
Figure 5.
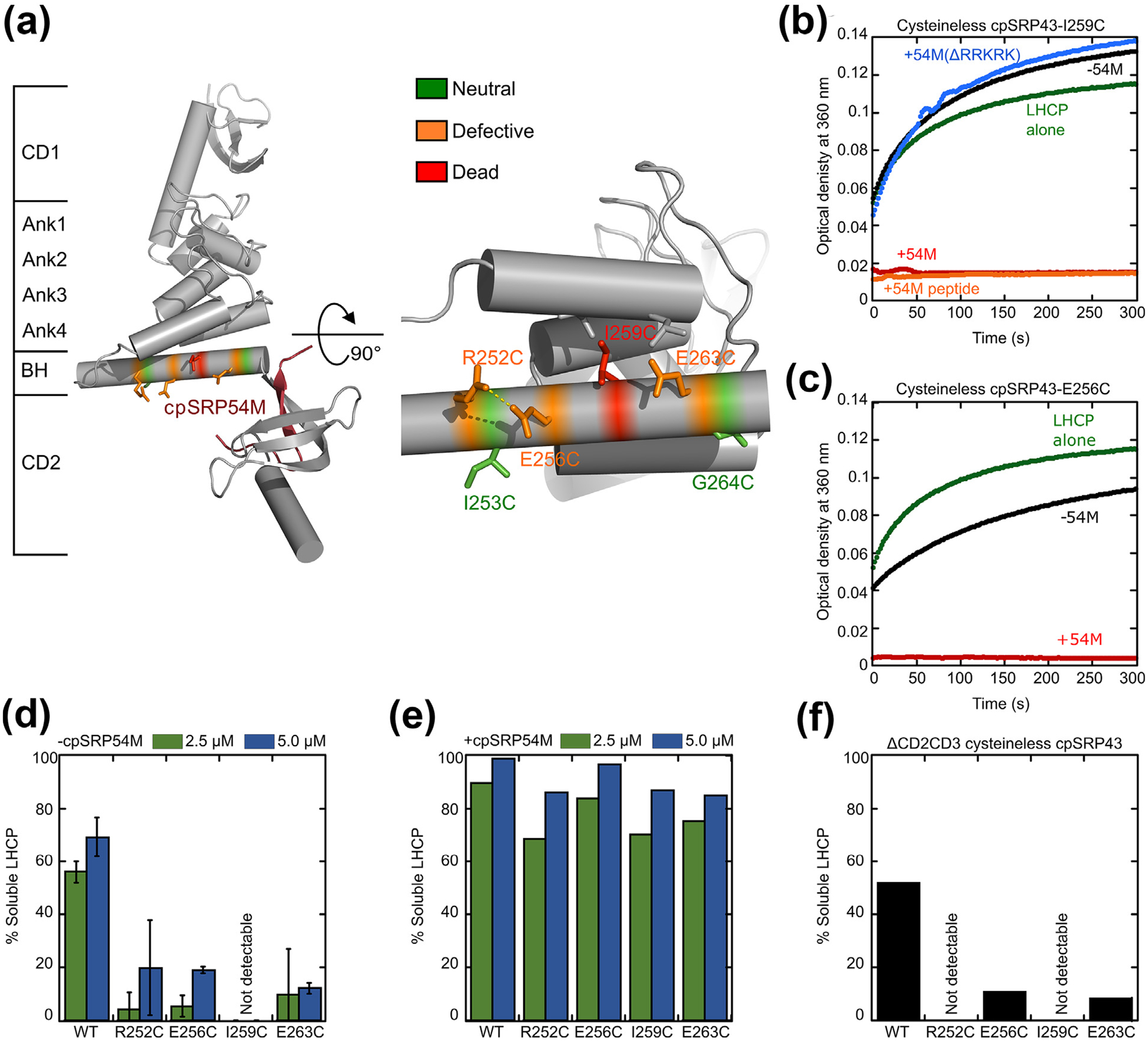
Disruption of intra- and inter-helical contacts in the bridging helix compromises chaperone activity and renders cpSRP43 more dependent on cpSRP54. (a) Structure of the SBD-CD2 fragment of cpSRP43 highlighting the residues in the BH subject to mutational analyses. Colors indicate different mutational effects on the chaperone activity of cpSRP43. (b) Time courses for the aggregation of LHCP, monitored by turbidity at 360 nm, upon dilution into buffer (green) or into solutions containing mutant cpSRP43(I259C) in the absence (black) and presence of the indicated regulatory factors: 54M (red); 54M peptide (orange); mutant 54M lacking the basic C-terminal residues that bind cpSRP43 (54M(ΔRRKRK); blue). (c) Time courses for aggregation of LHCP upon dilution into buffer (green), or into solutions containing mutant cpSRP43(E256C) in the absence (black) and presence (red) of the cpSRP54 M-domain. (d, e) Summary of the chaperone activities of indicated BH mutants in the absence (d) and presence (e) of the cpSRP54 M-domain, quantified from the degree of solubilization of LHCP at the indicated chaperone concentrations based on turbidity measurements. (f) Mutations in the BH disrupt chaperone activity even in the absence of CD2 and CD3. All mutations were constructed in cysteinless cpSRP43 (C175A, C297S), which is denoted as WT in this figure.