

Abstract
Background
Peritoneal metastases portend poor prognosis in the setting of standard chemotherapy. Cytoreductive surgery and hyperthermic intraperitoneal chemotherapy (CRS/HIPEC) improves outcomes, but relapse is common. We report a phase II trial evaluating the safety and efficacy of adjuvant αDC1 vaccination with chemokine modulation (CKM) after CRS/HIPEC.
Methods
Patients undergoing CRS/HIPEC for appendiceal cancer, colorectal cancer, or peritoneal mesothelioma were enrolled. In addition to standard adjuvant chemotherapy, patients received intranodal and intradermal injections of autologous tumor-loaded αDC1 vaccine. After each vaccine booster, patients received CKM over 4 days, consisting of celecoxib, interferon (IFN)-α, and rintatolimod.
Results
Forty-six patients underwent CRS/HIPEC followed by αDC1 treatment, including 24 appendiceal primaries, 20 colorectal, and 2 mesotheliomas. DC maturation was successful, with 97% expressing HLA-DR and CD86. Tumor cell recovery from peritoneal tumors was challenging, resulting in only 17% of patients receiving the target dose of αDC1. The αDC1 and CKM regimen was well tolerated. CKM successfully modulated serum inflammatory cytokine and chemokine levels. Median progression-free survival (PFS) for appendiceal primaries was 50.4, 34.2, and 8.9 months for grade 1, 2, and 3 tumors, respectively, while median PFS for colorectal cancer was 20.5 and 8.9 months for moderately and poorly differentiated tumors, respectively.
Conclusions
Adjuvant autologous tumor antigen-loaded αDC1 vaccine and CKM is well tolerated. The mucinous nature of peritoneal metastases limits the feasibility of obtaining adequate autologous tumor cells. The improvement in median PFS did not meet our predefined thresholds, leading us to conclude that αDC1 vaccination is not appropriate for patients undergoing CRS/HIPEC for peritoneal metastases.
Patients with peritoneal metastases from colon, appendiceal, and peritoneal mesothelioma have a poor prognosis and limited therapeutic options. The best outcomes have been reported using cytoreductive surgery and hyperthermic intraperitoneal chemotherapy (CRS/HIPEC) combined with systemic chemotherapy.1–5 Despite this aggressive treatment, relapse remains common, with a median progression-free survival (PFS) in randomized trials of 1 year in patients with colorectal primaries.3,5 Additional adjuvant therapy that addresses residual microscopic disease in the systemic circulation, nodal system, and peritoneal cavity is needed. Dendritic cell (DC) vaccine therapy may address that need and represents a promising approach to immune therapy due to its ability to activate T cells for immediate cytotoxic response and to induce memory T cells to prevent relapse. This approach improves survival in metastatic prostate cancer patients, leading to the first US FDA-approved DC vaccine, Sipuleucel-T (Provenge).6 We previously reported a patient with aggressive recurrent peritoneal metastases from colon cancer achieving long-term cure after CRS followed by autologous DC vaccine therapy as a participant in a previous DC trial.7 This led us to explore a trial of DC therapy in patients with peritoneal metastases from colon cancer, appendiceal cancer, and peritoneal mesothelioma.
DCs are potent antigen-presenting cells that present tumor-specific peptides for T cell activation. For effective vaccines, the principle of functional polarization is important with the induction of type-1 immunity—high co-stimulatory activity of T helper (Th) 1 and cytotoxic T lymphocytes (CTL). Ex vivo DC polarization using a combination of interferon (IFN)-α and polyinosinic polycytidylic acid (poly-I:C), in addition to tumor necrosis factor (TNF)-α, interleukin (IL)-1β/IL-6/prostaglandin E2, demonstrate high co-stimulatory activity and up to 30 times greater IL-12 production after stimulation by antigen.8–14 In addition, they can be primed for tumor-specific activation by loading with tumor antigen, resulting in the production of highly effective, tumor-specific CD8+ cytotoxic T cells (CTL) and T-cell expansion.15–20
We have previously demonstrated that a chemokine modulating (CKM) regimen consisting of INFα, poly I:C (toll-like receptor [TLR] 3 agonist), and celecoxib (a selective cyclooxygenase [COX]-2 prostaglandin inhibitor) serves to reverse the immune suppressive chemokine profile, increase the attraction of CTL, and improve effective homing of these cells into tumors.9,13,20–27 Thus, in patients undergoing CRS/HIPEC for peritoneal metastases, we hypothesized that adjuvant autologous tumor-pulsed αDC1 therapy plus CKM may address residual microscopic disease and prevent relapse. This phase II study evaluated the safety and efficacy of adjuvant autologous αDC1 vaccine with systemic CKM consisting of celecoxib, INFα, and rintatolimod (a TLR3 ligand derivative of poly-I:C) after CRS/HIPEC for patients with peritoneal metastases from appendiceal cancer, colorectal cancer, and peritoneal mesothelioma.
Methods
Patients
Patients were enrolled in the trial after informed consent approvals from the University of Pittsburgh Institutional Review Board for protocol UPCI 12-110 (NCT02151448). Eligibility included histologically proven peritoneal metastases from peritoneal mesothelioma, appendiceal cancer or colorectal cancers, undergoing CRS/HIPEC. After CRS/HIPEC, patients were screened for completeness of cytoreduction (CC) scores of 0 (no residual macroscopic disease), 1 (residual tumor nodules < 2.5 mm in diameter), or 2 (residual tumor nodules < 2.5 cm in diameter), Eastern Cooperative Oncology Group (ECOG) performance status of 0–2, ability to swallow pills, adequate laboratory parameters without evidence of renal or hepatic dysfunction, and ability to undergo plasmapheresis within 8 weeks of surgery. Patients with immunosuppression, autoimmune disorders, cardiac events within the preceding 3 months, and previous allergic reaction or hypersensitivity to celecoxib or non-steroidal anti-inflammatory drugs (NSAIDs) were excluded.
Sixty-three patients were enrolled, underwent CRS/HIPEC, and received the initial vaccine priming course. Of these patients, two had no clinical response data, seven were lost to follow-up, and eight received a suboptimal dose of INFα on the phase I lead-in portion of the trial. The remaining 46 patients are the subjects of this report.
Clinical Trial Design
This single-institution phase II study was designed to assess the toxicity of the αDC1 vaccine and CKM, evaluate induction of an inflammatory response, and determine clinical benefit (Fig. 1). Vaccines were delivered intradermally and intranodally using ultrasound guidance, rotating between the left and right inguinal lymph node clusters.7 After the patient recovered from CRS/HIPEC, a priming dose of αDC1 was administered on the first week of treatment. The priming dose consisted of both intradermal and intranodal injections of αDC1 vaccine alone. Within 4 weeks, patients received a booster cycle consisting of both intradermal and intranodal injections of αDC1 vaccine followed by 4 subsequent days of CKM. The CKM consisted of intravenous IFNα (20 mu/m2) on days 2–5, intravenous rintatolimod (200 mg) on days 3 and 5, and twice-daily oral celecoxib (200 mg) on days 2–5. In those without adverse events (AE), two additional booster cycles were offered every 4 weeks using the same αDC1/CKM regimen. Patients could receive standard-of-care adjuvant chemotherapy concurrently. Adjuvant chemotherapy was administered between vaccine therapies but no sooner than 5 days after completing a cycle of vaccine therapy. Variabilities in the timing of the vaccine and adjuvant therapy were allowed.
Fig. 1.

Intended course of adjuvant DC vaccination. Variabilities in the timing of vaccine and adjuvant therapy were allowed. CKM: IFNα on days 2–5, rintatolimod on days 3 and 5, and celecoxib on days 2–5. Vx DC vaccine injection, CRS/HIPEC cytoreductive surgery plus hyperthermic intraperitoneal chemotherapy, ChemoTx chemotherapy, DC dentritic cells, CKM chemokine modulation, IFN interferon
Toxicity Assessment and Stopping Rules
Patients were monitored for treatment-related adverse events (AE) using the National Cancer Institute Common Toxicity Criteria version 3.0. In the phase I cohort, dose-limiting toxicity (DLT) of CKM was evaluated for the likelihood of the following being due to the CKM treatment: grade 2 or higher hypersensitivity; grade 3 or higher non-hematologic/metabolic toxicity; and grade 3 or higher hematologic (except for lymphopenia) or metabolic toxicity that did not subside after 4 weeks’ cessation of the CKM.
Vaccine Preparation
Our technique for generation of αDC1 has been previously described.6 For this trial, patients underwent leukapheresis preoperatively or within 8 weeks after CRS/HIPEC for extraction of DC. Monocytes were isolated by the University of Pittsburgh Cancer Institute Immunologic Monitoring and Cellular Products Laboratory (IMCPL) using the Elutra Cell Separation System (TerumoBCT, Lakewood, CO, USA). These were cultured in antibiotic-free culture medium (CellGenix Technologie Transfer GmbH, Antioch, IL, USA) supplemented with 1000 U/mL IL-4 and 1000 U/mL granulocyte–macrophage colony stimulating factor (GM-CSF) for 6 days to generate immature DC. Immature DC were cryopreserved until vaccination.
Prior to vaccination, immature DC were recovered from cryopreservation and polarized with IL-1β (25 ng/mL; CellGenix GmbH, Freiburg, Germany), TNFα (50 ng/mL; CellGenix GmbH), IFNα (3000 U/mL; Merck & Co, Whitehouse Station, NJ, USA), poly-I:C (20 mg/mL; EMD Millipore, Philadelphia, PA, USA), and IFNγ (1000 U/mL, InterMune Inc, Brisbane, CA, USA) at 37 °C in 5% CO2 for 48 h.12
Tumor specimens were collected at the time of surgery. Fresh tissue was minced and digested with collagenase and DNase. Lysates were sterilized with ultraviolet B (UVB) and treated with γ-irradiation (20,000 Rads) to induce apoptosis. Apoptotic tumor cells were added to the immature DC cultures at day 6, during the induction of DC maturation into polarized DC1 at a target ratio of 30:1 monocytes to tumor cells. This corresponds to an estimated ratio of 10:1 αDC1 to tumor cells, since typically 10 monocytes yield 2–3 DCs.
Mature antigen-loaded αDC1 were harvested after 48 h and tested for sterility and viability (endotoxin < 5.0 EU/kg of body weight, DC viability > 70%). αDC1 purity was evaluated by flow cytometry for the presence of CD83 > 70% and CCR7 > 50%, and < 10% contaminating CD3+ , CD19+ , or CD14+ cells. The in vitro function of αDC1 was evaluated by the ability to release IL-12p70 following stimulation with CD40L-transfected J558 cells, as previously described.28,29
Cytokine and Chemokine Assays
Luminex-based assays were performed by UPCI Cancer Biomarkers Facility to evaluate in vivo activity of the αDC1 and CKM. Cytokine and chemokine expression were measured in frozen plasma samples drawn prior to priming with αDC1 (baseline), prior to the first booster of αDC1 and CKM (pre-αDC1+CKM), and after the booster (post-αDC1+CKM). Expression of the following cytokines were measured: CXCL10, CXCL11, CXCL12, IL-6, IL-8, IL-10, and TNFα (R&D High Sensitivity 10plex, R&D Luminex 4plex, R&D INFα, Millipore Human Immuno-Oncology Checkpoint Protein 1plex [TLR2]).
Clinical Endpoints
The primary clinical endpoint of the study was time to progression. Progression was defined as radiographic or clinical evidence of new lesions, recurrence at previous sites, or progression compared with baseline imaging. We also followed patients for overall survival.
Statistical Analysis
The aim of the study was to demonstrate a 25% improvement in PFS over the expected median PFS based on historical control patients within our institutional database. The expected and target median PFSs were 20 and 25 months for appendiceal cancer, 12 and 15 months for colorectal cancer, and 20 and 25 months for peritoneal mesothelioma, respectively. The target accrual was 168 patients over 3 years to detect a 25% improvement with 90% power.
Results
Patient Population
Patient demographics and the results of CRS/HIPEC are outlined in Table 1. The 46 patients included 15 females and 31 males with a mean age of 55 years. Twenty-four patients had appendiceal peritoneal metastases, 20 patients had colorectal peritoneal metastases, and 2 patients had peritoneal mesothelioma. Overall, 34 (74%) patients had recurrent disease at presentation, 32 (70%) had undergone previous CRS, and 20 (43%) had previously undergone HIPEC. On average, patients had been diagnosed with peritoneal metastases for 27 months prior to being recruited into the trial. The median Peritoneal Cancer Index (PCI) was 15, indicating a high burden of intraperitoneal disease. CC-0 was achieved in 35 patients, 9 patients had small-volume residual disease (CC-1), and 2 patients had moderate residual disease (CC-2).
Table 1.
Patient characteristics
| Trial patients [n = 46] | |
|---|---|
| Mean age, years (SD) | 55 (11) |
| Male sex | 31 (67) |
| Diagnosis | |
| Appendiceal | 24 (52) |
| LAMN | 7 (29) |
| MACA—moderately differentiated | 8 (33) |
| MACA—poorly differentiated | 8 (33) |
| Colon | 20 (43) |
| Moderately differentiated | 11 (55) |
| Poorly differentiated | 9 (45) |
| Mesothelioma | 2 (4) |
| Recurrent disease | 34 (74) |
| Previous CRS | 32 (70) |
| Previous HIPEC | 20 (43) |
| Mean time from diagnosis to trial, months (SD) | 27 (29) |
| Median PCI (IQR) | 15 (10–22) |
| CC score | |
| 0 | 35 (76) |
| 1 | 9 (20) |
| 2 | 2 (4) |
| Median operative time, mins (IQR) | 506 (392–668) |
| Median hospital stay, days (IQR) | 12 (8–16) |
| Complications [median (IQR)] | 22.6 (0–37) |
| Readmission | 21 (46) |
Data are expressed as n (%) unless otherwise specified
LAMN low-grade appendiceal mucinous neoplasms, MACA mucinous adenocarcinoma, CRS cytoreductive surgery, HIPEC hyperthermic intraperitoneal chemotherapy, SD standard deviation, PCI Peritoneal Cancer Index, IQR interquartile range
Vaccine Production
Vaccine production and administration data are outlined in Table 2. All patients had tumor harvested for antigen loading of DCs. The goal was for recovery of at least 8 g of tumor and for recovery of 1.2 × 107 viable tumor cells for optimal loading of DCs. Based on viable cell count, excluding white blood cells, we planned to load the DCs at a target ratio of 30:1 monocytes to tumor cells (corresponding to a target ratio of 101:DC to tumor cells). Overall, we were able to harvest a median of 37.5 g of tumor, ranging from 2 to 71 g. While we did not use CD326 (EpCAM) staining to determine the numbers of tumor cells (vs. stromal cells) for DC antigen loading, we did measure it in retrospect for patients with colon or appendiceal tumors to differentiate the tumor cells from stromal cells and estimate the number of tumor cells used for DC loading. A median of 1.8 × 106 viable, EpCAM + tumor cells were recovered for DC loading (ranging from 3.9 × 106 to 3.5 × 107). Only eight patients had the desired number (1.2 × 107) of EpCAM + tumor cells for DC loading. While we intended to load DCs with tumor-derived cells at a ratio of 30:1, when analyzing EpCAM + staining in retrospect, we actually loaded with a median ratio of 15 times lower (when calculating ratios of DCs to EpCAM + tumor cells, range 0.12–28). No patients reached the desired 30:1 ratio of DC precursor monocytes to EpCAM + tumor cells (or the target 10:1 ratio of DCs to tumor cells).
Table 2.
Tumor cell isolation, DC recovery and vaccine delivery
| Target | Actual | |
|---|---|---|
| Tumor cell isolation | ||
| Tumor recovery, median (g) | 8 | 37.5 (2–71) |
| Viable tumor cells needed for DC loading, median (× 106 cells) | 12 | 1.8 (3.9–35) |
| DC production (%) | ||
| DC maturation (+HLA-DR, +CD86) | 100 | 97 |
| CD40L-induced IL12p70 > 1.0 ng/mL/24 h | 100 | 54 |
| Vaccine administration [n (%)] | ||
| Patients receiving target dose αDC1 [3 × 106 cells] | 46 (100) | 9 (20) |
| Patients receiving all three boosters | 46 (100) | 32 (70) |
| Patients receiving all three target dose boosters [3 × 106] | 46 (100) | 8 (17) |
DC dentritic cell
DCs were able to be isolated and matured, with 97% of αDC1 expressing HLA-DR and CD86. CD40L-induced IL-12p70 production was, on average, 2.8 ± 3.9 ng/mL/2 × 104 αDC1/24 h, indicating adequate maturation.9 Fifty-four percent of patients had > 1.0 ng/mL/24 h IL-12p70. Production > 3.0 ng/mL has been previously correlated with clinical response, and 10 patients had IL-12p70 levels > 3.0, ranging up to 17.20
Because of low tumor cell recovery from operative tissue specimens, we did not always have enough primed αDC1 for optimal vaccination dosing and/or for completing the priming and three booster doses. This resulted in only 9 of the 46 patients receiving the target dose of 3 × 106 αDC1, and only 32 of the 46 patients received all three boosters—6 received two boosters, 7 received only one booster, and 1 patient received only the initial priming dose of DCs. Only 8 of the 46 patients received all three boosters at the target dose.
Summary of Adverse Events
Grade 1 flu-like symptoms were the most common AEs attributed to the DC vaccination and CKM, including chills (n = 36), fatigue (n = 19), and nausea (n = 19). They were usually self-limited to 24 h after each vaccine dose. Grades 1 and 2 injection-site reactions were relatively uncommon, affecting only two patients. Six patients had grades 3–5 AEs, possibly attributed to treatment. These included a decrease in lymphocyte count, diarrhea, transient blurry vision, stroke, heart failure, and gastrointestinal bleed while receiving blood thinners for personal history of stroke (Table 3). One patient experienced transient blurry vision that may be related to rintatolimod and was self-limited.
Table 3.
Adverse events related to vaccine and CKM interventionToxicity likely or possibly related to vaccine
| Frequency [n = 46] | |
|---|---|
| Grade 1 | |
| Flu-like symptoms: chills, fever, nausea | 36, 19, 19 |
| Grade 2 | |
| Injection-site reactions | 2 |
| Grade 3 | |
| Anemia | 1 |
| Lymphocyte count decrease | 1 |
| Diarrhea | 1 |
| Blurred vision—self-limited | 1 |
| Grade 4 | |
| Coronary heart failure and respiratory distress | 1 |
| Stroke | 1 |
CKM chemokine modulation
Serum Cytokines and Chemokines
We measured CXCL10, CXCL11, CXCL12, IL-6, IL-8, IL-10, and TNFα in serum at baseline and after the first booster with vaccine and CKM. All these inflammatory cytokines demonstrated an increase in serum concentrations after CKM treatment (Table 4).
Table 4.
In vitro inflammatory cytokine response to CKM
| Baseline concentration (pg/mL) | Post-booster concentration (pg/mL) | Mean difference | Standard error | p Value | |
|---|---|---|---|---|---|
| IL-6 | 12.8 | 35.4 | 22.5 | 6.3 | < 0.01 |
| IL-8 | 10.1 | 17.2 | 7.2 | 1.8 | < 0.01 |
| IL-10 | 1 | 7.7 | 6.7 | 1.9 | < 0.01 |
| TNF-α | 10.1 | 31.3 | 21.2 | 3.7 | < 0.01 |
| CXCL10 | 124.8 | 16841.6 | 16716.9 | 2974.2 | < 0.01 |
| CXCL11 | 368 | 12,223 | 11,862 | 988.6 | < 0.01 |
| CXCL12 | 1284.6 | 2204 | 919.6 | 57.7 | < 0.01 |
CKM chemokine modulation, IL interleukin, TNF tumor necrosis factor
Clinical Outcomes
The study was originally powered to determine a 25% improvement in PFS over historical controls from our own database, stratified by histology: colon cancer, appendiceal cancer, and mesothelioma with an accrual goal of 168 patients. The expected and target median PFSs were 20 and 25 months for appendiceal cancer, 12 and 15 months for colorectal cancer, and 20 and 25 months for peritoneal mesothelioma, respectively. The trial was terminated early after an interim analysis demonstrated futility due to a lack of improvement in PFS (colon cancer), variable tumor grade affecting results (appendiceal), or slow accrual (mesothelioma). In total, 46 patients were enrolled and had evaluable data prior to early termination.
PFS for colon cancer was 20.5 months for moderately differentiated tumors, and 8.9 months for poorly differentiated tumors (Fig. 2a). Two of 11 patients with moderately differentiated tumors have not recurred at 58 and 46 months after CRS/HIPEC. Both had PCI scores of < 12 and had all gross disease removed at the time of surgery. Overall survival for the moderately differentiated tumors was not reached, and for poorly differentiated tumors was 18.3 months (Fig. 2b). This is consistent with our prior reports of CRS/HIPEC for colon peritoneal metastases.
Fig. 2.
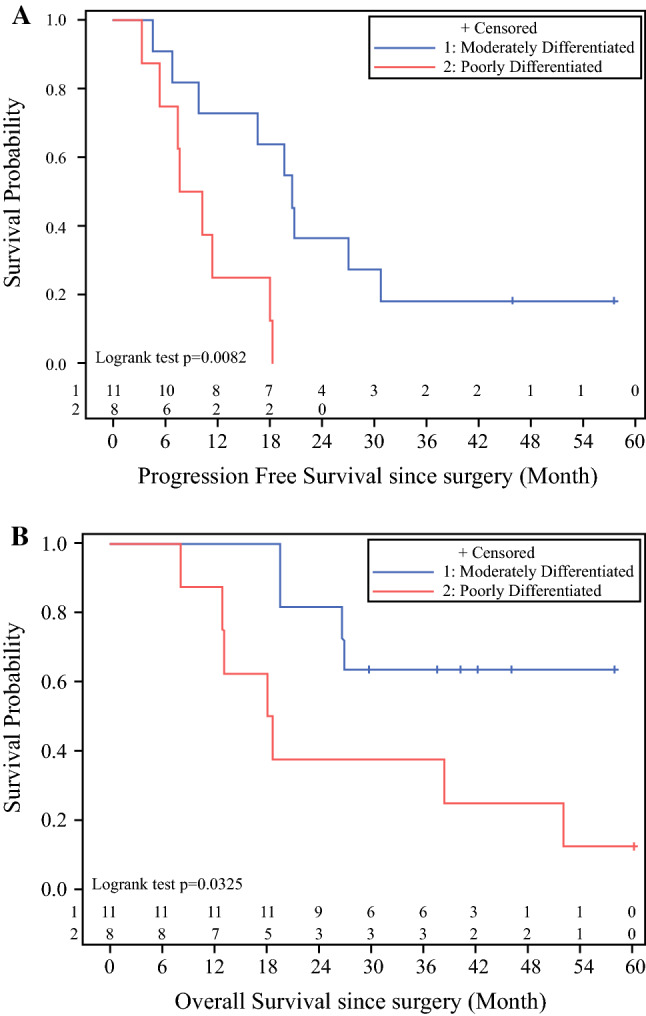
Kaplan–Meier colorectal cancer survival by differentiation: a progression-free survival, b overall survival
For appendiceal cancers, PFS was 50.4 months for low-grade (grade G1), 34.2 months for intermediate-grade (grade G2), and 8.9 months for high-grade/poorly differentiated/signet ring cell cancers (grade G3) (Fig. 3a). Three of eight patients with grade G2 tumors have not recurred at 55, 44, and 35 months of follow-up, with PCI scores of 8, 3, and 28, respectively, and all gross disease removed. Four of seven patients with grade G1 tumors have not recurred at 55, 51, 49, and 45 months, with PCI scores of 10, 14, 16, and 10, respectively, and all gross disease removed. Three of eight patients with grade G3 tumors have died of their disease at 6, 7, and 8 months after surgery; the remaining five patients are still alive 30–46 months after surgery. One of eight patients with grade G2 tumors had died at 44 months, with the remaining seven patients were alive 35–56 months after surgery. All patients with grade G1 tumors were still alive at a median follow-up of 50 months (Fig. 3b). These results are consistent with our previously reported results of survival for appendiceal cancer after CRS/HIPEC. The two patients with mesothelioma recurred at 25 and 8 months and died of their disease at 40 and 10 months, respectively. Both had biphasic mesothelioma, with PCI scores > 20 and complete cytoreduction.
Fig. 3.
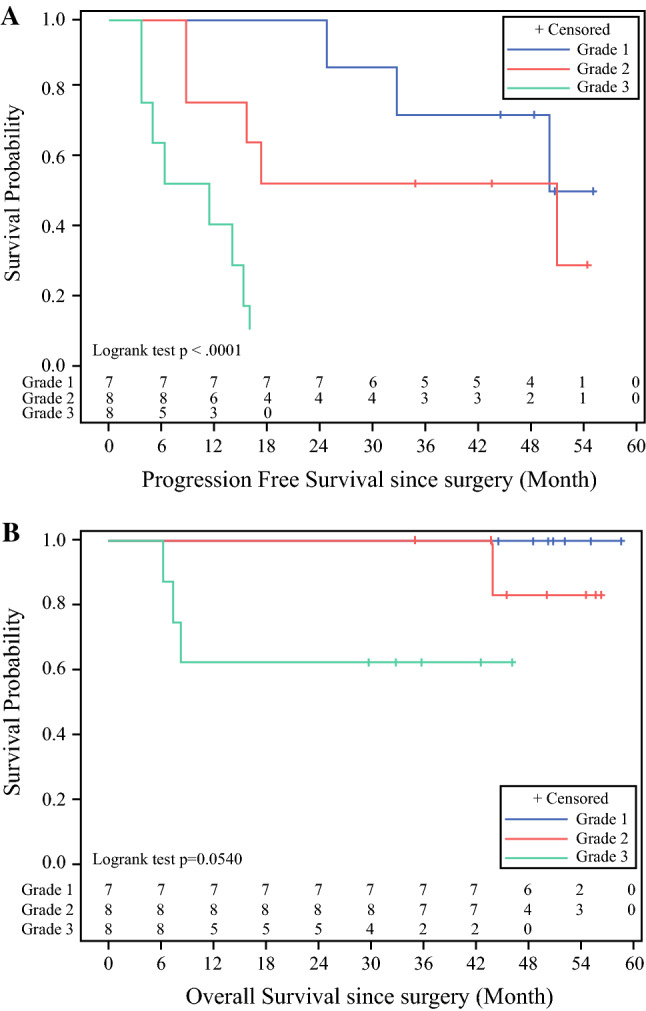
Kaplan–Meier appendiceal cancer survival by grade: a progression-free survival, b overall survival
As discussed above, 10 patients had IL-12p70 production > 3 ng/mL/24 h. No trend towards improved survival could be identified for those patients (data not shown). Two patients received all intended target-dose priming and booster injections of αDC1, with a high percentage of EpCAM+ cell loading. The first patient had high-grade appendiceal cancer, with a PCI score of 21 and minimal residual gross disease after CRS. He received all four doses of vaccine and three doses of CKM, and had 25.7% EpCAM+ cells loaded at a ratio of 30:1, with IL-12p70 production of 2.3 ng/mL/24 h. Unfortunately, the patient developed progressive disease 4.9 months after surgery and died of his disease 7.4 months after surgery. The second patient had poorly differentiated colon cancer, with a PCI score of 20 and no residual disease after CRS. He had 34.1% EpCAM+ cells and IL-12p70 production of 4 ng/mL/24 h. He developed a recurrence at 3.2 months and died 18 months after surgery.
Discussion
Patients with peritoneal metastases have limited therapeutic options, rendering a poor long-term prognosis. CRS/HIPEC combines complex surgical resection with concurrent heated intraperitoneal chemotherapy, and can render a patient free of disease and offer long-term remission in certain scenarios.1–5,30,31 However, in most cases, patients develop recurrence and thus there remains a need to discover effective adjuvant therapies for prolonging PFS. DC immune therapies have been investigated in several tumor types, however to the authors’ best knowledge, this is the first trial of DC therapy for peritoneal metastases.20,32,33
The αDC1 used in this trial is the result of a previously published culture method that generates mature DC with both high co-stimulatory activity and secretory activity.6–8 These αDC1 have been shown to induce production of CD8+ T cells and convert non-cytolytic CD8+ T cells into efficient effector T cells.34 In this trial, the αDC1s produced successfully expressed IL-12 at threshold levels in response to stimulation in 54% of patients, demonstrating the ability to activate CTL. IL-12 production from DCs after CD40L stimulation has been previously shown to correlate with clinical activity.9 We previously reported prolonged survival in patients with glioblastoma multiforme using antigenic peptide-pulsed DCs, with 1 of 22 patients demonstrating a sustained complete response.20 However, using apoptotic tumor cells in the current study, we experienced repetitive challenges isolating an adequate numbers of cells from the peritoneal tumor specimens to load the αDC1. The planned target 30:1 ratio of the plated monocytes to tumor cells is borne out of preclinical studies in an in vitro sensitization system, showing that effective induction of cancer cell-specific CTL is achieved when loading the DCs with cancer cells at ratios of 30:1 (one cancer cell gives rise to multiple apoptotic bodies and can feed antigen to multiple DCs). At lower ratios, the induction of tumor-specific CTL drops rapidly when fewer cancer cells are used,15–19 [un published data].
The cytokine microenvironment is pivotal in regulating the expression of various T-cell subtypes.13–21,35 This forms the rationale for administering CKM following the booster doses, whereby the DC vaccine is expected to induce tumor-specific CTL, and CKM is expected to direct CTL to tumor tissues. Based on preclinical data, our CKM consisted of IFNα, celecoxib, and rintatolimod. While we could not document intratumoral changes in the production of chemokines and cytokines due to the adjuvant nature of the treatment, we did examine changes in circulating levels. We measured an increase in proinflammatory cytokines and chemokines after CKM, including CXCL10, CXCL11, IL-6, IL-8, SDF1α, and TNFα. In addition, the anti-inflammatory cytokine IL-10 was increased, perhaps as a negative feedback response to TLR signaling.36–38 Unfortunately, with this trial and these data, we cannot confirm the prior ex vivo observations regarding the immune profile of the tumor microenvironment. A separate trial examining CKM therapy for metastatic colorectal cancer with pre- and post-treatment biopsies has finished accrual and is currently being analyzed.
One of the conclusions of this trial is the challenges associated with the optimal production of autologous tumor antigen-loaded αDC1 vaccine as intended in the study protocol. No patients received the target DC to EpCAM+ cancer cell ratio of 30:1 due to difficulties extracting tumor cells from resected tumor tissue. The low yields can be partially attributed to the specimens achievable at CRS. The diffuse, miliary pattern of peritoneal metastases can make extraction of tumor cells from discrete tissue specimens difficult, and, similarly, tumors with large deposits of mucin can demonstrate low cellularity within a defined specimen. These pose challenges to isolating tumor cells. Additionally, patients undergoing CRS/HIPEC are usually pretreated with chemotherapy, and, as a result, tumor specimens may demonstrate increased fibrosis and necrosis, which also adversely affects tumor yield. The less-than-target ratio likely adversely affects the maturing of αDC1 and the presentation of rejection antigens, limiting its clinical impact. In concert with less-than-target antigen loading, we faced challenges generating adequate vaccine to reach the target dose of 3 × 106 αDC1 per injection for each of the planned four injections. The target dose and four treatments were only achieved in 17% of patients, and only two patients received the complete treatment with a high percentage of EpCAM+ tumor cells (> 25%) and IL-12 levels > 2 ng/mL/24 h. These challenges illustrate the need for improved methods of generating adequate tumor antigen and vaccine in patients undergoing CRS.
This trial also serves to offer guidance into future investigations of DC therapy in gastrointestinal malignancies. The inadequacy of tumor antigen was the predominant technical shortcoming within this trial, with the αDC1 and CKM showing good biologic function. The low yields of tumor antigen is likely due to the mucinous predominance of the extracted tumor, leading us to conclude that use of autologous tumor for loading of DC is likely not a feasible strategy for tumors with mucinous predominance. Other strategies worth exploring include the use of synthetic tumor peptides, use of tumor-associated protein, and tumor-derived mRNA. These strategies may ensure better maturation of αDC1 and clinical effectiveness.
Conclusion
This study demonstrates the safety of CKM in combination with autologous tumor pulsed αDC1 vaccine, and shows that CKM elicits a heightened inflammatory state in the serum. It also illustrates the logistical challenges with isolating sufficient cancer cells to generate sufficient numbers of autologous tumor-pulsed αDC1 immune therapy in patients with peritoneal metastases.
Acknowledgments
This work was supported in part by David C. Koch Regional Cancer Therapy Center. This project has used the University of Pittsburgh shared facilities (Clinical Research Services and the Immunological Monitoring and Cellular Products Laboratory) that are supported in part by the National Institutes of Health award P30CA047904.
Disclosures
Pawel Kalinski has received a Grant from AIM (previously Hemispherx) to support the trial of Ampligen in patients with COVID-19. This latest development occurred after preparation of this manuscript. Rajesh Ramanathan, Haroon Choudry, Heather Jones, Mark Girgis, William Gooding, and David L. Bartlett have no disclosures to declare.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Magge D, Zenati MS, Austin F, et al. Malignant peritoneal mesothelioma: prognostic factors and oncologic outcome analysis. Ann Surg Oncol. 2014;21:1159–1165. doi: 10.1245/s10434-013-3358-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Polanco PM, Ding Y, Knox JM, et al. Outcomes of cytoreductive surgery and hyperthermic intraperitoneal chemoperfusion in patients with high-grade, high-volume disseminated mucinous appendiceal neoplasms. Ann Surg Oncol. 2016;23:382–390. doi: 10.1245/s10434-015-4838-z. [DOI] [PubMed] [Google Scholar]
- 3.Verwaal VJ, Bruin S, Boot H, et al. 8-year follow-up of randomized trial: cytoreduction and hyperthermic intraperitoneal chemotherapy versus systemic chemotherapy in patients with peritoneal carcinomatosis of colorectal cancer. Ann Surg Oncol. 2008;15:2426–2432. doi: 10.1245/s10434-008-9966-2. [DOI] [PubMed] [Google Scholar]
- 4.Kuijpers AMJ, Mirck B, Aalbers AGJ, et al. Cytoreduction and HIPEC in the Netherlands: nationwide long-term outcome following the Dutch protocol. Ann Surg Oncol. 2013;20:4224–4230. doi: 10.1245/s10434-013-3145-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mirnezami R, Mehta AM, Chandrakumaran K, et al. Cytoreductive surgery in combination with hyperthermic intraperitoneal chemotherapy improves survival in patients with colorectal peritoneal metastases compared with systemic chemotherapy alone. Br J Cancer. 2014;111:1500–1508. doi: 10.1038/bjc.2014.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 7.Radomski M, Zeh HJ, Edington HD, et al. Prolonged intralymphatic delivery of dendritic cells through implantable lymphatic ports in patients with advanced cancer. J Immunother Cancer. 2016;19:24. doi: 10.1186/s40425-016-0128-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mailliard RB, Wankowicz-Kalinska A, Cai Q, et al. Alpha-type-1 polarized dendritic cells: a novel immunization tool with optimized. Cancer Res. 2004;64:5934–5937. doi: 10.1158/0008-5472.CAN-04-1261. [DOI] [PubMed] [Google Scholar]
- 9.Obermajer N, Urban J, Wieckowski E, et al. Promoting the accumulation of tumor-specific T cells in tumor tissues by dendritic cell vaccines and chemokine-modulating agents. Nat Protoc. 2018;13:335–357. doi: 10.1038/nprot.2017.130. [DOI] [PubMed] [Google Scholar]
- 10.Vieira PL, de Jong EC, Wierenga EA, et al. Development of Th1-inducing capacity in myeloid dendritic cells requires environmental instruction. J Immunol. 2000;164:4507–4512. doi: 10.4049/jimmunol.164.9.4507. [DOI] [PubMed] [Google Scholar]
- 11.Wesa A, Kalinski P, Kirkwood JM, et al. Polarized type-1 dendritic cells (DC1) producing high levels of IL-12 family members rescue patient TH1-type antimelanoma CD4+ T cell responses in vitro. J Immunother. 2007;30:75–82. doi: 10.1097/01.cji.0000211316.15278.6e. [DOI] [PubMed] [Google Scholar]
- 12.Giermasz AS, Urban JA, Nakamura Y, et al. Type-1 polarized dendritic cells primed for high IL-12 production show enhanced activity as cancer vaccines. Cancer Immunol Immunother. 2009;58:1329–1336. doi: 10.1007/s00262-008-0648-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalinski P, Okada H. Polarized dendritic cells as cancer vaccines: directing effector-type T cells to tumors. Semin Immunol. 2010;22:173–182. doi: 10.1016/j.smim.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wieckowski E, Chatta GS, Mailliard RM, et al. Type-1 polarized dendritic cells loaded with apoptotic prostate cancer cells are potent inducers of CD8(+) T cells against prostate cancer cells and defined prostate cancer-specific epitopes. Prostate. 2011;71:125–133. doi: 10.1002/pros.21228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee J-J, Foon KA, Mailliard RB, et al. Type 1-polarized dendritic cells loaded with autologous tumor are a potent immunogen against chronic lymphocytic leukemia. J Leukoc Biol. 2008;84:319–325. doi: 10.1189/jlb.1107737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Connell PJ, Son Y-I, Giermasz A, et al. Type-1 polarized nature of mouse liver CD8alpha- and CD8alpha + dendritic cells: tissue-dependent differences offset CD8alpha-related dendritic cell heterogeneity. Eur J Immunol. 2003;33:2007–2013. doi: 10.1002/eji.200323379. [DOI] [PubMed] [Google Scholar]
- 17.Muthuswamy R, Urban J, Lee J-J, et al. Ability of mature dendritic cells to interact with regulatory T cells is imprinted during maturation. Cancer Res. 2008;68:5972–5978. doi: 10.1158/0008-5472.CAN-07-6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kato M, Nakamura Y, Suda T, et al. Enhanced anti-tumor immunity by superantigen-pulsed dendritic cells. Cancer Immunol Immunother. 2011;60:1029–1038. doi: 10.1007/s00262-011-1015-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang D-H, Kim M-H, Hong CY, et al. Alpha-type 1-polarized dendritic cells loaded with apoptotic allogeneic myeloma cell line induce strong CTL responses against autologous myeloma cells. Ann Hematol. 2010;89:795–801. doi: 10.1007/s00277-010-0931-3. [DOI] [PubMed] [Google Scholar]
- 20.Okada H, Kalinski P, Ueda R, et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J Clin Oncol. 2011;29:330–336. doi: 10.1200/JCO.2010.30.7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muthuswamy R, Corman JM, Dahl K, et al. Functional reprogramming of human prostate cancer to promote local attraction of effector CD8(+) T cells. Prostate. 2016;76:1095–1105. doi: 10.1002/pros.23194. [DOI] [PubMed] [Google Scholar]
- 22.Francis L, Guo ZS, Liu Z, et al. Modulation of chemokines in the tumor microenvironment enhances oncolytic virotherapy for colorectal cancer. Oncotarget. 2016;7:22174–22185. doi: 10.18632/oncotarget.7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li J, O’Malley M, Urban J, et al. Chemokine expression from oncolytic vaccinia virus enhances vaccine therapies of cancer. Mol Ther. 2011;19:650–657. doi: 10.1038/mt.2010.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muthuswamy R, Wang L, Pitteroff J, et al. Combination of IFNalpha and poly-I: C reprograms bladder cancer microenvironment for enhanced CTL attraction. J Immunother Cancer. 2015;3:6. doi: 10.1186/s40425-015-0050-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wong JL, Obermajer N, Odunsi K, et al. Synergistic COX2 induction by IFNgamma and TNFalpha self-limits type-1 immunity in the human tumor microenvironment. Cancer Immunol Res. 2016;4:303–311. doi: 10.1158/2326-6066.CIR-15-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu X, Fallert-Junecko BA, Fujita M, et al. Poly-ICLC promotes the infiltration of effector T cells into intracranial gliomas via induction of CXCL10 in IFN-alpha and IFN-gamma dependent manners. Cancer Immunol Immunother. 2010;59:1401–1409. doi: 10.1007/s00262-010-0876-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muthuswamy R, Berk E, Junecko BF. NF-κB hyperactivation in tumor tissues allows tumor-selective reprogramming of the chemokine microenvironment to enhance the recruitment of cytolytic T effector cells. Cancer Res. 2012;72:3735–3743. doi: 10.1158/0008-5472.CAN-11-4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kalinski P, Schuitemaker JH, Hilkens CM, et al. Final maturation of dendritic cells is associated with impaired responsiveness to. J Immunol. 1999;162:3231–3236. [PubMed] [Google Scholar]
- 29.Butterfield LH, Gooding W, Whiteside TL. Development of a potency assay for human dendritic cells: IL-12p70 production. J Immunother. 2008;31:89–100. doi: 10.1097/CJI.0b013e318158fce0. [DOI] [PubMed] [Google Scholar]
- 30.Radomski M, Pai RK, Shuai Y, et al. Curative surgical resection as a component of multimodality therapy for peritoneal metastases from goblet cell carcinoids. Ann Surg Oncol. 2016;23:4338–4343. doi: 10.1245/s10434-016-5412-z. [DOI] [PubMed] [Google Scholar]
- 31.Wagner PL, Austin F, Zenati M, et al. Oncologic risk stratification following cytoreductive surgery and hyperthermic intraperitoneal chemotherapy for appendiceal carcinomatosis. Ann Surg Oncol. 2016;23:1587–1593. doi: 10.1245/s10434-015-5037-7. [DOI] [PubMed] [Google Scholar]
- 32.Kandalaft LE, Chiang CL, Tanyi J, et al. A Phase I vaccine trial using dendritic cells pulsed with autologous oxidized lysate for recurrent ovarian cancer. J Transl Med. 2013;11:149. doi: 10.1186/1479-5876-11-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ai Y-Q, Cai K, Hu J-H, et al. The clinical effects of dendritic cell vaccines combined with cytokine-induced killer cells intraperitoneal injected on patients with malignant ascites. Int J Clin Exp Med. 2014;7:4272–4281. [PMC free article] [PubMed] [Google Scholar]
- 34.Watchmaker PB, Berk E, Muthuswamy R, et al. Independent regulation of chemokine responsiveness and cytolytic function versus CD8+ T cell expansion by dendritic cells. J Immunol. 2010;184:591–597. doi: 10.4049/jimmunol.0902062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zeytin HE, Patel AC, Rogers CJ, et al. Combination of a poxvirus-based vaccine with a cyclooxygenase-2 inhibitor (celecoxib) elicits antitumor immunity and long-term survival in CEA.Tg/MIN mice. Cancer Res. 2004;64:3668–78. 10.1158/0008-5472.CAN-03-3878 [DOI] [PubMed]
- 36.Randow F, Syrbe U, Meisel C, et al. Mechanism of endotoxin desensitization: involvement of interleukin 10 and transforming growth factor beta. J Exp Med. 1995;181:1887–1892. doi: 10.1084/jem.181.5.1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saraiva M, O’Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. 2010;10:170–181. doi: 10.1038/nri2711. [DOI] [PubMed] [Google Scholar]
- 38.Ouyang W, Rutz S, Crellin NK, et al. Regulation and functions of the IL-10 family of cytokines in inflammation and disease. Annu Rev Immunol. 2011;29:71–109. doi: 10.1146/annurev-immunol-031210-101312. [DOI] [PubMed] [Google Scholar]