo‐controlled, phase 3 trial. Lancet. 2013;381:303‐312. [DOI] [PubMed] [Google Scholar]
- 26. Siravegna G, Mussolin B, Buscarino M, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015;21:795‐801. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Table S1