

SUMMARY
CELF6 is a CELF-RNA-binding protein, and thus part of a protein family with roles in human disease; however, its mRNA targets in the brain are largely unknown. Using cross-linking immunoprecipitation and sequencing (CLIP-seq), we define its CNS targets, which are enriched for 3′ UTRs in synaptic protein-coding genes. Using a massively parallel reporter assay framework, we test the consequence of CELF6 expression on target sequences, with and without mutating putative binding motifs. Where CELF6 exerts an effect on sequences, it is largely to decrease RNA abundance, which is reversed by mutating UGU-rich motifs. This is also the case for CELF3–5, with a protein-dependent effect on magnitude. Finally, we demonstrate that targets are derepressed in CELF6-mutant mice, and at least two key CNS proteins, FOS and FGF13, show altered protein expression levels and localization. Our works find, in addition to previously identified roles in splicing, that CELF6 is associated with repression of its CNS targets via the 3′ UTR in vivo.
Graphical Abstract
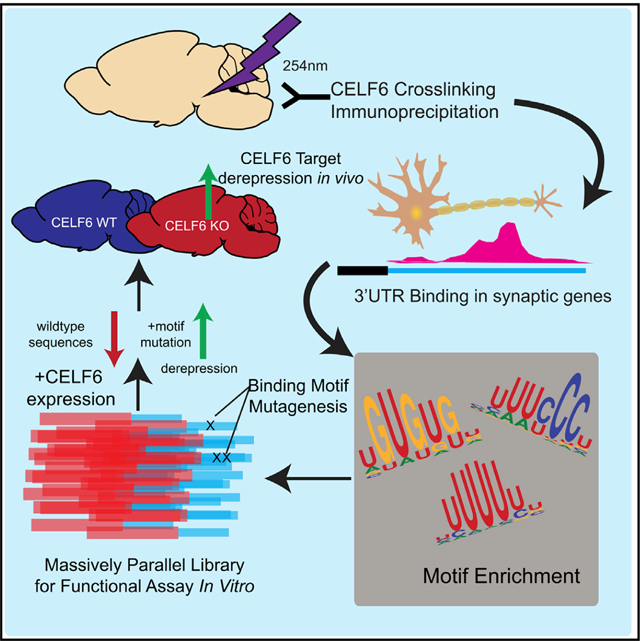
In Brief
Rieger et al. assay the function of the RNA-binding protein CELF6 by defining its targets in the brain. They show that CELF6 largely binds 3′ UTRs of synaptic mRNAs. Using a massively parallel reporter assay, they further show that CELF6 and other CELFs are associated with lower mRNA abundance and that targets are derepressed in Celf6-knockout mice in vivo.
INTRODUCTION
RNA-binding proteins (RBPs) regulate the life cycle of messenger RNAs (mRNA), from transcription, splicing, and nuclear export to localization, maintenance, translation, and degradation (Vindry et al., 2014; Xiong et al., 2015). The CUGBP and ELAV-like factor (CELF) family of RBP has six members (CELF1–6), which can be divided into two groups based on sequence homology: CELF1–2, which are expressed ubiquitously, and CELF3–6, which show enrichment in the CNS (Dasgupta and Ladd, 2012). The most studied, CELF1, was first characterized in relation to myotonic muscular dystrophy pathogenesis (Lee and Cooper, 2009). It binds CUG- and (U)GU-rich motifs (Timchenko et al., 1996; Wang et al., 2015) and has been shown to promote exon skipping (Wang et al., 2015) as well as mRNA degradation via recruitment of deadenylases (Moraes et al., 2006). CELF3–6, however, have not been as well characterized.
We identified CELF6 to be both enriched in serotonin-producing neurons and disrupted in an individual with autism (Dougherty et al., 2013). CELF6 showed expression in the hypothalamus and in several monoaminergic cell populations commonly targeted in psychiatry (Maloney et al., 2016). Likely as a consequence of its role in those cells, it is also associated with conditioned behaviors: Celf6-knockout (KO) mouse pups show altered ultrasonic vocalization, while adults exhibit deficits in fear conditioning and loss of conditioned place preference to cocaine (Dougherty et al., 2013; Maloney et al., 2019). Although capable of regulating splicing in vitro (Ladd et al., 2004), CELF6 has recently been shown to interact with the 3′ untranslated region (UTR) as well, allowing it to stabilize p21 mRNA in colorectal cancer cells (Liu et al., 2019). Its targets and function in the CNS, however, are not well described. Here, we sought to explore the molecular role of CELF6 in the brain to better understand how its disruption may alter behavior.
To define the function of CELF6 in vivo, we performed cross-linking immunoprecipitation and sequencing (CLIP-seq) in the brain and found CELF6 to be overwhelmingly associated with 3′ UTRs of target mRNAs, many coding for proteins involved in synaptic transmission. UTRs showed enrichment for UGU-containing motifs, consistent with prior in vitro findings (Ray et al., 2013), as well as other motifs. We cloned UTR elements under CLIP-seq peaks into the 3′ UTR of a reporter construct and measured reporter library expression and abundance on translating ribosomes while modifying CELF6 expression and sequence integrity. We found CELF6-associated UTR sequences were repressive in general, CELF6 could function to enhance this reduction to mRNA levels, and this effect could be abrogated by motif mutation. We also observed this effect across CELF3–6. In the Celf6-KO mouse brain, we find CELF6 targets to be generally derepressed, validating our findings in vivo. Taken together, we show that CELF6 is largely associated with reductions to transcript abundance of its neuronal targets and thus may have an important role regulating cellular functions in the brain.
RESULTS
CELF6 Associates with Conserved Elements in 3′ UTRs of Target mRNAs In Vivo
We first sought to use CLIP to identify CELF6 targets in vivo. Unfortunately, available antibodies against CELF6 protein do not function well for immunoprecipitation against endogenous protein (data not shown). We previously developed a BAC transgenic mouse expressing epitope-tagged CELF6-YFP/HA (78 kDa), which exhibits endogenous patterns of anatomical and subcellular (cytoplasmic and nuclear) expression, with only slight overexpression compared with native CELF6 (Maloney et al., 2016). Because we have previously validated antibodies for GFP (and related YFP) pull-down (Doyle et al., 2008), we designed our CLIP studies using that line.
To establish the in vivo RNA-binding properties of CELF6, we first performed CLIP with anti-EGFP antibodies on CELF6-YFP/hemagglutinin (HA) mice followed by radiolabeling of nucleic acid (Figure 1A). Controls included an immunoprecipitate (IP) from uncrosslinked samples and IP from wild-type (WT) tissue. As expected, there was a lack of detectable RNA in IP from WT tissue and uncrosslinked YFP+ tissue. To capture the targets of CELF6 in vivo, we purified a region of 60–200 nucleotides in size (80–150 kDa) from lysates of CELF6-YFP/HA+ brains. Similar to the Vidaki et al. (2017) study of Mena, we found that the stringent IP conditions of standard CLIP protocols were incompatible with our anti-GFP IP (not shown). Thus, to enable statistical background estimation, we also collected samples from size-selected input RNA (controlling for differences in RNA abundance), as well as pull-down from WT littermate brains (to control for non-specific binding). We defined high-confidence targets as those showing statistical enrichment in CLIP samples compared with both controls and we defined lower-confidence targets as showing enrichment against only one control.
Figure 1. CELF6 Primarily Associates with 3′ UTRs of Target mRNAs In Vivo.
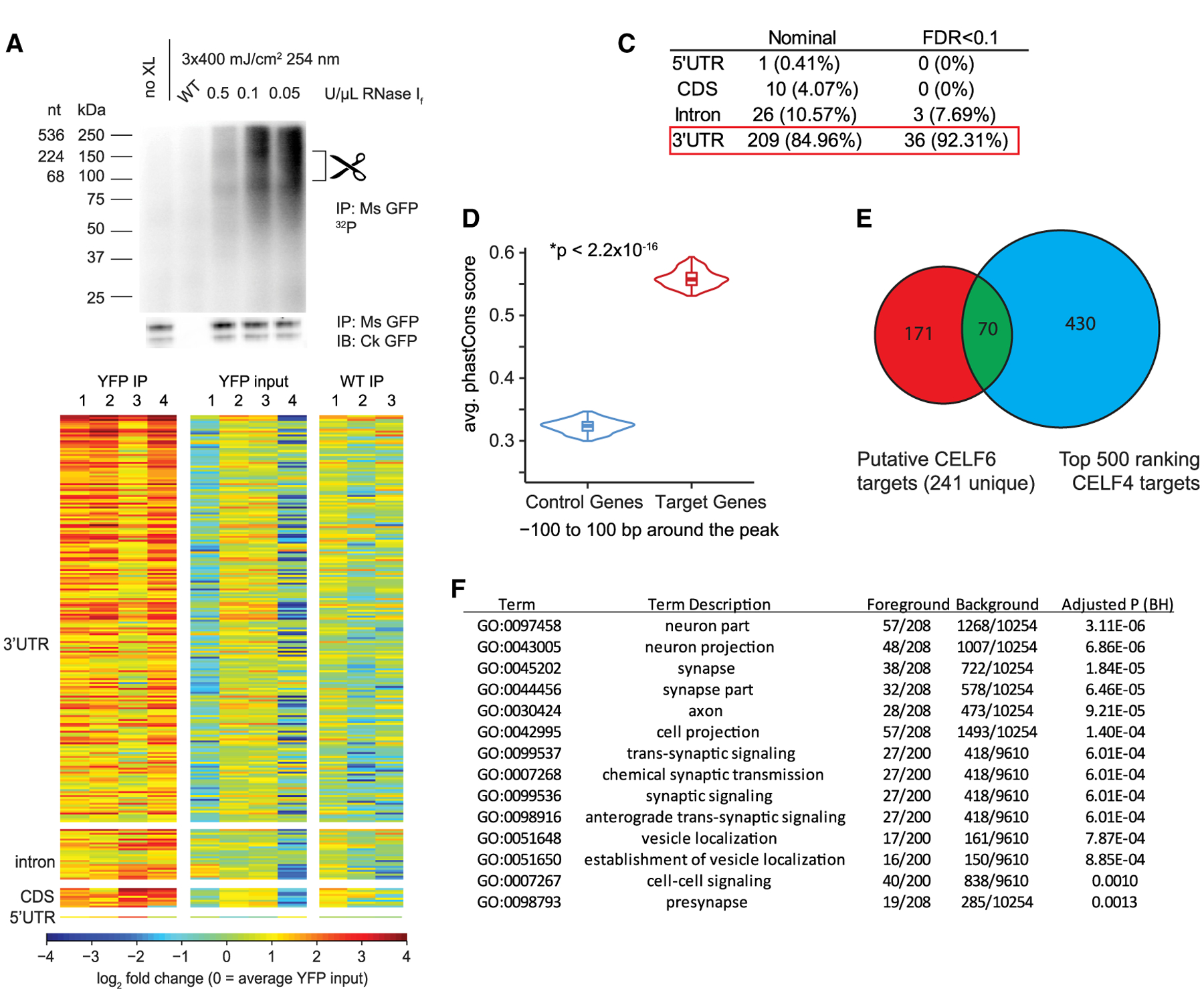
(A) CELF6 CLIP, lanes: no crosslink (no XL), wild-type (WT) control, and three concentrations of RNase If. Top: RNA 32P autoradiogram. Bottom: anti-GFP immunoblot. Scissors mark region (80–150 kDa) above CELF6-HA/YFP (78 kDa) isolated for sequencing. Immunoblot detects two bands, corresponding to sizes of known isoforms of CELF6.
(B) Log2 counts-per-million (CPM) RNA in four CELF6-HA/YFP+ replicates and three WT replicates differentially abundant regions. Heatmaps show enrichment in HA/YFP+ immunoprecipitate (IP) samples relative to input and WT controls. CPM is normalized to mean YFP input by row.
(C) Summary of differential enrichment analysis of genes showing nominally significant (p < 0.05), and Benjamini-Hochberg (FDR < 0.1) enrichment in HA/YFP+ IP samples relative to both HA/YFP+ input and WT IP controls. No peaks were found in intergenic regions.
(D) Average phastCons score for ±100 bp around CLIP peaks in targets compared with control genes not exhibiting enrichment in CLIP (p < 2.2 × 10−16, two-tailed t test).
(E) Overlap of CELF6 and CELF4 targets identified in Wagnon et al. (2012).
(F) DAVID gene ontology analysis of CELF6 CLIP targets (241 unique genes).
We prepared sequencing libraries from those samples using an adaptation of the eCLIP workflow (Van Nostrand et al., 2017) (Methods S1). All samples were sequenced to a similar depth, and, on average, 94% of reads mapped to genic regions (Table S1). Next, to define specific sites of CELF6 binding, we called peaks genome-wide using Piranha (Uren et al., 2012) and performed differential enrichment analysis using edgeR comparing CLIP to controls for read counts under peaks (Robinson et al., 2010). In the event that genome-wide background count estimation in Piranha would limit discovery, we also summed reads mapping to smaller subgenic features: 5′ UTR, coding sequence (CDS), intron, or 3′ UTR and performed differential enrichment analysis restricted to those regions. In total, we identified significant enrichment across 241 genes combined across analysis methods (Table S2).
CELF proteins have been shown to function both in splicing and post-transcriptional regulation. To understand how CELF6 may function in the brain, we examined the distribution of CLIP reads across genes. We hypothesized that splicing-related functions would correspond to increased density in internal coding exons and introns, whereas post-transcriptional regulation would be reflected as increased density in UTRs. Figure 1B shows a heatmap of CLIP targets across all samples relative to controls. Most enriched regions are in 3′ UTRs (Figure 1C; >80%). Additionally, UTRs showing CELF6 binding were more conserved than expected by chance (Figure 1D). We also compared our CLIP hits to the top-ranking targets of the related protein CELF4 (Wagnon et al., 2012). Figure 1E shows this overlap—with 70/241 CELF6 target genes in common with CELF4 target genes (also noted in Table S2). The Wagnon et al. (2012) study was performed on different brain regions, cell types, and at different ages, and thus, this overlap is notable. CELF6 targets were also disproportionately involved in synaptic transmission (Figure 1F). Taken together, these data suggest CELF6 may regulate neuronal function by binding UTRs of synaptic gene transcripts, altering their stability or translation.
CELF6-Bound 3′ UTRs Are Enriched for U-, UGU-, and CU-Containing Motifs
Previous study in vitro has identified CELFs as preferring UGU-rich sequences (RNACompete; Ray et al., 2013) and manual inspection under CELF6 clip peaks found matches to those motifs (Figure 2A). To scan CLIP targets systematically and potentially identify other preferred sequences in the brain, we used MEME-SUITE to search for motif enrichment. We analyzed 50 nucleotide segments centered under 544 peaks. Those included 25 3′ UTR peaks (25 unique genes) identified using Piranha genome wide. However, we performed a second pass of peak calling in those UTRs identified via summation across the UTR (as described in the preceding section). Doing so enabled us to discover an additional 519 peaks in 191 UTRs, including new peaks in 10 genes identified in the initial genome-wide pass. We suspect the additional discovery is due to incorrect background estimation when using read counts across an entire gene or genome wide versus more local background-count estimation. UTRs varied between 1 and 9 peaks with a median of 2, with 75% of the UTRs containing 1–3 peaks. We compared those sequences to 544 control sequences sampled randomly from UTRs of brain-expressed genes showing no CLIP enrichment.
Figure 2. CELF6-Associated 3′ UTRs Are Enriched for U-, UG-, and CU-Containing Motifs.
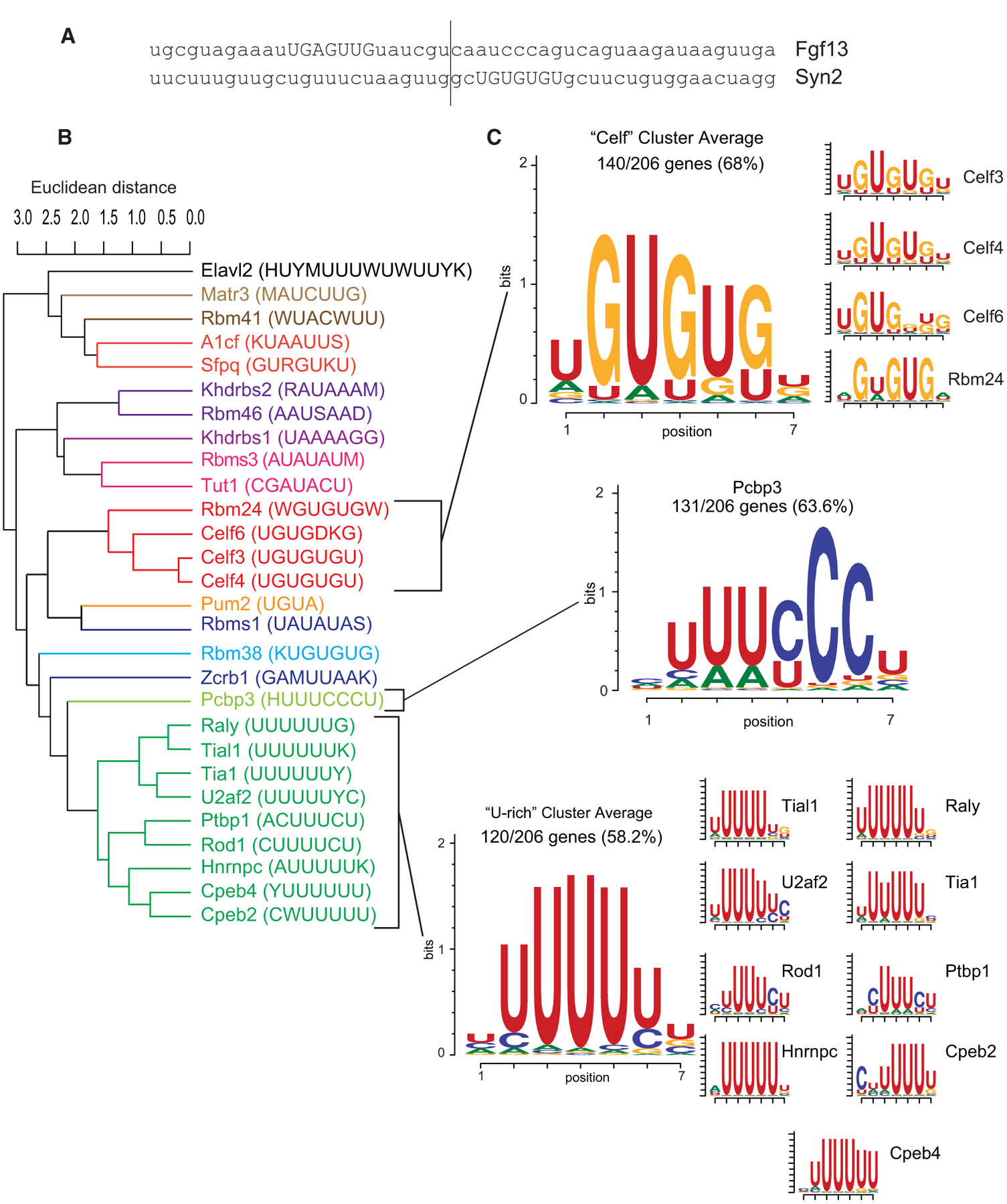
(A) Example 50-nt regions under CLIP peaks.
(B) Clustering of enriched motifs identified with MEME-SUITE. Color coding shows cluster membership.
(C) Motif logos for the clusters found under the CLIP peak. Logos represent the average of position probability matrices (PPMs) for each motif cluster. Side panels show individual PPMs making up each cluster’s membership.
We found 29 enriched motifs at a false discovery rate (FDR) < 0.05, including the RNACompete binding motifs for CELFs. We also found U-rich motifs of the TIA/CPEB families to be enriched, among others. Because RBPs can exhibit degenerate preferences (Auweter et al., 2006; Harvey et al., 2018), for a more holistic summary, we clustered enriched motifs by the Euclidean distance between their position probability matrices (PPMs) in the CISBP-RNA database. As expected, enriched motifs clustered partially by RBP family (Figure 2B), and motifs within a cluster were highly similar. After clustering, we scanned our sequences again to determine which showed matches to the cluster-average PPMs, as “meta-motifs.” We found that 519/544 sequences showed significant matches to at least one cluster (Tables S3 and S4).
A few of these clusters were highly recurrent (Figure 2C; Tables S3 and S4); 140/206 unique genes’ 3′ UTRs (68%) showed at least one match to the “CELF”-cluster ([U/A] GUGU[G/U][UGA]), and 131/206 UTRs (63.6%) showed at least one match to the PCBP3 motif, which forms its own cluster with a central UUU[C/U]CC sequence. PCBP3 binds both double- and single-stranded nucleic acids, known primarily as a transcription factor (Choi et al., 2009), though a related protein (PCBP4) has also been shown to regulate mRNA stability (Scoumanne et al., 2011). 120/206 (58.2%) genes showed at least one match to the “U-rich” cluster, whose members all possess a central stretch of 4–5 Us. Proteins in this cluster include TIA1, involved in stress granule localization (Gilks et al., 2004); CPEBs, involved in polyadenylation (Villalba et al., 2011); and HNRNPC, involved in both mRNA stability and localization (Nakielny and Dreyfuss, 1996; Shetty, 2005). Moreover, 61/206 genes (29.6%) had ≥1 match to both the CELF cluster and the U-rich cluster, and 65/206 genes (31.5%) had ≥1 match to both the CELF and PCBP3 clusters. Thus, CELF6 binding appears to be associated with combinations of known motifs for both CELFs and other RBPs, and not just UGU-rich sequences.
We also tested whether CELF6 peaks had location biases within UTRs that might suggest function, such as an enrichment near a polyadenylation signal (PAS). We found a small increase in the number of predicted PASs per transcript compared with length-matched UTRs from control genes (p = 0.00023; Figure S1C), as well as decreased distance between CLIP peaks and a PAS (p = 5.3E–6; Figure S1D). In the case of UTRs with only one predicted PAS, this was true only for the PAS [ATTAAA], but not [AATAAA] (p = 0. 00154; Figure S1E). Although these findings suggest that CELF6’s proximity may influence choice of PAS, an examination of alternative polyadenylation in vivo in Celf6-KO mice did not show CELF6 to alter the probability of alternative 3′ UTR usage (not shown, available at GEO: GSE160293).
Massively Parallel Reporter Assay Defines the Function of CELF6-Bound Motifs
Motif analysis indicates that sequence specificity mediates interactions between CELF6 and RNA in vivo, so we next sought to understand the consequence of that association, such as on translation or transcript stability. We used a massively parallel reporter assay framework for post-transcriptional regulatory element sequencing (PTRE-seq) to evaluate a large number of target UTR sequences at once (Cottrell et al., 2018). For PTRE-seq, we sub-cloned 410 independent, CLIP-defined UTR elements, each 120 bp long and centered under CLIP peaks, into the 3′ UTR of a tdTomato expression plasmid (Figure 3A). That library included 254 high-confidence targets showing enrichment against both CLIP controls (input and WT/non-specific pull-down). We also included cloneable elements from 156 additional elements under peaks that only met the input comparison criterion (lower stringency, highlighted in green dots going forward). As these performed similarly, we included both sets in subsequent figures. For reproducibility, each UTR element was included in the library six times, with unique 9-bp barcodes. As a comparison group, we also included 410 elements with all significant motif matches mutated. Sequences and their mutations are shown in Table S5.
Figure 3. CELF6-CLIP-Enriched Motif Sequences Represent a Set of Repressive Elements.
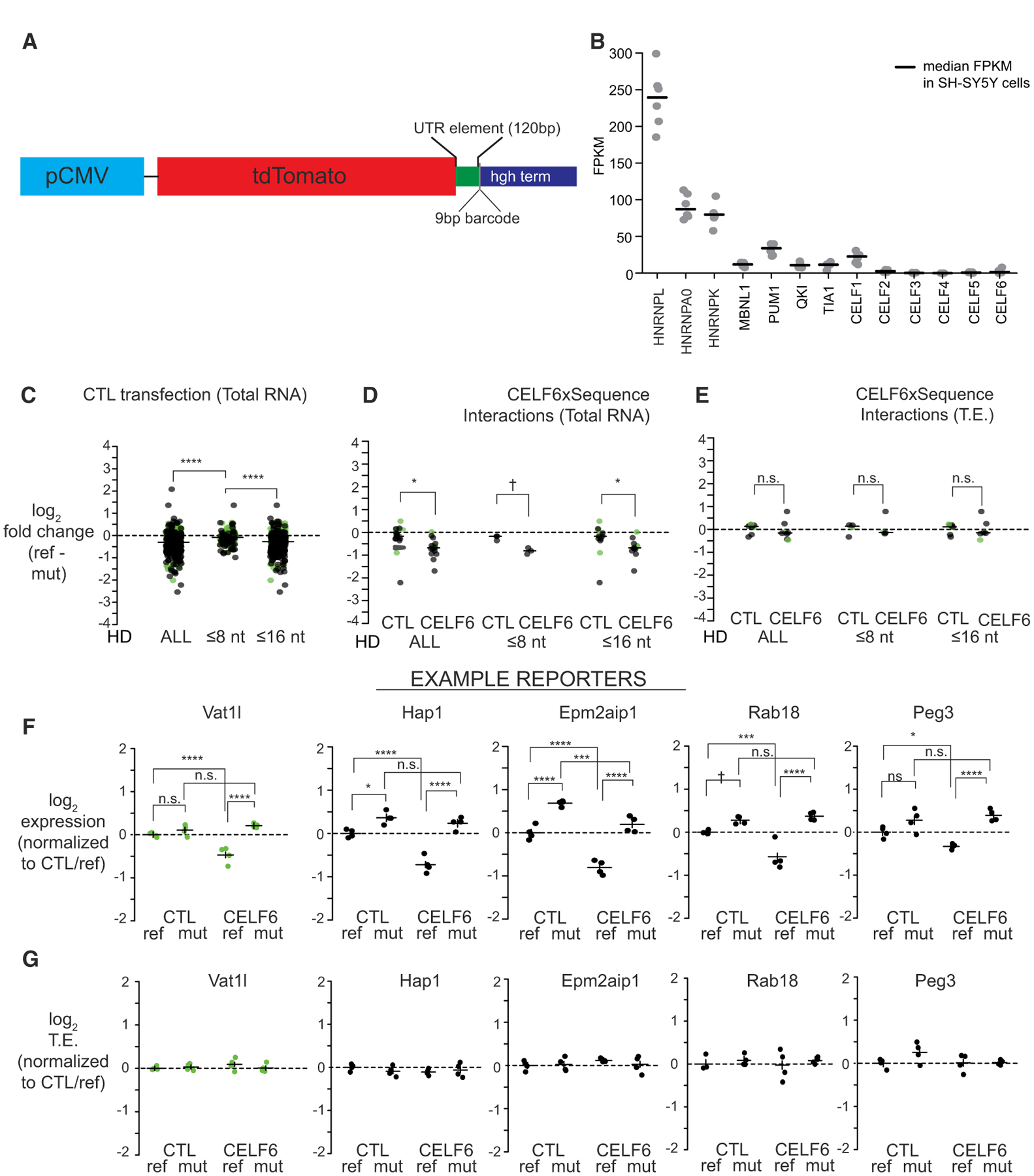
(A) PTRE-seq reporter (pmrPTRE_AAV).
(B) Fragments per kilobase of transcript per million reads (FPKM) levels are shown for several RNA-binding proteins in SH-SY5Y cells (n = 8). Lines represent median FPKM.
(C) Reporter RNA log2-fold changes in expression (reference versus mutant) in CTL transfections, for all element pairs, as well as Hamming distance (HD) ≤ 8 or ≤ 16.
(D) Reporter RNA log2-fold changes between reference and mutant in CTL and CELF6 expression conditions, from elements showing significant condition X sequence interactions.
(E) Reporter RNA log2-fold changes in translation efficiency (TE) between reference and mutant in CTL and CELF6 conditions, for elements in (D). Data points in (C)–(E) are log2-fold changes (reference versus mutant) per condition, averaged by replicate over barcodes and then over replicates. Lines represent medians. Comparisons between conditions or sequence mutation were assessed by Mann-Whitney U tests.
(F) Log2 expression across five example reporter library elements in CTL, CELF6, reference, and mutant conditions.
(G) Log2 TE across example reporters in (F). Data points in (F) and (G) are averaged across barcodes for each biological replicate.
Lines in (F) and (G) represent average ± 95% confidence intervals, normalized to CTL/reference condition. Post hoc pairwise comparisons in (F) and (G) computed using the multcomp package in R. n.s., p > 0.1; †p < 0.1, *p < 0.05, **p < 0.001, ***p < 0.0001, ****p < 1E–5. Green points represent library elements selected from less-stringent CELF6 target criteria (enrichment over input alone).
Scale bar in (A) represents 0.1 mm as indicated.
We assayed the effect of each element on both transcript abundance and final translation levels, as assessed by ribosome occupancy of those reporters. We used transient cotransfection of the library with an EGFP/RPL10A construct that tags ribosomes with GFP enabling translating ribosome affinity purification (TRAP). This method allows assessment of RNA abundance on ribosomes by anti-GFP pull-down and has previously been shown to be sensitive to UTR elements that regulate translation (Heiman et al., 2008). To assess transcript abundance, we also collected total RNA from the same cells. We opted for an in vitro culture system with largely undetectable levels of CELF6 expression: SH-SY5Y neuroblastoma cells (Figure 3B), so that we could control CELF6 expression exogenously, confirmed by RT-PCR (Figure S2).
We assessed expression of PTRE-seq library sequences by counting barcodes in sequencing libraries prepared from transient transfection replicates. Pearson correlation in barcode counts between replicates was >0.9, indicating good reproducibility. To account for any differences in starting abundance within the library itself, log2 counts per million (CPM) in RNA barcodes were further normalized to log2 CPM from sequencing the starting DNA plasmid pool (normalized count data hereafter termed “log2 expression”). After removing barcodes that were absent or poorly measured across all samples, the final analyzed library contained 379 UTR element pairs (reference and mutant, 229 high-confidence targets and 150 lower-stringency targets); all of which were represented by three to six barcodes per element, across 153 total genes (104 high-confidence genes and 49 lower-stringency genes).
To determine the relationship between CELF6 expression and element sequence, we analyzed log2 TRAP levels using a 2 × 2 factorial design linear mixed-effects model, fitting fixed effects of the element sequence (“reference” or “mutant”) and the CELF6 expression condition (control [CTL] or CELF6 expression) and the interaction of the condition X sequence, with a random intercept term for replicate sample, treating an element’s barcodes within a sample as a repeated measure. Individual models were fitted for each UTR element pair. A summary of effects across all library elements and estimates of R2 are shown in Table S6. At nominal p < 0.05, 293/379 (77.3%) of elements showed some significant effect (main effects of either sequence or CELF6 expression or a sequence-X CELF6-expression interaction, 283/379 [74.7%] at FDR < 0.1).
CELF6-Associated Motif Sequences Decrease RNA Abundance and Corresponding Ribosome Occupancy
We first examined the role of the element sequence itself on ribosome occupancy by comparing the UTR elements with and without motif mutations in the TRAP data in the empty-vector condition. Among the 77.3% of elements showing any significant effect, 88% (258/293 elements) showed a main effect of sequence, regardless of CELF6 expression. Looking at the distribution of log2-fold changes in expression between reference and mutant sequences under CTL transfection conditions, 220/258 (85.3%) of them were repressive when compared with their mutated counterparts (median log2-fold change: −0.47; 1.39-fold decrease). CELF6-bound elements decreased ribosome occupancy, even in the absence of CELF6 expression.
There are two primary mechanisms by which a sequence can alter ribosome occupancy—altering translation efficiency (TE), usually reflecting the loading of mRNAs onto ribosomes, or altering mRNA stability (Garneau et al., 2007; Hinnebusch et al., 2016; Ivshina et al., 2014; Piqué et al., 2008). To assess TE, log2 expression for each barcode in the TRAP samples was normalized to input RNA to account for their overall abundance (log2 TE). We then fit our model independently for both input RNA expression and TE. A summary of those effects is shown in Tables S7 and S8.
To assess relative influence of either mechanism, we first looked at input RNA expression; 285/379 elements (75.2%) showed any significant effect (282/379, 74.4% at FDR < 0.1). Similar to what was found in the analysis of TRAP RNA levels, 89.8% of them (256/285) showed a main effect of sequence mutation, regardless of CELF6 expression. Similar to ribosome occupancy, 86.3% had fold changes less than 0, indicating that reference sequences are repressive when compared with their mutated counterparts (Figure 3C) (median log2-fold change: −0.47; 1.39-fold decrease). We did not observe a similar trend in TE. Only 34/379 sequences (8.97%) showed any nominally significant effect on TE, and 0 showed any significant effect at FDR < 0.1; 15/34 showed a significant effect of sequence; however, six showed negative log2-fold changes, and nine showed positive fold changes. This indicates that alteration of transcript abundance is the primary mechanism leading to decreased ribosomal occupancy, rather than changes in TE.
Significant changes to expression could be incurred because of large numbers of mutated bases. Thus, we looked at changes between reference and mutant for elements with smaller amounts of mutation (Hamming distance [HD] of ≤8, the approximate size of a motif, or ≤16 nucleotides; maximum HD = 25 for any element). Expression of reference sequences was generally lower than mutant sequences for these subsets, with 72.9% at <0 and 84.8% at <0 for HD ≤8 and HD ≤16, respectively (Figure 3C). Thus, even a modest amount of mutation is capable of elevating the expression levels of most elements.
CELF6 Decreases RNA Abundance in a Sequence-Dependent Manner
We next assessed interactions between CELF6 expression and sequence, focusing on RNA expression; 16 elements (15 unique genes) showed a nominally significant interaction (p < 0.05) between CELF6 expression and sequence. Figure 3D plots the average log2-fold change between reference and mutant sequence for both the CTL and CELF6 conditions across each subsetted level of sequence mutation. The fold changes between reference and mutant sequences were more negative (median: 1.4-fold) under CELF6 expression. That is driven by repression of the reference sequence, rather than an elevation of the mutant sequence by CELF6 (median log2-fold change CELF6-CTL reference = −0.37, CELF6-CTL mutant −0.05; p = 9E–4 Mann-Whitney U test). This indicates the CELF6 affects those elements by further decreasing mRNA levels, and that effect is sequence dependent.
As mentioned earlier, few effects were observed on TE by comparison. Only 8/34 elements with any effect on TE showed interactions with CELF6 expression at p <0.05, with 0 showing interactions at FDR <0.1 (Figure 3E). Additionally, we did not detect a consistent direction of CELF6 effect on TE. Looking at changes to reference sequence TE when CELF6 is overexpressed, only two elements showed a trend to difference compared with CTL, one showing a decrease (Lin7c, log2-fold change: −0.45; p = 0.05) and one showing an increase (Enc1, log2-fold change: +0.71; p = 0.052). Other elements had different sources of interaction, e.g., a difference between CELF6 and CTL only in mutated elements (e.g., Prkacb, log2-fold change in mutated sequence only: +0.29; p = 0.002). By contrast, looking at input RNA expression, 14/16 elements with nominally significant interactions showed log2-fold changes in the reference between CELF6 and CTL, changes that were negative. Examples of element expression are shown in Figures 3F, and Figure 3G shows TE. These findings indicate that, for elements showing interactions between CELF6 and element sequence, there is a repression of the reference sequence via a decrease in RNA abundance with CELF6 expression, and that effect is abolished after mutation of the conserved motifs. Therefore, we conclude that, overall, when CELF6 exerts a measurable effect in our system, that effect is to decrease RNA abundance. Where impact on TE occurs, it is not generalizable in the direction of the effect.
To further validate these findings and confirm an effect on final protein levels, we overexpressed five individual reporters, with and without mutation, along with EGFP-tagged CELF6 or EGFP alone, then measured reporter expression by tdTomato fluorescence (Figure 4). For 4/5 cases, repression upon CELF6 expression was the same direction and magnitude to that observed by PTRE-seq. For 3/4, the repression observed by CELF6 was significantly reversed by motif mutation. This shows that this finding ultimately affects target protein levels.
Figure 4. Individual mtdTomato UTR Element Constructs Replicate Interactions of CELF6 Expression and Sequence.
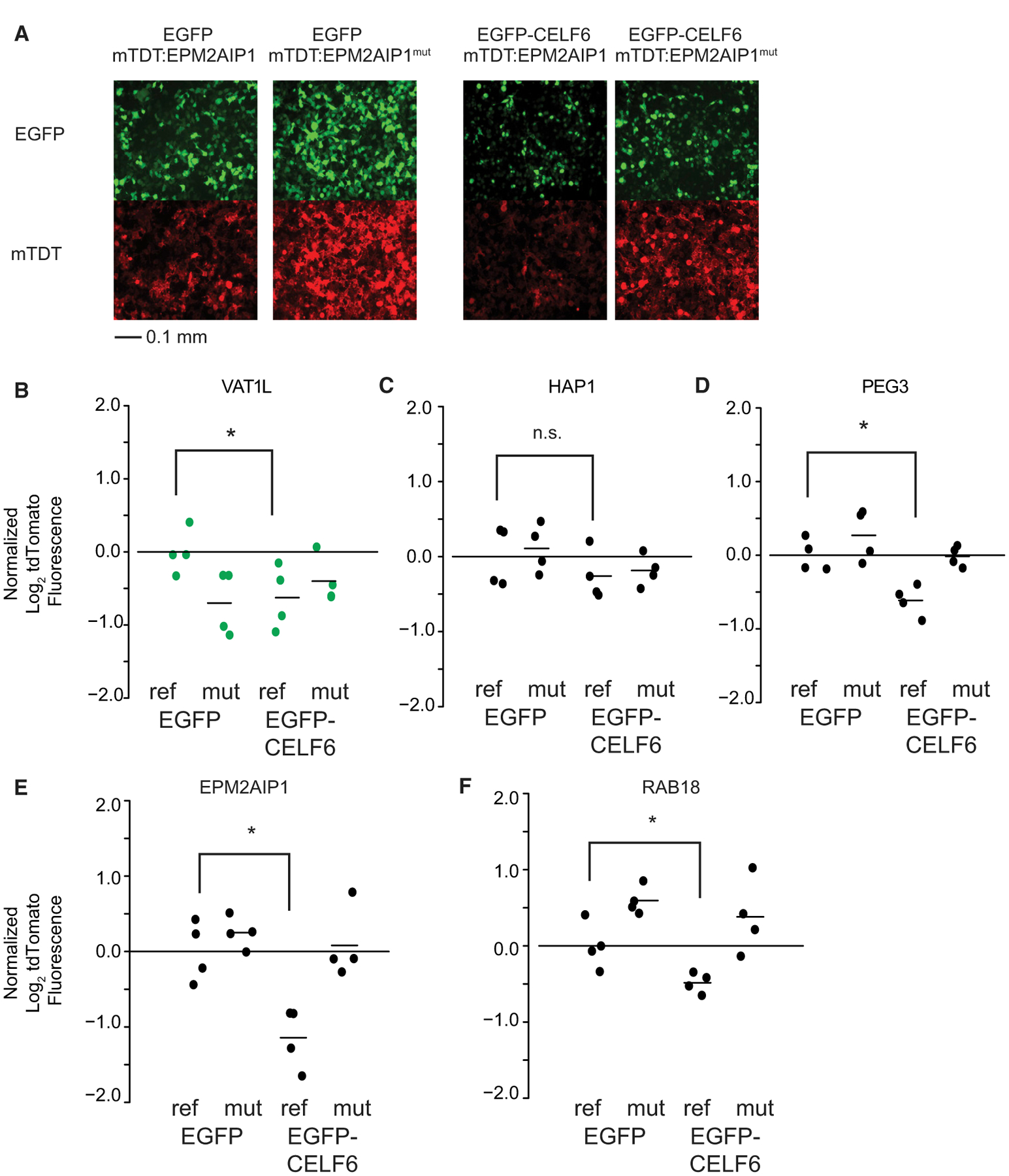
Individual UTR elements were cloned as in Figure 3A and transfected along with either EGFP or EGFP-CELF6
(A) Example of live-cell epifluorescent images from a transfection experiment showing EGFP or EGFP-CELF6 expression (top panels) and either tdTomato:EPM2AIP1 or tdTomato:EPM2AIP1(mutant) (bottom panels).
(B–F) tdTomato log2 fluorescence, with lines representing means normalized to EGFP/reference sequence.
Significance was assessed by two-way ANOVA, with post hoc pairwise comparisons using the multcomp package in R. n.s., p > 0.1; †p < 0.1, *p < 0.05, **p < 0.001. Green points represent library elements selected from less-stringent CELF6 target criteria (enrichment over HA/YFP input alone). Scale bar in (A) represents 0.1 mm as indicated.
CELF3–5 Also Decrease RNA Abundance
CELF3, CELF4, and CELF6 binding preferences determined by RNAcompete are highly similar (Figure 2), and as a group, CELF3–6 are more similar in amino acid identity than CELF1 or CELF2 are (Dasgupta and Ladd, 2012). Therefore, we hypothesized that the repression we observed with CELF6 would also be true of CELF3–5. We transiently transfected our PTRE-seq library along with His/Xpress-tagged human CELF3, CELF4, or CELF5 used previously to study these proteins (Figure S2) (Ladd et al., 2001, 2004). We then performed the analysis of mRNA abundance described in the preceding section, refitting the linear mixed models to include data from these new expression conditions (Figure 5).
Figure 5. CELF3–5 Show Redundancy in Ability to Enhance Repression of CELF6-CLIP-Enriched UTR Elements.
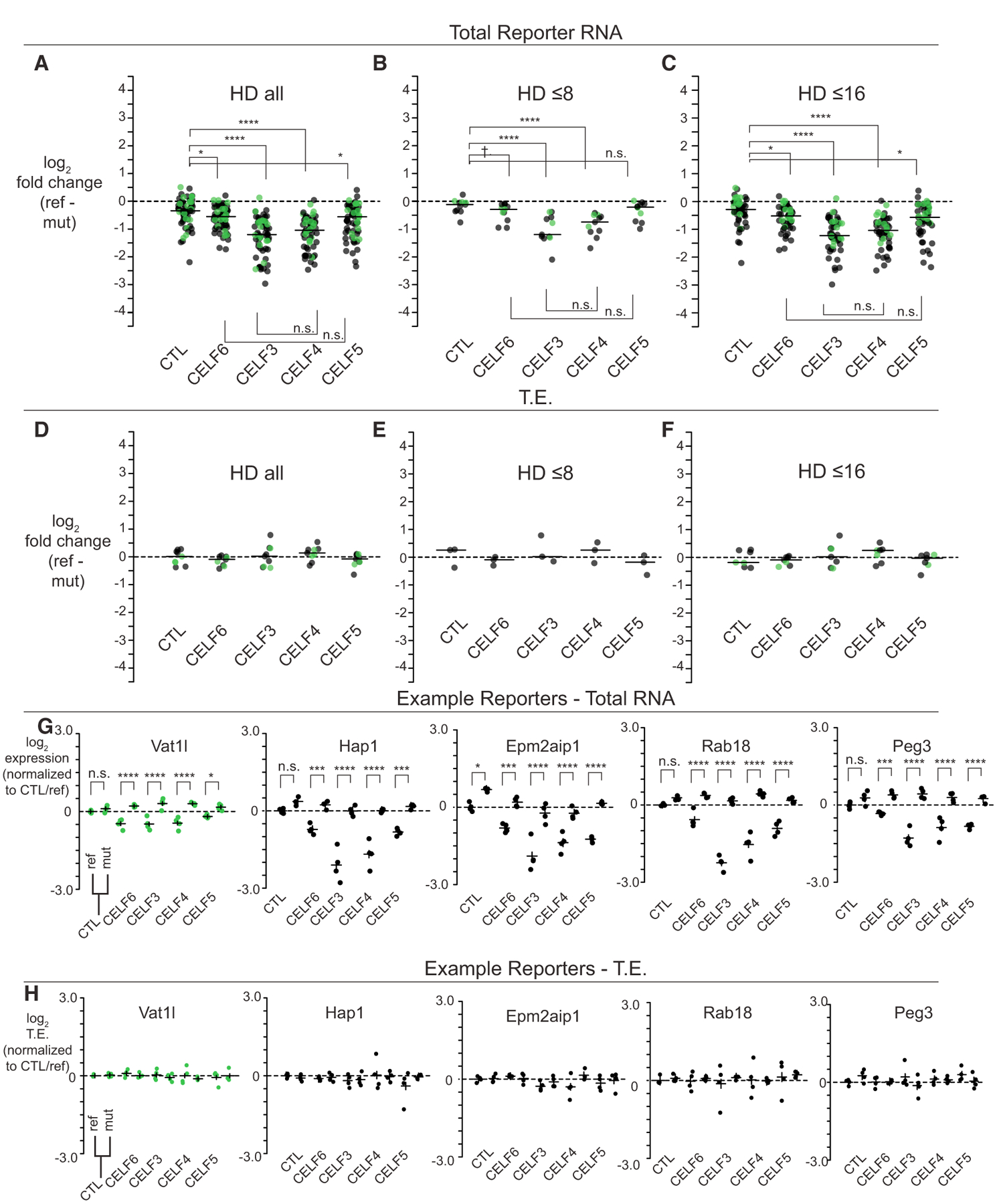
(A–C) Log2-fold change across CELF conditions considering all reporter elements, or reporter elements with HD ≤ 8 or ≤ 16, for any element with significant condition X sequence interactions.
(D–F) Log2-fold change in TE for conditions, as in (A)–(C). Data points in (A)–(F) were averaged for redundant barcodes, then, across replicates, with lines representing medians. Statistical comparisons in (A)–(F) were assessed by Mann-Whitney U tests.
(G) Log2 expression across five example reporter library elements across CELF, reference, and mutant conditions.
(H) Log2 TE across example reporters in (G). Data points in (G) and (H) are averaged across barcodes, with each dot representing a replicate.
As in Figure 3, horizontal lines in (G) and (H) represent average expression or TE ± 95% confidence intervals, normalized to the CTL/reference. Post hoc pairwise comparisons in (G) and (H) computed with multcomp in R. n.s., p > 0.1; †p < 0.1, *p < 0.05, **p < 0.001, ***p < 0.0001, ****p < 1E–5. Green points represent library elements selected from less-stringent CELF6 target criteria (enrichment over HA/YFP input alone).
There were 63/379 (p < 0.05; 42/379 with FDR < 0.1) elements representing 63/153 genes showing a nominally significant sequence by CELF-expression interactions in the total reporter RNA. Looking at the log2-fold change in the reference versus the mutant sequence, CELF3 and CELF4 showed the largest differences (Figure 5A). The median log2-fold change in the reference versus the mutant sequence in the CELF3 expression condition was −1.22, with comparable change under CELF4 expression (−1.06) (CTL median log2-fold change: −0.36; CTL versus CELF3 p = 9.5E–13; CTL versus CELF4 p = 1.2E–11; and CELF3 versus CELF4 p = 0.31; Mann-Whitney U tests). CELF6 and CELF5 also showed comparable fold changes, and those were intermediate between the CTL condition and CELF3 or CELF4 conditions (CELF5: −0.58, CELF6: −0.57; CELF5 versus CTL, p = 0.0025; CELF6 versus CTL, p = 0.0016; CELF5 versus CELF6, p = 0.57). These rankings were consistent at both low (HD ≤ 8) and higher (HD ≤ 16) levels of sequence mutation (Figures 5B and 5C). Thus, expression of any of these CELFs was able to reduce abundance of reference reporters, an effect that could be abolished by mutation. Furthermore, CELF3 and CELF4 were associated with the strongest effects, whereas CELF5 and CELF6 showed more moderate effects. Thus, within these CELF proteins, CELFs 5/6 and CELFs 3/4 appear to form distinct subgroups with respect to their effects on mRNA abundance.
When looking at TE, there were fewer significant effects. Nine elements showed significant interactions of condition by element sequence (p < 0.05, 0/379 at FDR < 0.1). However, when plotted in terms of their fold change between reference and mutant sequence, we were again unable to generalize a direction of effect on TE across elements. Median log2-fold changes in TE were near 0, with no significant differences across conditions (Figures 5D–5F). Thus, CELF expression decreased overall ribosome occupancy of reference elements, but largely by disrupting reporter transcript abundance. Total reporter expression for key examples are shown in Figure 5G with their respective TE shown in Figure 5H. Among significant CELF by sequence interactions, 6/63 showed significant reductions between a CELF condition and CTL in the mutant sequence (Slc39a6, Olfm1, Slc25a1, Pcdh17, Zbtb5, and Socs5). However, even though these mutant sequences appeared to respond to the CELF condition, their respective reference sequences were still more negative (Table S7). Thus, we find, in general, the effect of CELF by sequence interaction to be a CELF-associated decrease in reporter levels for elements with intact motifs.
Because expression of various CELFs may overlap in the brain, we next tested combinatorial effects of co-expressing CELF proteins. We transfected CELF6 construct with one of CELF3–5 in equimolar proportions. Among elements showing significant sequence-by-condition interactions, expression of CELF3 and CELF6 together resulted in repression similar to CELF3 by itself (median CELF3/CELF6 log2-fold change reference versus mutant: −1.23, CELF3 alone: −1.22; p = 0.37). This was also true of CELF4 (median CELF4/CELF6 log2-fold change: −1.13, CELF4 alone: −1.06; p = 0.34). Thus, the effect of CELF4 and CELF3 appears to be dominant, or at least maximal, in these co-transfections, not because of differences in the level of each CELF’s expression (Figure S2). When CELF5 and CELF6 were expressed together, the median log2-fold change of reference versus mutant sequence was more negative in the CELF5/CELF6 condition compared with CELF6 alone by approximately 20% (median log2-fold change in CELF5/CELF6: −0.72, CELF6 alone: −0.57; p = 0.037), although that comparison did not reach significance compared with CELF5 alone (CELF5 alone: −0.58; p = 0.19). Nevertheless, that may indicate that CELF5 and CELF6 can act additively. Overall, our reporter assays show that CELF proteins suppress mRNA abundance of targets containing specific sequence motifs.
CELF6 Regulates Transcript Abundance in the Brain
We next determined whether CELF6 affects target transcript abundance in vivo when CELF6 is absent. We examined targets by microarray analysis, comparing eight WT and eight Celf6-KO brains. Microarray probes measuring Celf6 are reduced, confirming loss of transcript, in the KO mouse (Figure 6B). Effects of CELF6 loss on total brain RNA abundance are modest: whereas several hundred genes show a nominally significant change in RNA abundance (Figure 6A), the median log2-fold change is 0.24, and only the Celf6 probes survive multiple testing corrections. However, examining the distribution of fold changes in CLIP targets compared with randomly selected probes, showed increased abundance in KO brains (p < 1.11E–09, Welch’s t test) (Figure 6C). This is consistent with a role for CELF6 in decreasing target stability in vivo. Given that CELF6 expression is restricted regionally (Maloney et al., 2016) and brain CELFs may have overlapping activity, modest changes are unsurprising. This experiment also combines direct effects on CELF6 targets and indirect effects on gene expression because of development in the KO background. However, despite those limitations, we see nominally significant (p < 0.05) regulation of several high-confidence CELF6 CLIP targets, including important neuronal signaling genes, such as Reln and Fgf13, as well as genes meeting lower-stringency criteria, such as Fos and Mecp2 (Figure S3); 20/21 measurement CELF6 targets change in the expected direction, showing increased mRNA levels in the KO, a result highly unlikely to be due to chance (p < 0.0005, χ2 test). This is driven by the 3′ UTR CLIP targets, (p < 2.2E–08; Figure 6C) because we did not detect changes in target genes identified via 5′ UTR or intronic binding. A few targets identified via binding in the CDS show a similar magnitude of increase in the KO; however, that did not reach statistical significance. Thus, overall, our analysis indicates CELF6 deletion is generally associated with an increase in the RNA abundance of its targets, in vivo, as it does in reporter assays in vitro.
Figure 6. CELF6 Targets Identified by CLIP Show Increased Expression in Celf6-Knockout Brains.
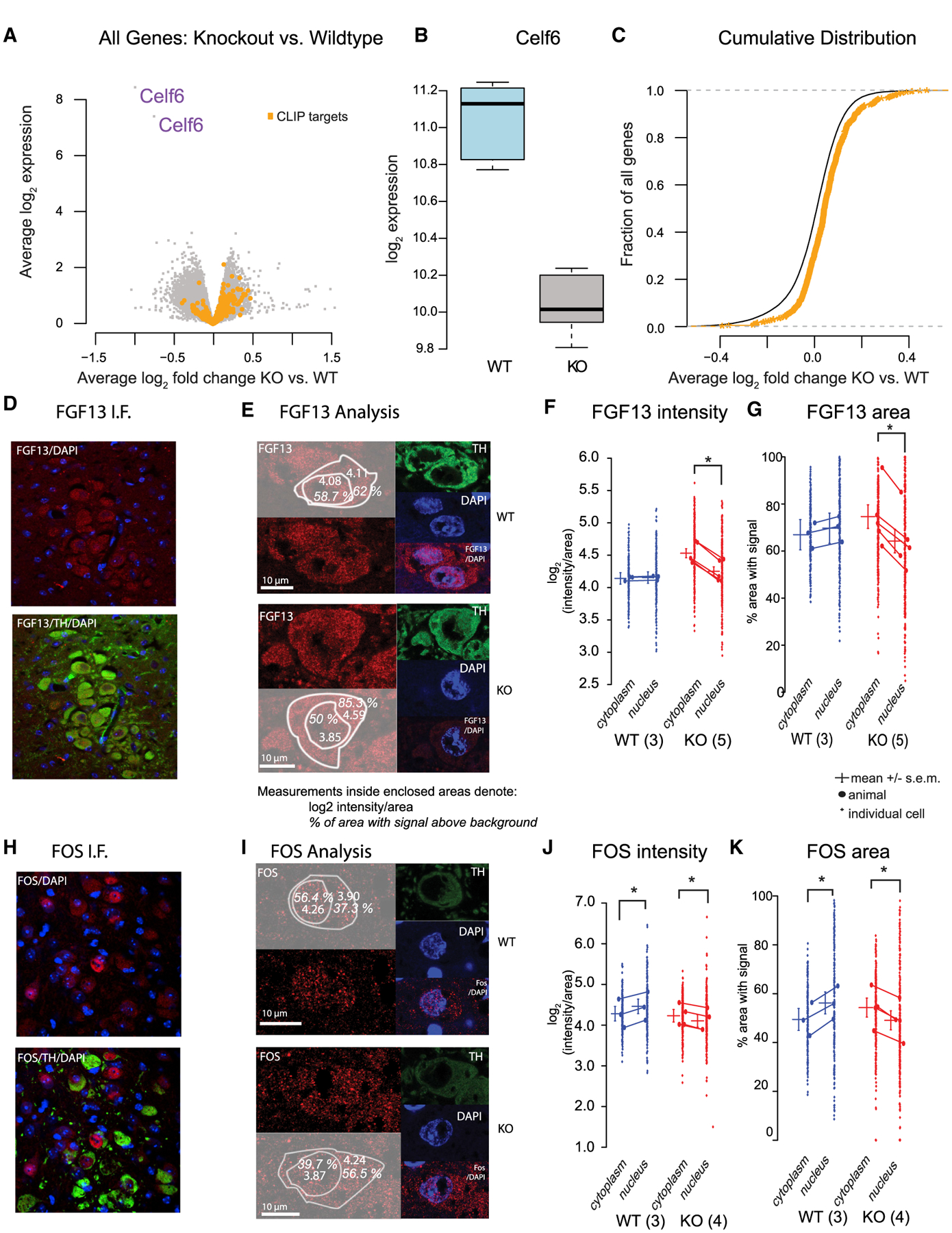
(A) Volcano plot showing gene-expression changes in Celf6 KO versus WT mouse brains.
(B) Celf6 mRNA is decreased in KO brains as expected (p < 1 × 10−6).
(C) CLIP targets (orange) are derepressed in KO brains compared with other genes (p < 1.1 × 10−9, Welch’s t test).
(D) Example of anti-FGF13 staining in LC.
(E) IF Analysis, left: FGF13 staining (red) with overlay showing quantification. Example images were selected to be near the mean of the data. Right: tyrosine hydroxylase (TH) and DAPI staining.
(F) FGF13 fluorescence intensity (log2 pixel intensity/area), with small dots representing cells and large dots averaged by animal (WT: n = 3, 92, 45, and 71 cells; KO: n = 5, 69, 19, 41, 99, and 69 cells). Lines represent grand means ± 95% confidence intervals.
(G–K) FGF13 area (percentage of region with an antibody signal above background) for the same cells as in (E). (G)–(I), as in (H)–(K), but for FOS immunostaining (WT: n = 3, 68, 70, and 62 cells; KO: n = 4, 94, 53, 44, and 98). Intensity and area with signal were analyzed using linear mixed models fitting fixed effects of subcellular localization (cytoplasm, nucleus), genotype (WT, KO), and their interaction, using individual cells per animal as a repeated-measure/random effect. Significant effects were determined by ANOVA with Tukey post hoc multiple comparisons between means computed in R using the lsmeans package. Significant comparisons in (D)–(K) are for p < 0.001. Scale bars in (E) and (I) represent 10 μm as indicated.
A systems analysis of the consequences of CELF6 loss is also intriguing (Figure S3C). Gene Ontology of the 200 most-upregulated genes in the KO brains (p < 0.05), regardless of CLIP status, reveals significant enrichment for terms for transcription regulators (transcription factor complex, p < 0.8E–3 with Benjamini-Hochberg [B-H] correction) and other components of the nucleus, as well as synaptic genes (growth cone, p < 3.47E–4, and dendritic spine, 4.02E–3). Downregulated genes showed no such consistent enrichment, with only a single category showing downregulation (flagellum, p < 9.62E–3). Thus, loss of CELF6 appears to dysregulate both nuclear transcription and synaptic genes in vivo.
CELF6 Regulates FOS and FGF13 Protein Abundance and Localization in the Locus Coeruleus
Finally, we tested the effect of Celf6 KO on target protein levels and localization in vivo. CELF6 is expressed sparsely in the brain, and immunoblots would be uninformative. We, therefore, began with CLIP targets in which antibodies had been previously validated for immunofluorescence in the brain. We focused on fibro-blast growth factor 13 (FGF13), a non-canonical FGF previously shown to bind intracellularly to voltage-gated sodium channels and regulate their subcellular spread (Pablo et al., 2016). Additionally, we looked at FOS (a lower-stringency target), an immediate early gene stimulated by neuronal activity and involved in downstream synaptic changes required for phenomena such as long-term potentiation (Abraham et al., 1991). We assayed expression in the locus coeruleus (LC), a stress-responsive neuromodulatory nucleus, identifiable by location and tyrosine hydroxylase (TH) staining, with robust CELF6 expression in WT mice (Maloney et al., 2016). To enable detection of FOS, animals were first subjected to restraint stress to activate the LC. We confirmed the antibodies produced a detectable signal in the LC under those conditions (Figures 6D and 6H). Because localization is important to the function of both proteins, the FGF13 or FOS signal was quantified for both fluorescence intensity and subcellular compartment (cytoplasmic or nuclear), as determined by DAPI or TH staining, respectively (Figures 6E–G and 6I–6K).
In the case of FGF13, we observed a significant increase (~20%) in the cytoplasmic KO mean expression (Figure 6E; log2 (intensity/area) cytoplasm = 4.53, nucleus = 4.25; p < 0.001; log2-fold change, cytoplasm − nucleus = 0.28), whereas we did not observe a significant change between WT cellular compartments (log2-fold change = −0.013). Additionally, we observed a significant increase (~10%) in KO cytoplasmic FGF13 spread (Figure 6F) quantified as the percentage of cytoplasmic area containing a signal above background (KO cytoplasmic mean = 74.6%, KO nuclear mean = 64.2%; p < 0.0001). In the WT, we did not detect a significant difference, although we observed an opposing trend with ~3% change in the area with a signal (WT cytoplasm = 66.9%, WT nucleus = 64.2%; p = 0.066).
In the case of FOS, we observed a small increase in signal (~10%) in the cytoplasm compared with the nucleus (Figure 6H; KO cytoplasmic log2 intensity/area = 4.23, KO nucleus = 4.1; p < 0.0001; log2-fold change cytoplasm − nucleus = 0.13) and the opposing trend (~12% change) in the WT (WT cytoplasm = 4.28, WT nucleus = 4.46; p < 0.0001, log2-fold change = −0.18.). The percentage of area containing a FOS signal was enriched in the nucleus of WT animals (Figure 6I; ~7% higher in the nucleus; p < 0.0001; cytoplasm = 49.4%, nucleus = 56.2%) but enriched in the cytoplasm of KO animals (~5% higher in the cytoplasm; p < 0.0001; cytoplasm = 54.3%, nucleus = 49.1%) The effects that are small in magnitude, however, are both consistent with the model in which CELF6 represses mRNA levels and protein expression in WT animals, which are de-repressed in the cytoplasm in KO animals.
DISCUSSION
In this study, we identify the in vivo targets of CELF6 in the brain. Our methods required us to use a tagged protein, but that protein exhibits the endogenous expression pattern and is only modestly overexpressed (Maloney et al., 2016). We found that CELF6 primarily binds to 3′ UTRs of mRNAs, with many targets shared by CELF4 (Wagnon et al., 2012). UTR elements under CELF6 CLIP peaks are enriched for several motifs previously identified (Ray et al., 2013) and are similar to CELF1 preferences (Wang et al., 2015). Using PTRE-seq (Cottrell et al., 2018), we evaluated the effect of CELF6 across many binding elements. Although our in vitro assays are not direct binding assays, we show that CELF6 and other CELFs are associated with downregulation of target 3′ UTRs, and this can be mitigated by motif mutation. Future work on this protein will involve understanding its binding affinity and effect on mRNA half-life. Although CELF6 has been shown to regulate alternative splicing (Ladd et al., 2004), we find very few significant targets outside of 3′ UTR regions, suggesting that CELF6 in the brain may be more involved with post-transcriptional regulation. UTR binding is consistent with recent work showing CELF6 is able to regulate the stability of p21 (Liu et al., 2019) and FBP (Yang et al., 2020) in cancer cells. We found very few changes to splicing in Celf6 KO mice (not shown; available at GEO: GSE160293) and few effects to TE, suggesting that CELF6 acts on its brain targets primarily to enhance mRNA degradation. We show these same targets are regulated in vivo and that loss of CELF6 can alter both protein intensity and subcellular distribution.
Our work here raises a number of biological questions. Although most of the PTRE-seq library was repressive, many elements were not further sensitive to CELF6 expression. This suggests that either CELF6 has additional functions on these transcripts not assayed (e.g., RNA localization) or that its functional effect on these sequences depends on cellular context. Although CELF2–6 RNA levels were largely undetectable in SH-SY5Y cells, the complement of other RBPs certainly differs from that of neurons. We did detect levels of MBNL1 in SH-SY5Y cells, an RBP that has been shown to antagonize CELF1 (Wang et al., 2015) leading to mRNA stabilization. It has also been shown that CELF1, CELF2, and CELF6 can all antagonize MBNL1’s splicing functions (Ohsawa et al., 2015). If MBNL1 and CELF6 can act antagonistically, then the resistance to CELF6 of some constructs in culture may be due to MBNL1 expression. Antagonistic activity of RBPs on mRNA translation and stability has also been observed for CELF2 and HUR (Sureban et al., 2007) and ELAVL1 and ZFP36 (Mukherjee et al., 2014). Such activity between RBPs may be mediated by binding-site proximity, and when RBP levels are altered, access to sites changes, and so does regulation (see Plass et al., 2017, study of AU-rich elements). In our analysis of CELF6 CLIP targets, we found enriched binding motifs across several RBP families. Future work will explore how interactions between sequence elements affects target CELF sensitivity.
It may be that CELF6-association downregulation of targets is due to enhancement of mRNA decay. Indeed, CELF1 has been shown able to recruit poly(A) ribonuclease (PARN) to RNA targets to facilitate mRNA decay, and Moraes et al (2006) found that CELF1 could associate with PARN in vitro. We tested co-immunoprecipitations of CELF6 with PARN but did not find evidence of an association in vitro (data not shown). However, it may associate with other proteins that mediate decay. The Xenopus homolog of the CELF proteins, EDEN-BP, has also been shown to regulate deadenylation, and oligomerization of the protein is required for that activity (Cosson et al., 2006). Future work will also explore whether CELF interactions (homo- or heterodimers) exist and can affect regulation. Finally, structure-function analysis mapping the domains involved in any such interactions will be of great interest in future studies.
CELF6 target enrichment for synaptic mRNAs and altered localization of FOS and FGF13 make it interesting to speculate that there is a role for CELF6 in regulating neuronal mRNA localization. Neurons are known to carefully regulate localization near activated spines to allow local translation to influence the development and strength of synaptic connections. However, because CELF6 expression is limited to sparse populations of neuronal cells poorly characterized with respect to local mRNA regulation, foundational work will be required before that can be fully assessed.
In sum, we have presented identification of CELF6 CNS binding targets in vivo and show that CELF6 can regulate mRNA abundance, both in vitro and in vivo. Some of these targets may mediate the behavioral phenotypes of the CELF6 KO mouse, which include communicative, exploratory, and reward system deficits (Dougherty et al., 2013; Maloney et al., 2019). Robust changes in response to more modest changes in RNA abundance indicate that mRNA translation must be carefully tuned for normal brain function and that subtle disruption of mRNA levels can substantially change the organism. Our data present an opportunity for further investigation into which targets, in which cell types, can regulate behavior.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Requests for information, resources, and reagents should be directed to Joseph D. Dougherty (jdougherty@genetics.wustl.edu).
Materials Availability
Materials are available as found above in the Key Resources Table. Plasmids generated in this study (pmrPTRE-AAV, pmrPTRE-AAV with CELF6 target UTR library), please see Lead Contact above. Raw sequencing data, as well as processing and analysis code, can be found under Data and Code Availability.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-EGFP(1) | MSKCC | Clone 19F7; RRID: AB_2716736 |
| Mouse anti-EGFP(2) | MSKCC | Clone 19C8; RRID: AB_2716737 |
| Chicken anti-GFP | AVES | GFP-1020; RRID: AB_10000240 |
| Mouse anti-TH | MilliporeSigma | MAB318; RRID: AB_2201528 |
| Rabbit anti-FGF13 | Geoffrey Pitt lab | N/A |
| Goat anti-cFOS | Santa Cruz | sc-52-g; RRID: AB_2629503 |
| Alexa Fluor 488 Donkey anti-Mouse | ThermoFisher | A21202; RRID: AB_141607 |
| Alexa Fluor 546 Donkey anti-Rabbit | ThermoFisher | A10040; RRID: AB_2534016 |
| Alexa Fluor 647 Donkey anti-Goat | ThermoFisher | A21447; RRID: AB_2535864 |
| Horseradish Peroxidase Goat anti-chicken IgY | AVES | H-1004; RRID: AB_2313517 |
| Bacterial Strains | ||
| NEB® 5-alpha Competent E. coli (High Efficiency) | New England Biolabs | C2987 |
| Deposited Data | ||
| CLIP-Seq raw data | This paper | GEO: GSE118623 |
| PTRE-Seq raw data | This paper | GEO: GSE118623 |
| Experimental Models: Cell Lines | ||
| Human: SH-SY5Y (female, age 4 years) | ATCC | CRL-2266 |
| Experimental Models: Organisms/Strains | ||
| Mouse: CELF6-HA/YFP | Dougherty Lab | JD2078 |
| Mouse: CeH6−/− (“KO”) | Dougherty Lab | Brj56 |
| Oligonucleotides | ||
| See Table S9 | Integrated DNA Technologies | N/A |
| Recombinant DNA | ||
| His/Xpress-CELF3 plasmid | Thomas Cooper lab | N/A |
| His/Xpress-CELF4 plasmid | Thomas Cooper lab | N/A |
| His/Xpress-CELF5 plasmid | Thomas Cooper lab | N/A |
| His/Xpress-CELF6 plasmid | Thomas Cooper lab | N/A |
| EGFP-CELF6 | This paper | N/A |
| pEGFP-C1 | Clontech | N/A |
| pcDNA3.1 His-A | Thermo Fisher | V38520 |
| EGFP-RPL10a | Heiman et al., 2008 | N/A |
| pQC membrane TdTomato IX | Addgene | 37351 |
| pmrPTRE-AAV | This paper | N/A |
| pmrPTRE-AAV with CELF6 CLIP target UTR library | This paper | N/A |
| Software and Algorithms | ||
| Trimmomatic | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| STAR | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| FGBio | Fulcrum Genomics | https://github.com/fulcrumgenomics/fgbio |
| Picard Tools | Broad Institute | https://broadinstitute.github.io/picard |
| Samtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
| Bedtools | Quinlan and Hall, 2010 | https://github.com/arq5x/bedtools2/blob/master/docs/content/overview.rst |
| Piranha | Uren et al., 2012 | http://smithlabresearch.org/software/piranha/ |
| Subread (featureCounts for read counting) | Liao et al., 2013 | http://subread.sourceforge.net/ |
| R | R Core Team, 2014 | https://www.r-project.org/ |
| edgeR (R package) | Robinson et al., 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| Discriminative Regular Expression Motif Elicitation (DREME) | Bailey, 2011 | http://meme-suite.org/tools/dreme |
| Analysis of Motif Enrichment (AME) | McLeay and Bailey, 2010 | http://meme-suite.org/tools/ame |
| Find Individual Motif Occurences (FIMO) | Grant et al., 2011 | http://meme-suite.org/tools/fimo |
| CISBP-RNA Database: Catalog of Inferred Sequence Binding Preferences of RNA binding proteins | Ray et al., 2013 | http://cisbp-rna.ccbr.utoronto.ca/index.php |
| lme4 (R package) | Bates et al., 2015 | https://cran.r-project.org/web/packages/lme4/index.html |
| multcomp (R package) | Hothorn et al., 2008 | https://cran.r-project.org/web/packages/multcomp/index.html |
| car (R package) | Fox and Weisberg, 2019 | https://cran.r-project.org/web/packages/car/index.html |
| limma (R package) | Ritchie et al., 2015 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| ImageJ | NIH | https://imagej.nih.gov/ |
| lsmeans (R package) | Lenth, 2016 | https://cran.r-project.org/web/packages/lsmeans/ |
Data and Code Availability
All software packages can be found in Key Resources Table. Raw and processed sequencing data from CLIP-Seq can be accessed through the National Center for Biotechnology Information’s Gene Expression Omnibus under accession number GSE118623. UCSC Genome Browser session showing CLIP-Seq data associated with NCBI GEO Accession GSE118623 can be viewed following this link: http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=mrieger&hgS_otherUserSessionName=celf6_clip_mm10
Code used to call peaks, perform MEME-SUITE motif enrichment clustering, and analyze the PTRE-Seq library data can be found at: https://github.com/clevermizo/CELF6CLIPMS
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal Models
All protocols involving animals were approved by the Animal Studies Committee of Washington University in St. Louis. Cages were maintained by our facility on a 12 hr: 12 hr light:dark schedule with food and water supplied ad libidum.
Genotyping
Genotyping of all mice was performed using a standard protocol. Animals were genotyped from toe clip tissue lysed by incubation at 50°C in Tail Lysis Buffer for 1 h to overnight (0.5 M Tris-HCl pH 8.8, 0.25 M EDTA, 0.5% Tween-20) containing 4 μL/mL 600 U/mL Proteinase K enzyme (EZ BioResearch), followed by heat denaturation at 99°C for 10 min. 1 μL Crude lysis buffer was used as template for PCR with 500 nM forward and reverse primers, as specified, using 1X Quickload Taq Mastermix (New England Biolabs) with the following cycling conditions: 94°C 1 min, (94°C 30 s, 60°C 30 s, 68°C 30 s) x 30 cycles, 68°C 5 min, 10°C hold. All referenced PCR primer sequences listed below are found in Table S9.
Mice for CLIP
Mice used in CLIP experiments derived from 4 litters of CELF6-HA/YFP x C57BL6/J crosses. Each sample used for sequencing was generated by pooling tissue from 3–4 CELF6-HA/YFP+ animals or WT animals from each litter. Pooling tissue was required to generate sufficient material for successful CLIP library generation. Genotyping was performed using HA-F/YFP-R pair (presence or absence of HA-YFP cassette) and Actb-F/Actb-R pair (internal PCR control). Genotyping was performed on post-natal day 7 to select animals for pools, and then performed again on tissue collected after sample processing on post-natal day 9 to confirm genotypes. Sex of animals was noted but not controlled in statistical analysis of CLIP-Seq data as all pools contained animals of both sexes.
-
Litter 1: 8 total animals.
CELF6-HA/YFP Pool 1: 4 animals: 1 male and 3 females.
CTL Pool 1: 4 animals: 3 males and 1 female.
-
Litter 2: 8 total animals.
CELF6-HA/YFP Pool 2: 4 animals: 2 males and 2 females.
CTL Pool from this litter was excluded because upon confirmatory regenotyping it was determined that 1 CTL animal in the pool was actually CELF6-HA/YFP+.
-
Litter 3: 6 total animals.
CELF6-HA/YFP Pool 3: 3 animals: 2 females and 1 male.
CTL Pool 2: 3 animals: 2 males and 1 female.
-
Litter 4: 8 total animals.
CELF6-HA/YFP Pool 4: 4 animals: 2 females, 2 males.
CTL Pool 3: 4 animals: 3 males, 1 female.
Mice for Agilent Expression microarray
8 Celf6+/+ (WT) (3 males and 5 females) and 8 Celf6−/− (KO) animals (4 males and 4 females) derived from 13 litters of Celf6+/− X Celf6+/− crosses were used to generate tissue for the RNA microarray experiment used to assay expression of CELF6 CLIP targets. Animals were genotyped as above using Celf6genoF/Celf6genoR primer pair and age when tissue was harvested ranged between 3.5–9 months.
Cell Culture
SH-SY5Y neuroblastoma cells (ATCC CRL-2266) were maintained at 5% CO2, 37°C, 95% relative humidity in 1:1 Dulbecco Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F12 GIBCO) supplemented with 10% fetal bovine serum (FBS Sigma). Under maintenance conditions, cells were also incubated with 1% Penicillin-streptomycin (Thermo), but antibiotics were not used during transient transfections. Cell passage was performed with 0.25% Trypsin-EDTA (Thermo).
METHOD DETAILS
CLIP
Our CLIP procedure is modeled after the procedure of Wang et al. (2015). Post-natal day 9 mice were euthanized by rapid decapitation, and brains were dissected. Cortices and cerebella were removed, retaining basal forebrain, striatum, diencephalon, colli-culi, and hindbrain regions, which are the brain regions with highest CELF6 expression (9). Dissected tissue was flash frozen in liquid nitrogen and then powdered with a mortar and pestle cooled with liquid nitrogen and kept on dry ice in 10 cm Petri dishes until use. Crosslinking was performed using 3 rounds of 400 mJ/cm2 dosage of 254 nm ultraviolet radiation, with Petri dishes on dry ice, in a Stratalinker UV crosslinker. After each round of crosslinking, powder in the dishes was redistributed to allow for even crosslinking. After crosslinking, powders were kept on wet ice and incubated with 1mL lysis buffer (50 mM Tris-HCl pH 7.4, 100 mM NaCl, 1X cOmplete EDTA-free protease inhibitor (Sigma), 0.04 U/μL recombinant RNasin (Promega), 10 mM activated sodium orthovanadate, 10 mM NaF). Recombinant RNasin does not inhibit RNase I which was used for RNase digestion in CLIP and was added to prevent other environmental RNase activity. To obtain both cytoplasmic and nuclear fractions in the lysate, lysis buffer was supplemented with NP40 (Sigma CA630) detergent (final concentration 1%) and subjected to mechanical homogenization in a teflon homogenizer 10 times, and lysates were allowed to incubate on ice for 5 min. For RNase digestion, RNase If (New England Biolabs) was diluted to final concentration 0.5, 0.1, or 0.05 U/mL per lysate for radiolabeling. For control radiolabeled samples (no crosslink and WT tissue immunoprecipitates), the highest (0.5 U/mL) concentration of RNase was used. For samples used for sequencing, 0.05 U/mL final concentration was used. RNase-containing lysates were incubated in a thermomixer set to 1200 RPM at 37°C for 3 min and then clarified at 20,000 xg for 20 min. 2% input lysate was saved for input samples for sequencing. Per immunoprecipitation, 120 μL of Dynabeads M280 streptavidin (Thermo) were incubated with 17 μL 1 mg/mL biotinylated Protein L (Thermo), and 36 μg each of mouse anti-EGFP clones 19F7 and 19C8 antibodies (MSKCC) for 1 h. Beads were prepared in batch for all immunoprecipitations and then washed five times with 0.5% IgG-free bovine serum albumin (Jackson Immunoresearch) in 1X PBS, followed by three washes in lysis buffer. Clarified lysates were incubated with coated, washed beads for 2 h at 4°C with end-over-end rotation and then washed in 1 mL of wash buffer (50 mM Tris-HCl pH 7.4, 350 mM NaCl, 1% NP-40, 0.04U/μL RNasin) four times, for 5 min with end-over-end rotation at 4°C. For radiolabeling experiments, 60% of washed bead volume was reserved for immunoblotting and added to 20 μL of 1X Bolt-LDS non-reducing sample buffer (Thermo), and 40% proceeded to radioactive labeling. Beads for radioactive labeling were subsequently washed 3×200 μL in PNK wash buffer (20 mM Tris-HCl pH 7.4, 10 mM MgCl2, 0.2% Tween-20, 2.5 U/μL RNasin) and then incubated with 10 μL PNK reaction mixture (1X PNK reaction buffer (New England Biolabs), 4 μCi γ32P-ATP (Perkin Elmer), 10 U T4 PNK (New England Biolabs)) for 5 min, at 37°C. After labeling, samples were washed in 3×200 μL PNK wash buffer to remove unincorporated label, and then added to 10 μL of 1X Bolt LDS non-reducing sample buffer. All samples in sample buffer were heated for 10 min at 70°C and then separated on a 4%–12% gradient NuPAGE Bis/Tris gels (Thermo) and then transferred to PVDF membranes with 10% methanol for 6 h at constant 150 mA. Samples for immunoblot were blocked for 1 h in block solution (5% nonfat dried milk in 0.5% Tween-20/1X TBS), and then overnight with 1:1000 chicken anti-GFP antibodies (AVES) with rocking at 4°C. Blots were washed 3 times for 5 min in 0.5% Tween20/1X TBS and then incubated with 1:5000 anti-chicken HRP secondary antibodies (AVES) for 1 h at room temperature and treated with Biorad Clarity enhanced chemiluminescence reagents for 5 min and chemiluminescent data acquired with a Thermo MyECL instrument. Radioactive signal was acquired using an Amersham Typhoon Imaging System and a BAS Storage Phosphor screen (GE Healthcare Life Sciences).
CLIP-Seq Sequencing Library Preparation
For CLIP-Seq, EGFP immunoprecipitated WT and HA-YFP+ tissue and 2% input samples were purified from PVDF membranes as follows. Membrane slices were cut with a clean razor according to the diagram in Figure 1A, from unlabeled samples as has been performed in eCLIP (14). PVDF membrane slices were incubated in 1.7 mL microcentrifuge tubes with 200 μL Proteinase K buffer (100 mM Tris-HCl pH 7.4, 50 mM NaCl, 10 mM EDTA, 1% Triton X-100) containing 40 μL of 800 U/mL Proteinase K (NEB) and incubated in a horizontal shaker at 250 RPM, 37°C for 1 h. Horizontal shaking reduces the need to cut the membrane into small pieces per sample which is seen in many protocols, and 1% Triton X-100 in the Proteinase K buffer facilitates increased yield from the membrane by preventing binding of Proteinase K to the membrane. 200 μL of fresh 7M Urea/Proteinase K buffer was then added to slices, and tubes were incubated an additional 20 min with horizontal shaking at 250 RPM, 37°C. RNA was purified by addition of 400 μL of acid phenol/chloroform/isoamyl alcohol and shaken vigorously for 15 s and allowed to incubate 5 min on the bench. RNA samples were centrifuged at 20,000 xg for 10 min. Aqueous layers were purified using a Zymo RNA Clean and Concentrator-5 column. Output from CLIP RNA samples was estimated for total concentration using an Agilent Bioanalyzer, and approximately 0.5 ng of RNA was used to prepare next generation sequencing libraries. The full protocol for sequencing library preparation is given in Methods S1. Although this protocol is based on eCLIP, Methods S1 is generalized for any RNA-Seq preparation.
Total RNA-Seq of SH-SY5Y cells
For preliminary RNA-Seq of SH-SY5Y cells, eight replicate sub-confluent (approximately 80%) 10 cm dishes (TPP) were harvested by dissociation from the plate by pipetting and centrifugation at 500 × g, 5 min at room temperature. Pellets were lysed according to TRAP protocol lysis conditions (Methods S2), and RNA was purified using Trizol LS, followed by treatment for 15 min at 37°C with DNase I (NEB) and cleanup using Zymo RNA Clean and Concentrator-5 columns (Zymo Research). RNA samples were assessed by Agilent TapeStation with RINe values between 8–10. RNA was fragmented and prepared into libraries for next generation sequencing using Methods S1 as for CLIP-Seq samples.
Total RNA-Seq and CLIP-Seq Raw Data Processing
As all total CLIP-Seq and Total RNA-Seq samples were prepared using a unified library preparation procedure, raw data processing used the same set of methods and tools (see Key Resources Table) for strand-specific quantification of sequencing reads containing unique molecular identifiers (See Methods S1) to collapse amplification duplicates. Briefly, CLIP libraries were sequenced in a 2 × 40 paired-end mode on an Illumina Next-Seq. Unique Molecular Identifier (UMI) sequences were extracted from Illumina Read 2, and reads were trimmed for quality using Trimmomatic. Using STAR, remaining reads were aligned to ribosomal RNA, and unalignable reads corresponding to non-rRNA were aligned to the mm10 mouse reference genome and assembled into BAM formatted alignment files. BAM files were annotated with UMI information using the FGBio Java package. PCR duplicates assessed by their UMIs were removed from the BAM files using Picard.
Peak Calling and Read Counting
To call peaks genome-wide using Piranha, the genome was windowed into 100 bp contiguous windows with 50% overlap using Bedtools, and strand-specific read counts were determined for each window. To ensure that called peaks would have the same boundaries for each replicate sample, peaks were called on a merged BAM file across all YFP-HA+ immunoprecipitated CLIP samples. Background counts for Piranha were estimated on a gene-by-gene basis to control for differences in overall level of expression. Piranha p values for significant peaks were adjusted for multiple testing using Benjamini-Hochberg, and all peaks called with False Discovery Rate < 0.1 were kept for further analysis and stored as a Gene Transfer Format (GTF) file. In practice we found that Piranha peaks varied widely, with some peaks called with widths on the order of several kilobases despite having a clear local maximum. To more narrowly count reads near peak maxima in a consistent way across all peaks, Piranha peak boundaries were truncated to a width of 100 base pairs around the peak maximum. As an alternate approach for discovery, we also wanted to consider total read counts across subgenic annotated regions (5′ UTR, 3′UTR, coding sequence (CDS) and introns), for which annotations were retrieved from the UCSC Genome Browser, in the event that CELF6 bound promiscuously without much sequence specificity though perhaps restricted to particular region of a gene. In order to ensure correct mapping of reads to splice sites, the table of intron annotations was allowed to overlap the surrounding exons by 10 bases. In the end, this procedure produced 5 GTF files:
Genome-wide peaks calls (100bp around called maxima from merged BAM file)
3′UTR
CDS
5′UTR
Introns
Each of these GTFs was used as a template for strand-specific feature counting in individual samples using the featureCounts program in the Subread package. For sums across regions (e.g., 3′UTRs) that showed enrichment in CLIP (see Quantification and Statistical Analysis below), we repeated the peak calling procedure in significantly enriched regions to determine whether any peaks missed in the genome-wide calling could be identified locally in enriched regions. We found most significantly enriched regions, such as 3′UTRs, had clearly definable peaks which were not determined in genome-wide peak calling. This may be due to misestimation of correct background in genome-wide calling. In genome-wide calling, background was estimated on a per-gene basis, but it may be that peaks are still missed because a transcript model is needed for most accurate calling. Determining enriched regions and then calling peaks on a small, localized region, seemed to ameliorate this issue.
SH-SY5Y Total RNA-seq samples were counted based on a GTF of all UCSC-annotated gene exons to derive total gene counts.
CLIP Motif Enrichment Analysis
50 bp regions under the maxima of peaks in CLIP target 3′UTRs were used in MEME Suite and compared to 50 bp regions sampled randomly from the 3′UTRs of non-targets – genes which exhibited 0 or negative fold enrichment in YFP-HA+ CLIP samples compared to input and WT controls. The AME tool was used to search motif enrichment against the CISBP-RNA database (Ray et al., 2013), using the maximum match score with the ranksum test.
The position probability matrices (PPMs) for CISBP-RNA motifs showing significant enrichment in CLIP 3′UTR peaks were hierarchically clustered (Figures 2B and 2C) by using vectorized PPMs for each motif and the hclust() function in R (using the “complete” method). To determine whether or not there were significant matches to a cluster, rather than an individual CISBP-RNA motif PPM, average PPMs were computed by averaging the PPMs across all cluster members. These averaged PPMs were then used with the MEME-Suite FIMO tool to determine whether individual peaks had significant matches to each of the clusters (Tables S3 and S4).
PTRE-Seq Reporter Library Preparation
All oligos used for library preparation are provided in Key Resources Table. We generated the pmrPTRE-AAV backbone from an existing mtdTomato construct by PCR amplification and subcloning of the following elements: CMV promoter and a T7 promoter, PCR amplified and subcloned into the MluI restriction site of pQC membrane TdTomato IX. CMV and T7 promters were amplified from pcDNA3.1 using pCMV_T7-F/pCMV_T7-R and Phusion polymerase (NEB). Then, in order to add a NheI-KpnI restriction enzyme cassette into the 3′UTR, the entire pmrPTRE-AAV plasmid was amplified (pmrPTRE_AAV_Full_F/R) and recircularized using Infusion HD (Clontech). The correct backbone sequence of pmrPTRE-AAV was confirmed by Sanger sequencing.
Originally, 462 sequences of 120 bp were considered for cloning into the library across significant genes. These included 295 peaks in high confidence targets (meeting enrichment against both input and non-specific IP controls, see Quantification and Statistical Analysis), and 167 peaks in lower confidence targets showing enrichment only over input. Mutations to motifs found by AME were made as follows. The location of significant matches to motifs were determined using FIMO. Next, for each matched motif, the PPM representing this motif in CISBP-RNA was used to determine the choice of mutation at each base. Bases showing PPM probability of 0.8 or greater were mutated to the base showing the minimum value of PPM at that position. If all other three bases showed equal probability, a base was randomly selected from the three. The procedure was repeated at each position for each motif match, before moving to the next matching motif. Where motifs overlapped, lower ranking motifs (based on FIMO score) did not override mutations already made based on higher ranking motifs. After completion, this generated a set of 473 mutant elements which ranged between 1 – 25 mutated nucleotides. Subsequently, sequences were scanned for poly A signals and restriction enzyme sites which would interfere with cloning (NheI, KpnI), which were removed. A final set of 410 sequences (254 high confidence, 156 meeting only input control criterion) and their paired mutant sequences were synthesized by Agilent Technologies with 120bp of UTR sequence in addition six unique 9 bp barcodes per sequence and priming sites for amplification and cloning (final length 210 bp). These sequences and their mutated forms are given in Table S5.
Obtained synthesized sequences were amplified with 4 cycles of PCR using Phusion polymerase with primers GFP-F and GFP-R. We selected these priming sites as these are standard primers in our laboratory used for genotyping that result in robust amplification. The library was PAGE purified and concentration of recovered library was estimated by Agilent TapeStation. The library was digested with NheI and KpnI enzymes and ligated into pmrPTRE-AAV with T4 Ligase (Enzymatics). In order to ensure high likelihood of obtaining all library elements, we prepared our plasmid pool from approximately 40,000 colonies.
PTRE-Seq Reporter Library Transfection
His/Xpress-tagged CELF3,4,5,&6 were obtained from the laboratory of Thomas Cooper. For the four-plasmid experiments, 2500 ng containing equimolar amounts of two His/Xpress-CELF constructs, an EGFP-RPL10a construct, and the CELF6 PTRE-Seq library were prepared with Lipofectamine 2000 in Opti-MEM I (GIBCO). For three-plasmid experiments, remaining mass was substituted with empty pcDNA3.1-His. SH-SY5Y cells were trypsinized and incubated in 10 cm dishes with Lipofectamine/DNA complexes overnight in DMEM/F12 supplemented with 10% FBS. The following day media was replaced with fresh DMEM/F12 supplemented with 10% FBS. Cells were pelleted for Translating Ribosome Affinity Purification (TRAP) and total RNA extraction 40 h post-transfection. TRAP and total RNA extraction were performed according to the Methods S2. RNA quality was assessed by Agilent TapeStation, and all samples had RINe values > 8. The procedure for TRAP in Methods S2 is based on (Heiman et al., 2008) with additional modifications that have been optimized in our laboratory. Five replicates per condition were generated in batches balanced for all conditions. In each case, replicates were transfected from newly thawed aliquots of cells passaged once before transfection to control for cell passage. Read counts from one batch were found to cluster separately from all others after sequencing, and data from this batch were excluded. The final data were analyzed from four replicates per condition.
PTRE-Seq Sequencing Library Preparation
PTRE-Seq sequencing libraries were prepared by cDNA synthesis using pmrPTRE-AAV antisense oligo for library specific priming, and Superscript III Reverse Transcriptase (Thermo) according to the protocol shown in Methods S3. After cDNA synthesis, cDNA libraries were enriched with PCR using Phusion polymerase (Thermo), and pmrPTRE-AAV antisense and sense oligos using 18 cycles. In parallel, plasmid pool DNA was also amplified for sequencing the original plasmid pool. Purified PCR products were digested with NheI and KpnI enzymes and ligated to 4 equimolar staggered adapters to provide sequence diversity for sequencing on the NextSeq. Ligated products were amplified with Illumina primers as in CLIP-Seq library preparation (Methods S1) and subjected to 2×40 paired-end next generation sequencing on an Illumina NextSeq.
RT-PCR and qRT-PCR
To confirm exogenous expression of CELF constructs used in PTRE-Seq experiments, 20 ng of total RNA was converted into cDNA using qScript cDNA synthesis kit (Quantabio, kit employs a mix of both oligo-dT and random hexamer priming) and diluted 4-fold. For RT-PCR, 4 μL of diluted cDNA was used in PCR reactions with 500 nM forward and reverse primers, (Figure S2; Table S9), using 1X Quickload Taq Mastermix (New England Biolabs) with the following cycling conditions: 94°C 1 min, (94°C 30 s, 60°C 30 s, 68°C 30 s) × 25 cycles, 68°C 5 min, 10°C hold, and then separated by 2% agarose and stained with ethidium bromide. For qRT-PCR, 4 μL of diluted cDNA was combined with 500 nM His/Xpress-pcDNA-F and His/Xpress-pcDNA-R primers or HsActb-F/HsAcb-R, and 1X PowerUP SYBR Green Master Mix (Thermo). Each sample/primer combination was run in 4 technical replicates on a Viia7 Real Time PCR System (Thermo) using the following cycling program: 50°C 2 min, 95°C 2 min (95°C 1 s, 60°C 30 s) × 40 cycles, followed by dissociation step: 95°C 15 s 1.6°/s ramp,60°C 1 min 1.6°/s ramp, 95°C 15 s 0.15°/s ramp. Samples were run alongside no template and no reverse transcription controls to ensure reactions were free of non-target contamination, and dissociation curves were inspected to ensure the absence of non-target amplicons. CT values for each sample were averaged across technical replicates and transformed by first computing the ΔCT: = CTHis/Xpress - CTHsActb, and then computing relative log2 expression (“ΔΔCT”): = − (ΔCTsample - ΔCTreference), where the reference was taken to be the average ΔCT across the CELF6 expression condition.
PTRE-Seq Reporter Validation
For validation of individual library element reporters (Figure 4), CELF6 CDS was subcloned from His/Xpress-CELF6 into pEGFP-C1 using EcoRI and BamHI restriction sites. Individual Vat1l, Hap1, Peg3, Epm2aip1, and Rab18 reference and mutant sequences were synthesized using Integrated DNA Technologies (IDT) gBlocks Gene Fragments and cloned into pmrPTRE-AAV with NheI and KpnI as above.
50 ng equimolar library element reporters with either pEGFP-C1 (EGFP alone) or EGFP-CELF6 was transiently transfected into SH-SY5Y cells in 96-well plates (TPP). 40 h post-transfection, media was removed and replaced with warm PBS (1.8 mM KH2PO4, 10 mM Na2HPO4, 2.7 mM KCl, 137 mM NaCl, pH 7.4). tdTomato and GFP fluorescence were determined by BioTek Instruments Cytation 5 Cell Imaging Multi-Mode Reader with internal temperature maintained at 37°C. 96-well plates were prepared in four replicate batches, where each 96-well plate contained one replicate well for all 20 conditions (five reference, five mutant reporters, in both EGFP-CELF6 and EGFP-only transfections). Log2-transformed fluorescence intensity measurements were z-score normalized on each plate to account for batch-to-batch differences in transfection efficiency and fluorescence intensity. Data in Figure 4 are shown further normalized to the average value for each reporter in the EGFP-only, reference sequence condition. Example epifluorescent images were obtained using a Leica DMI3000 B microscope with 20X magnification. Monochromatic images were acquired with QCapture software (QImaging), using gain = 1, offset = 1, exposure = 205 ms for both red and green fluorescent filter sets. 16-bit grayscale images were converted to RGB color and minimally brightness-adjusted for presentation using Adobe Photoshop CS2.
Agilent Gene Microarray
Brains from eight WT and eight Celf6 mutant mice (see) were extracted, frozen in liquid nitrogen, and crushed into a fine powder, from which RNA was extracted using QIAGEN RNEasy columns on a Qiacube robot, following manufacturer’s protocol. RNA was DNase I treated, and RNA quantity and integrity were confirmed using Agilent Bioanalyzer. cDNA was prepared and chemically labeled with Kreatech ULS RNA labeling kit (Kreatech Diagnostics) and Cy5-labeled cDNAs were hybridized to Agilent Mouse v2 4×44K microarrays (G4846A-026655). Hybridization of the labeled cDNAs was done in Agilent 2x gene expression hybridization buffer, Agilent 10x blocking reagent and Kreatech Kreablock onto Agilent 4×44K V2 microarrays at 65°C for 20 min. Slides were scanned on an Agilent C-class Microarray scanner. Gridding and analysis of images was performed using Agilent Feature Extraction V11.5.1.1.
FGF13 and FOS Immunofluorescence
3 Celf6+/+ (WT) (2 males and 1 female) and 3 Celf6 −/− (KO) animals (2 males and 1 female) derived from 2 litters of Celf6+/− × Celf6 ± crosses were used to generate tissue for immunofluorescence. Mice were perfused with PBS followed by 4% paraformaldehyde(PFA) in PBS. Brains were dissected then post-fixed overnight in 4% PFA, cryoprotected in increasing concentrations of sucrose over 48 h, and frozen in blocks of OCT (Tissue-Tek), sectioned on a cryostat to 40 microns, and stored at 4°C in PBS/0.25% Sodium Azide until use. Sections were blocked with 5% serum for 1h at room temperature, immersion-stained with primary antibodies overnight at room temp, washed 3x with PBS, then incubated with appropriate Alexa Fluor-labeled secondary antibodies for 90 min, counterstained with DAPI, and sections mounted on slides. Primary antibodies included rabbit anti-FGF13 (a gift from Geoffrey Pitt) and mouse anti-TH. Primary antibodies for FOS stain included goat anti-cFos, and mouse anti-TH (see Key Resources Table). Immunofluorescent imaging was conducted by an experimenter blind to genotype, focusing on the locus coeruleus (as identified by TH staining) using a Zeiss LSM 510 laser-scanning confocal microscope and accompanying software.
QUANTIFICATION AND STATISTICAL ANALYSIS
Defining CELF6 CLIP Targets by Differential Expression Analysis
Currently there is no standard statistical approach for identification of targets from CLIP data. Methods typically include clustering aligned sequences within individual CLIP RNA samples or replicate averages, with varying probabilistic modeling approaches for assessing signal-to-noise in read density, but rarely take into account variance across replicates or differential abundance compared to control samples (Ascano et al., 2012; Corcoran et al., 2011; Kishore et al., 2011; Lebedeva et al., 2011; Memczak et al., 2013; Moore et al., 2014; Mukherjee et al., 2011; Sugimoto et al., 2012; Ule et al., 2003; Wang et al., 2015; Zhang and Darnell, 2011). Furthermore, CLIP studies frequently fail to account for differences in the starting abundance of possible target mRNAs, thus reported targets are frequently biased toward highly expressed genes (Ouwenga and Dougherty, 2015). Here, we adopted a strategy of counting reads (as described above) and using standard differential expression analysis tools (edgeR) to make statistical inference on counted features in target immunoprecipitated samples compared to controls, with the hypothesis being that true targets will be enriched in target HA-YFP CLIP samples over both WT controls (representing non-specific immunoprecipitation pull down) and input samples (accounting for differences in starting abundance of possible target mRNAs). WT controls are a key recommendation in discussion of CLIP methods using engineered tags (Van Nostrand et al., 2017).
Samples counted using Subread were imported into R using edgeR. To be included in differential analysis, counted regions were required to possess a minimum of 10 read counts in at least 3 samples. Differential testing was then performed in edgeR against read counts deriving from WT samples or YFP-HA+ input samples. We defined CLIP targets as those having positive fold change enrichment in YFP-HA+ CLIP samples compared to both WT CLIP and input samples, with edgeR p values < 0.05 from the edgeR two sample negative binomial exact test. To test less stringent cut-offs, we also examined some targets meeting p < 0.05 for just the YFP-HA versus Input as noted in figure legends. Target peaks and gene information are found in Table S2. Analysis of enrichment for gene ontology terms was performed in DAVID (Huang et al., 2009a, 2009b) using genes of known expression in the brain as the background list rather than the entire mouse genome.
PTRE-Seq Barcode Counting and Normalization
Barcode counts from sequencing read FASTQ files for each element were determined using a Python script. Read counts were imported into R using edgeR and converted to counts-per-million (CPM) to normalize for differences in library size. Elements showing no counts in the DNA plasmid pool sequencing were removed. Expression was then computed as:
Translation efficiency was computed as:
We required that analyzed barcodes have at least 10 counts in at least four samples to be included in analysis. Additionally, we required that all elements in the library have a minimum of three out of the original six barcodes present, and present for both reference and mutant sequences.
PTRE-Seq Linear Mixed Model Analysis
The final set of elements after filtering on expression and numbers of barcodes was 379 across 153 unique gene UTRs, comprising 229 high confidence (meeting both input and wt/non-specific IP controls) and 150 lower confidence (meeting input control only) CLIP targets. Individual linear mixed models were performed using lme4 fitting the following:
where barcodes were used as repeated-measures for each sample, per element. Fixed effect terms of condition referred to either: (a) CTL or CELF6 expression for analyses in Figure 3, or (b) CTL, CELF6, CELF3, CELF4, CELF5, CELF3/6, CELF4/6, CELF5/6 for analyses related to Figure 5. The fixed effect term of sequence was either (a) reference or (b) mutated sequence. Omnibus Analysis of Deviance tests for significant effects of fixed effect terms were computed using type II ANOVA R with the car package with Satterthwaite degrees of freedom. Estimates of R2 were determined according to the procedure of Nakagawa and Schielzeth (2013) which provides a simple method for obtaining these estimates from non-linear and empirical model fits in a “percentage of variance explained”-interpretative sense. Omnibus p values for fixed effects are also reported in Tables S6, S7, and S8 for models alongside Benjamini-Hochberg adjusted False Discovery Rates for TRAP, input expression, and translation efficiency (TE) respectively.
Figures 3 and 5 show analysis of these across all individual models and for subsets of elements with smaller numbers of mutations introduced (≤8 or ≤ 16) to discern whether radically mutating elements has exerted a strong effect. Because these subsets are nested and because this bird’s-eye view analysis is pooling independently fitted models, we have assessed significance between them using non-parametric Mann-Whitney U tests for differences between medians. Post hoc multiple comparisons significance and confidence intervals reported for individual elements in Figures 3 and 4, in order to identify the sources of interaction, were computed using the multcomp R package.
PTRE-Seq Validation Analysis
Validation of reporters in Figure 4 was assessed using the car package in R to compute Type II two-way ANOVA with main effects of sequence and CELF6 exogenous expression and their interaction, with post hoc pairwise multiple comparisons determined using multcomp.
Agilent Expression Microarray Analysis
Features were extracted with Agilent Feature Extraction software and processed with the limma package in R to background correct (using normexp) and quantile normalize between arrays. Genotype of knockout samples was confirmed via decrease of Celf6 mRNA expression level, and differential expression analysis was conducted using limma with empirical bayes (eBayes) analysis. Log2 fold change of CLIP targets was compared to an equivalent number of randomly sampled probes by Welch’s t test. Gene Ontologies Analysis was conducted in Cytoscape using the BiNGO Module (Maere et al., 2005), with Benjamini-Hochberg correction and a display cutoff of p < 0.01. Cellular component results are displayed.
Immunofluorescence Quantification
Positive FOS and FGF13 immunofluorescent signal was quantified using the NIH Image analysis software, ImageJ. Cell bodies and nuclei were demarcated via freehand outlining by experimenter blind to genotype based on TH and DAPI stain of cells. Intensity of signal was measured in each cell, as well as within the nucleus of each cell, and normalized by total pixel area. Fraction of area containing positive signal in each cell and within each nuclei were also measured using a standardized threshold for positive signal. Signal intensity/area as well as fraction of area containing signal for each protein was analyzed using a linear mixed effects model in R with lme4, with individual quantified regions of interest as a repeated-measure for each animal, with fixed effects of region (cytoplasm versus nucleus) and genotype (WT versus KO). Significance was determined using Type II ANOVA with Satterthwaite degrees of freedom using the car package in R, with post hoc multiple pairwise comparisons determined using the multcomp package.
Supplementary Material
Highlights.
CELF6 primarily associates with 3′ UTRs of synaptic genes in the mouse brain
CELF6:sequence interaction is assayed using a massively parallel reporter assay
CELF3–6 all result in lower mRNA levels with few changes to translation efficiency
CELF6 targets are derepressed in vivo in Celf6-knockout mice
ACKNOWLEDGMENTS
This work was supported by the NIH (5R00NS067239, 5R21MH099798, 5R01HG008687, and R01MH116999) and the Simons Foundation. We thank Eric Wang, Eric Van Nostrand, Jean Schaffer, and Joshua Langmade for advice and training on CLIP-seq; Kyle Cottrell and Sergej Djuranovic for help with PTRE-seq assays; and Hemangi Chaudhari, Kristina Sakers, and Bernard Mulvey for consultation. We also thank Allison Lake, Susan Maloney, Affua Akuffo, Jasbir Dalal, and Nilambari Pisat, who were involved in generation of mice, data collection, and advice; the Thomas Cooper laboratory for CELF expression constructs; and Gregory Pitt for the FGF13. Finally, we thank Tomás Lagunas, Jr., Anthony Fischer, Stephen Plassmeyer, Sarah Koester, and Kayla Nygaard for reviewing this manuscript.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108531.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abraham WC, Dragunow M, and Tate WP (1991). The role of immediate early genes in the stabilization of long-term potentiation. Mol. Neurobiol 5, 297–314. [DOI] [PubMed] [Google Scholar]
- Ascano M, Hafner M, Cekan P, Gerstberger S, and Tuschl T (2012). Identification of RNA-protein interaction networks using PAR-CLIP. Wiley Interdiscip. Rev. RNA 3, 159–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auweter SD, Oberstrass FC, and Allain FH-T (2006). Sequence-specific binding of single-stranded RNA: is there a code for recognition? Nucleic Acids Res. 34, 4943–4959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL (2011). DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics 27, 1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates D, Mächler M, Bolker B, and Walker S (2015). Fitting linear mixed-effects models using lme4. J. Stat. Softw 67, 1–47. [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HS, Hwang CK, Song KY, Law P-Y, Wei L-N, and Loh HH (2009). Poly(C)-binding proteins as transcriptional regulators of gene expression. Biochem. Biophys. Res. Commun 380, 431–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran DL, Georgiev S, Mukherjee N, Gottwein E, Skalsky RL, Keene JD, and Ohler U (2011). PARalyzer: definition of RNA binding sites from PAR-CLIP short-read sequence data. Genome Biol. 12, R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosson B, Gautier-Courteille C, Maniey D, Aït-Ahmed O, Lesimple M, Osborne HB, and Paillard L (2006). Oligomerization of EDEN-BP is required for specific mRNA deadenylation and binding. Biol. Cell 98, 653–665. [DOI] [PubMed] [Google Scholar]
- Cottrell KA, Chaudhari HG, Cohen BA, and Djuranovic S (2018). PTRE-seq reveals mechanism and interactions of RNA binding proteins and miRNAs. Nat. Commun 9, 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta T, and Ladd AN (2012). The importance of CELF control: molecular and biological roles of the CUG-BP, Elav-like family of RNA-binding proteins. Wiley Interdiscip. Rev. RNA 3, 104–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty JD, Maloney SE, Wozniak DF, Rieger MA, Sonnenblick L, Coppola G, Mahieu NG, Zhang J, Cai J, Patti GJ, et al. (2013). The disruption of Celf6, a gene identified by translational profiling of serotonergic neurons, results in autism-related behaviors. J. Neurosci 33, 2732–2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JP, Dougherty JD, Heiman M, Schmidt EF, Stevens TR, Ma G, Bupp S, Shrestha P, Shah RD, Doughty ML, et al. (2008). Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell 135, 749–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox J, and Weisberg S (2019). An R Companion to Applied Regression (Sage). [Google Scholar]
- Garneau NL, Wilusz J, and Wilusz CJ (2007). The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol 8, 113–126. [DOI] [PubMed] [Google Scholar]
- Gilks N, Kedersha N, Ayodele M, Shen L, Stoecklin G, Dember LM, and Anderson P (2004). Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell 15, 5383–5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, and Noble WS (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey RF, Smith TS, Mulroney T, Queiroz RML, Pizzinga M, Dezi V, Villenueva E, Ramakrishna M, Lilley KS, and Willis AE (2018). Transacting translational regulatory RNA binding proteins. Wiley Interdiscip. Rev. RNA 9, e1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suárez-Fariñas M, Schwarz C, Stephan DA, Surmeier DJ, et al. (2008). A translational profiling approach for the molecular characterization of CNS cell types. Cell 135, 738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch AG, Ivanov IP, and Sonenberg N (2016). Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science 352, 1413–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hothorn T, Bretz F, and Westfall P (2008). Simultaneous inference in general parametric models. Biom. J 50, 346–363. [DOI] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009a). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009b). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivshina M, Lasko P, and Richter JD (2014). Cytoplasmic polyadenylation element binding proteins in development, health, and disease. Annu. Rev. Cell Dev. Biol 30, 393–415. [DOI] [PubMed] [Google Scholar]
- Kishore S, Jaskiewicz L, Burger L, Hausser J, Khorshid M, and Zavolan M (2011). A quantitative analysis of CLIP methods for identifying binding sites of RNA-binding proteins. Nat. Methods 8, 559–564. [DOI] [PubMed] [Google Scholar]
- Ladd AN, Charlet N, and Cooper TA (2001). The CELF family of RNA binding proteins is implicated in cell-specific and developmentally regulated alternative splicing. Mol. Cell. Biol 21, 1285–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladd AN, Nguyen NH, Malhotra K, and Cooper TA (2004). CELF6, a member of the CELF family of RNA-binding proteins, regulates muscle-specific splicing enhancer-dependent alternative splicing. J. Biol. Chem 279, 17756–17764. [DOI] [PubMed] [Google Scholar]
- Lebedeva S, Jens M, Theil K, Schwanhäusser B, Selbach M, Landthaler M, and Rajewsky N (2011). Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol. Cell 43, 340–352. [DOI] [PubMed] [Google Scholar]
- Lee JE, and Cooper TA (2009). Pathogenic mechanisms of myotonic dystrophy. Biochem. Soc. Trans 37, 1281–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenth RV (2016). Least-squares means: the R package lsmeans. J. Stat. Softw 69, 1–33. [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Sub-group (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2013). The subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 41, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Zhang Q, Xia L, Shi M, Cai J, Zhang H, Li J, Lin G, Xie W, Zhang Y, and Xu N (2019). RNA-binding protein CELF6 is cell cycle regulated and controls cancer cell proliferation by stabilizing p21. Cell Death Dis. 10, 688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maere S, Heymans K, and Kuiper M (2005). BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21, 3448–3449. [DOI] [PubMed] [Google Scholar]
- Maloney SE, Khangura E, and Dougherty JD (2016). The RNA-binding protein Celf6 is highly expressed in diencephalic nuclei and neuromodulatory cell populations of the mouse brain. Brain Struct. Funct 221, 1809–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloney SE, Rieger MA, Al-Hasani R, Bruchas MR, Wozniak DF, and Dougherty JD (2019). Loss of CELF6 RNA binding protein impairs cocaine conditioned place preference and contextual fear conditioning. Genes Brain Behav. 18, e12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeay RC, and Bailey TL (2010). Motif enrichment analysis: a unified framework and an evaluation on ChIP data. BMC Bioinformatics 11, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, et al. (2013). Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495, 333–338. [DOI] [PubMed] [Google Scholar]
- Moore MJ, Zhang C, Gantman EC, Mele A, Darnell JC, and Darnell RB (2014). Mapping argonaute and conventional RNA-binding protein interactions with RNA at single-nucleotide resolution using HITS-CLIP and CIMS analysis. Nat. Protoc 9, 263–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraes KCM, Wilusz CJ, and Wilusz J (2006). CUG-BP binds to RNA substrates and recruits PARN deadenylase. RNA 12, 1084–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee N, Corcoran DL, Nusbaum JD, Reid DW, Georgiev S, Hafner M, Ascano M Jr., Tuschl T, Ohler U, and Keene JD (2011). Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol. Cell 43, 327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee N, Jacobs NC, Hafner M, Kennington EA, Nusbaum JD, Tuschl T, Blackshear PJ, and Ohler U (2014). Global target mRNA specification and regulation by the RNA-binding protein ZFP36. Genome Biol. 15, R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa S, and Schielzeth H (2013). A general and simple method for obtaining R2 from generalized linear mixed-effects models. Methods Ecol. Evol 4, 133–142. [Google Scholar]
- Nakielny S, and Dreyfuss G (1996). The hnRNP C proteins contain a nuclear retention sequence that can override nuclear export signals. J. Cell Biol 134, 1365–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand EL, Gelboin-Burkhart C, Wang R, Pratt GA, Blue SM, and Yeo GW (2017). CRISPR/Cas9-mediated integration enables TAG-eCLIP of endogenously tagged RNA binding proteins. Methods 118–119, 50–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsawa N, Koebis M, Mitsuhashi H, Nishino I, and Ishiura S (2015). ABLIM1 splicing is abnormal in skeletal muscle of patients with DM1 and regulated by MBNL, CELF and PTBP1. Genes Cells 20, 121–134. [DOI] [PubMed] [Google Scholar]
- Ouwenga RL, and Dougherty J (2015). Fmrp targets or not: long, highly brain-expressed genes tend to be implicated in autism and brain disorders. Mol. Autism 6, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pablo JL, Wang C, Presby MM, and Pitt GS (2016). Polarized localization of voltage-gated Na+ channels is regulated by concerted FGF13 and FGF14 action. Proc. Natl. Acad. Sci. USA 113, E2665–E2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piqué M, López JM, Foissac S, Guigó R, and Méndez R (2008). A combinatorial code for CPE-mediated translational control. Cell 132, 434–448. [DOI] [PubMed] [Google Scholar]
- Plass M, Rasmussen SH, and Krogh A (2017). Highly accessible AU-rich regions in 3′ untranslated regions are hotspots for binding of regulatory factors. PLoS Comput. Biol 13, e1005460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2014). R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing; ). [Google Scholar]
- Ray D, Kazan H, Cook KB, Weirauch MT, Najafabadi HS, Li X, Gueroussov S, Albu M, Zheng H, Yang A, et al. (2013). A compendium of RNA-binding motifs for decoding gene regulation. Nature 499, 172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scoumanne A, Cho SJ, Zhang J, and Chen X (2011). The cyclin-dependent kinase inhibitor p21 is regulated by RNA-binding protein PCBP4 via mRNA stability. Nucleic Acids Res. 39, 213–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty S (2005). Regulation of urokinase receptor mRNA stability by hnRNP C in lung epithelial cells. Mol. Cell. Biochem 272, 107–118. [DOI] [PubMed] [Google Scholar]
- Sugimoto Y, König J, Hussain S, Zupan B, Curk T, Frye M, and Ule J (2012). Analysis of CLIP and iCLIP methods for nucleotide-resolution studies of protein-RNA interactions. Genome Biol. 13, R67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sureban SM, Murmu N, Rodriguez P, May R, Maheshwari R, Dieckgraefe BK, Houchen CW, and Anant S (2007). Functional antagonism between RNA binding proteins HuR and CUGBP2 determines the fate of COX-2 mRNA translation. Gastroenterology 132, 1055–1065. [DOI] [PubMed] [Google Scholar]
- Timchenko LT, Timchenko NA, Caskey CT, and Roberts R (1996). Novel proteins with binding specificity for DNA CTG repeats and RNA CUG repeats: implications for myotonic dystrophy. Hum. Mol. Genet 5, 115–121. [DOI] [PubMed] [Google Scholar]
- Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, and Darnell RB (2003). CLIP identifies Nova-regulated RNA networks in the brain. Science 302, 1212–1215. [DOI] [PubMed] [Google Scholar]
- Uren PJ, Bahrami-Samani E, Burns SC, Qiao M, Karginov FV, Hodges E, Hannon GJ, Sanford JR, Penalva LOF, and Smith AD (2012). Site identification in high-throughput RNA-protein interaction data. Bioinformatics 28, 3013–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidaki M, Drees F, Saxena T, Lanslots E, Taliaferro MJ, Tatarakis A, Burge CB, Wang ET, and Gertler FB (2017). A requirement for mena, an actin regulator, in local mRNA translation in developing neurons. Neuron 95, 608–622.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalba A, Coll O, and Gebauer F (2011). Cytoplasmic polyadenylation and translational control. Curr. Opin. Genet. Dev 21, 452–457. [DOI] [PubMed] [Google Scholar]
- Vindry C, Vo Ngoc L, Kruys V, and Gueydan C (2014). RNA-binding protein-mediated post-transcriptional controls of gene expression: integration of molecular mechanisms at the 3′ end of mRNAs? Biochem. Pharmacol 89, 431–440. [DOI] [PubMed] [Google Scholar]
- Wagnon JL, Briese M, Sun W, Mahaffey CL, Curk T, Rot G, Ule J, and Frankel WN (2012). CELF4 regulates translation and local abundance of a vast set of mRNAs, including genes associated with regulation of synaptic function. PLoS Genet. 8, e1003067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Ward AJ, Cherone J, Wang TT, Giudice J, Treacy D, Freese P, Lambert NJ, Saxena T, Cooper TA, et al. (2015). Antagonistic regulation of mRNA expression and splicing by CELF and MBNL proteins. Genome Res. 25, 858–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RKC, Hua Y, Gueroussov S, Najafabadi HS, Hughes TR, et al. (2015). RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 347, 1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Zhao L, Pei J, Wang Z, Zhang J, and Wang B (2020). CELF6 modulates triple-negative breast cancer progression by regulating the stability of FBP1 mRNA. Breast Cancer Res. Treat 183, 71–82. [DOI] [PubMed] [Google Scholar]
- Zhang C, and Darnell RB (2011). Mapping in vivo protein-RNA interactions at single-nucleotide resolution from HITS-CLIP data. Nat. Biotechnol 29, 607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All software packages can be found in Key Resources Table. Raw and processed sequencing data from CLIP-Seq can be accessed through the National Center for Biotechnology Information’s Gene Expression Omnibus under accession number GSE118623. UCSC Genome Browser session showing CLIP-Seq data associated with NCBI GEO Accession GSE118623 can be viewed following this link: http://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_otherUserName=mrieger&hgS_otherUserSessionName=celf6_clip_mm10
Code used to call peaks, perform MEME-SUITE motif enrichment clustering, and analyze the PTRE-Seq library data can be found at: https://github.com/clevermizo/CELF6CLIPMS