

Abstract
Breast cancer in woman is the most common cancer and in 2018 there were around 2 million new cases recorded. The maximum rate of breast cancer is reported in Belgium followed by Luxembourg. It is the second most general cancer, Lung cancer being the first. If the cancer tumor is located only in the breast, the survival rate would be 99%. If the tumor has wide to lymph nodes around the survival rate would be 85% and if the tumor had extend to distant parts, the survival rate would come down to 27%. Mammary gland is an important organ in mammals which has potential function to secrete, synthesize and deliver milk to the infants for nourishment, improvement and protection. Generally, cancer is named after the body part in which it originated; thus, breast cancer refers to the erratic development and proliferation of cells that originate in the breast tissue (7). There are some kinds of tumors that may grow within various areas of the breast. Most tumors are the outcome of benign (non-cancerous) alters within the breast. The estrogen receptors (ER) in ordinary and diseased states are significant for the improvement of relevant therapeutic strategies. Two main forms of ER exist, ERα and ERβ, which are encoded by separate genes. Estrogens play a central role in breast cancer improvement with ERα status being the mainly significant predictor of breast cancer prognosis. The potent lead molecule binding mode, residue-interaction patterns and docking energy were examined by molecular docking and binding free energy studies. The lead compounds and 3ERT complex structural stability and dynamic behavior were monitored by molecular dynamics analysis. The drug-likeness properties of lead compounds were predicted ADME analysis.
Keywords: Estrogen protein, Breast cancer, Pharmacophore, Virtual screening
1. Introduction
Cancer is a chronic abnormal cell disorder or a lethal disease demonstrates by immortality and unrestricted cell division. Cancer cells may be invasive, aggressive and metastatic and generally spread into various organs. Breast cancer affects the function of normal mammary epithelial cells and is highly heterogenous in nature. Breast cancer is one of the leading causes of disease among female individuals throughout the world [Jemal et al., 2011]. About 25% of breast cancer patients were reported from developed countries [Park et al., 2005]. Breast cancer is one of the common types of cancer contributing more than 27% in total cancer patients in Indian population [Torre et al., 2015]. In the year 2012 alone more than 144,937 breast cancer cases have been reported with 48.45% mortality. The incidence of breast cancer cases increased dramatically in patients at the age group of 50–64 [Nandakumar, 2001]. In cities, one breast cancer case was reported in every 22 women and in the rural area this concentration slashed to 1 in 60 women. However breast cancer cases have also been reported among male individuals rarely. In United States, more than 1500 new cases are reported every year [Giordano et al., 2002]. In western countries, this cancer is one of the second most leading causes of death worldwide. In Asian countries, more than 60% breast cancer cases are diagnosed as estrogen receptor alpha positive (ERα) cancers. In normal mammary gland development, ERα plays a significant role in breast cancer development, however, ERα specifically mediate the proliferation of estrogen-induced cells in an autocrine mode of action in ERα breast cancer cell lines in vitro [Tan et al., 2009]. Breast cancer diagnostic methods with the maximum positive predicted value (88%) with estrogen are progesterone hormone receptor and mammaglobin in a stepwise manner [Maria et al., 2012].
The hyper production of estrogen is one of the leading causes for the development of breast cancer. It was reported that estrogen receptor is a receptor of nuclear and effectively activated by binding of 17β- estradiol legand and also described as estrogen. Naturally human population contains ER-α and ER-β estrogen receptors, of these two receptors, ER-α is mainly expressed in the mammary gland and uterus. In women, estrogen receptor plays a critical role in apoptosis, inflammation, homeostasis, differentiation, metabolism, maturation and proliferation in breast cancer [Bai and Gust, 2009]. The receptor, ER-α is well known to take part immune surveillance, resistance to apoptosis, metastasis and cell growth [Jiang et al., 2006]. The hyper activity of estrogen hormone may potentially lead to the multiplication of the ER-α in the mammalian cells which lead to maintenance and growth of types of breast cancers, and also holds various molecular targets for the investigation of cancer drug.
In recent years, hormonal treatment, chemotherapy, radiotherapy form a various combination which must be highly organized for every individual therapy. Approximately, 60% of the pre-menopausal women and about 75% of the post-menopausal women have suffered by estrogen-dependent breast cancer, and ER-α activity was effectively inhibited by cancer therapy [Peng et al., 2009]. In the case of breast cancer, endocrine therapy is widely recommended for metastatic/recurrent breast cancer or the beginning stage. Presently, the available treatment for estrogen-based breast cancer contains fulvestrant - a selective estrogen receptor down regulator and selective estrogen receptor modulators that induce degradation and destabilization of ER, letrozole as aromatase inhibitors which generally reduce the production of estrogen in peripheral cells and in the tumour by the activity of aromatase. Although there are many anticancer drugs and potential inhibitors against various targets, effectively increase in resistance coupled with various side effects suggests that there is an urgent need for novel cancer therapy.
The presence of ER-α receptor shows that virtual screening (VS) may be used as an effective tool to identify and screen the potential compounds from various natural sources. Many VS methods containing negative image-based screening, molecular docking, and general pharmacophore hypothesis have been used for the identification of ligands against ER-α receptor [Niinivehmasa et al., 2016]. About 50% of patients diagnosed for cancer for both progesterone and ER-α receptor respond to various treatments, including, tamoxifen, that is mainly designed to disrupt the function of ER-α. ER-coregulatory proteins are very tightly regulated protein under natural conditions with absence of expression was diagnosed in cancer [Suganya et al., 2014]. The estrogen receptor has the ability to bind with DNA. In human, alpha and beta were estrogen receptor was reported and many studies focused on these receptors [McDonnell et al., 2015]. Estrogen is an important steroid hormone, which controls differentiation, improvement and function of the specific organ. The Hellenic mechanism of ERs shows binding of estrogen to its receptor in the nucleus. ERs bind specifically with Estrogen Receptor Elements (EREs) and EREs are mainly represented in the promoters of specific target genes [Nilsson et al., 2001]. EREs bind with ERs effectively stimulates conformational changes in the ligand binding domain of the receptors.
2. Materials and methods
2.1. Protein structure preparation
In this study, the structure of the Estrogen receptor protein (PDB ID – 3ERT) was retrieved from the protein Data Bank, RCSB. The structure of PDB has various missing information on specific connectivity, along with the bond orders and formal changes. The structure of protein is imported from PDB into Maestro in the protein preparation wizard. The protein preparation wizard provides the display hydrogen polar only option. This option represents to shows only the polar hydrogen atoms.
2.2. Pre-processing and analyzing the structures
This pre-processing is significant for further structure preparation actions, such as generating heteroamorous states, H-bond assignment and also the level of minimization. In protein preparation wizard, the pre-processing step consists of several options. They are assigning bond orders, add hydrogen, delete water molecules and fill loops.
2.3. Optimization of the hydrogen bond assignment
This Assignment is an automated optimization, in which the optimization runs on all H-bonds (with or without the inclusion of water molecules) and an interactive optimization in which various clusters of hydrogen-bonded species can be effectively optimized. Reducing the structure has been acted specifically to refine the structure, with a restrained reduction. The RMSD value of the atom was specifically minimized in Angstrom. Also, the partial atomic charges were computed effectively with the use of optimized potentials for Liquid Simulations_2005 (OPLS_2005) force field.
2.4. Ligand structure preparation
Ligand structure preparation (LigPrep) is a strong collection of tools was specially designed to make high quality, all atom 3D structures for large numbers of drug-like molecules, starting with two dimensional (2D) or three Dimensional (3D) structures is SDF (Structures Data File) or Maestro format. The tautomer’s and ionization states were generated. The force field geometry is optimized in the macro model and partial atomic charges were perfectly computed by utilizing the OPLS_2005 force field.
2.5. Setting ionization states
The ionization states are generated by removing or adding the protons from the ligand. Ionization states study can be highly helpful to generate various ranges of ionization states that are populated with a selected pH range. The Generated possible states are significantly populated in the specified particular pH range. Tautomer was generated for the input ligand. LigPrep perform KetoEnoltautomerization and the analogous nitrogen and sulfur tatutomerization and DNA base and histidine tatutomerizations.
2.6. Setting stereoisomer options
Setting stereoisomer has three steps i.e. 1 Retain specific chiralities (It retrieve the information about the chirality from the input structures files and retain these chirality for the entire calculations) 2. Chirality includes parities and bond directions from SD files and 3. Atom numbering chirality properties of Maestro files
2.7. Receptor grid generation
The grid box can be generally generated by utilizing the receptor grid generation. Docking of ligand cannot be performed before the grid generation step. Receptor grid generation required a prepared protein structure with an appropriate bond order and formal charges. The receptor grid generation has four tabs, which are utilized to generate a grid. They are Receptor, Site, Constraints and Rotatable groups.
2.8. Receptor
It explains the part of the system in the workspace is considered as the receptor. The ligand can be identified either as an entry or a molecule in the workspace. The marked green color should be displayed after the ligand (or site) molecule is selectively picked in the workspace. Van der waals radius scaling of 14 non-polar atoms, which reduces the penalities for closely contacts, and it can be utilized to model the receptor and the legand. The partial charge cut off default value is 0.25 and the scaling factor default value is 0.30. The per atom scale factors consists of three options. The first choice does not utilized per atom scale factors. The second choice is read from the input structure file option will acted to read the charge and radius scaling factors from the input file for the receptor and the third one is specified for selected residues define to specify scaling factors for choosing residues. The Van der waals radius scale factor and also the charge scale factor default values is 1.0.
2.9. Site
This is defined as the set of site points on a specific grid that are contact by very small gaps in the solvent-exposed regions and describes where in the scoring grids are located and how to prepare from the structure in the workspace. The enclosing box or outer box is defined as the grid themselves are evaluated within the particular space. Ligand atoms contained within the box.
2.10. Constraints
To generate the receptor grid Glide constraints was used. The constraints have three sub tabs, one is positional, another one is H-bond or Metal and the last one is Hydrophobic. Constraints settings may be useful to hold display of the most of the receptor, leaving only the residues with a short distance of the ligand visible.
2.11. Rotatable groups
Rotatable groups of Glide used to adopt different orientation of hydroxyl groups with different ligands. The rotatable hydroxyl groups should be treated flexibly in the receptor grid generation.
2.12. GLIDE
GLIDE (Grid-based Ligand Docking with Energetic) is one of the Schrödinger suites, which is used for the docking analysis. GLIDE searches for favourable interaction regions between the ligands and protein. It uses a hirerachical series of filters to search for particular locations of the ligand in the active-site of the protein molecule. For each core conformation has orientations and search of possible locations is carried out over the active-site of the protein of interest. GLIDE XP is designed for using only on good ligand poses. XP mode has docked only the top-scoring ligands. GLIDE docking can be done by using Protein structure preparation, Receptor Grid generation, Ligand structure preparation, and Ligand Docking.
2.13. Structure based virtual screening
Virtual screening is an effective tool for identifying active compounds or lead molecules has combined with the pipeline of drug research in various pharmacological companies. These screening studies at the active sites were carried out using, Schrodinger suite, Glide and virtual screening work flow docking program.
GLIDE offers accuracy and speed from high-throughput virtual screening of numbers of compounds to exact binding mode predictions, giving good results in every level. This screening method is used to screen various compounds against a single target. The protocol also includes, preparation of ligand using LigPrep. High Throughput Virtual Screening (HTVS) docking is highly useful for the screening of number of ligands. In HTVS, advanced settings are not available. Standard-precision (SP) docking is widely useful to screen ligands with unknown quality in very large numbers. Docking by Extra-Precision (XP) docking method and score analysis were useful and shows distinct procedure which generally takes more time to run than SP docking.
Ligands were taken from database such as UNPD, NPACT, FDA and SANDB. In the input tab, the ligands source were specified and ensured that all ligands are identified. Filtering tab is use do filter the ligands and to run Qikprop. The virtual screening protocol includes, pre-filtering by Lipinski's Rule of 5, removing ligands and pre-filtering using a custom filter.
2.14. Ligand based pharmacophore mapping of estrogen receptor
Pharmacophore modelling consists of two features, virtual screening of ligand database and pharmacophore features generation. Phase is useful for activity prediction, structure alignment, pharmacophore perception and searching of 3D database. Phase is useful for for lead expansion, lead optimization and SAR development. The core of the UNPD, NPACT, FDA and SANDB were useful allowing logical SAR development, medicinal chemistry and optimization of Hit-to-Lead. These four databases offer various ranges of chemistry services and products designed in the field of drug discovery. Experimentally proved Estrogen Receptor were drawn and saved in .sdf format. Drawn compounds was loaded to develop Pharmacophore Model panel from file and the ligand geometries were cleaned, followed by the generation of ligand conformers. The ligand was identified from the pharmacophore sites using the create sites option, on the second tab of the Develop Pharmacophore Model panel. Once the pharmacophore site creation completed, it takes the user to next step of find common pharmacophore process. The common pharmacophore hypothesis was identified by selecting the find option. Maximum common hypothesis was analyzed and score was analyzed for the ligand, which shows the common pharmacophoric characters and it was further analyzed before being subjected to database ligand against the UNPD, NPACT, FDA and SANDB database through HTVS (High Throughput Virtual Screening) mode.
2.15. Binding free energy calculation
Prime /MM-GBSA was utilized to analyze free binding energy between the receptor and the set of ligands. The free energies were calculated using the optimized potential for liquid simulation (OPLS-AA) and GB/SA method.
Where ΔEMM shows the reduced energy variations between the sum of the energies of the protein and ligand and the complex structure of protein–ligand. ΔGsolv indicates the variation in the GBSA solvation energy of the complex structure of ligand–protein and sum of the salvation energies for the ligand and protein. ΔGSA represents the variation in the energies for the surface area of protein–ligand and the sum of the surface area of the ligand and protein.
2.16. DFT calculation
Density functional theory (DFT) was applied to the most active compound as well as top screened compounds obtained from virtual screening studies. DFT mainly calculates the features namely, LUMO, HOMO and electron density to predict the molecular properties and biological activities of the lead molecule. Hybrid DFT is useful for obtaining detailed geometric features of the molecule.
Jaguar v8.7 module was used to calculate various molecular electrostatic properties namely, Unoccupied Molecular Orbital (LUMO) energy, Highest Occupied Molecular Orbital (HOMO), dipole moment, and MESP. The energy of the electrostatic potential of the compounds of interest was evaluated and negative and regions in the compounds were described by colour difference.
3. Result
3.1. Protein preparation of estrogen protein
Estrogen receptor protein crystal structure was accessed from Protein Data Bank (ID: 3ERT) and it was designed using Schrodinger module. Hydrogen atom was assigned to the protein heavy atoms and optimized. Water molecules and cofactors were removed. Protein was also effectively minimized using OPLS-3 force field with atoms attached in the backbone (Fig. 1).
Fig. 1.
Three dimensional structure of prepared Estrogen protein (PDB ID: 3ERT).
3.2. Virtual screening of estrogen protein
In this study, the active site was formed based on the ligand that was available in the structure of estrogen. From the active site, the residues such as, LEU_387, ARG_394, GLU_353 and THR_347 were determined (Table 1 and Fig. 2). From the results obtained it is shown that NPACT search of analyzed compound showed good binding efficiency on Estrogen hormone with docking score varied from −14.248 kcal/mol to −13.925 kcal/mol along with SANDB compounds has showed docking score between −13.892 kcal/mol and −13.257 kcal/mol. UNPD database leads has showed scores ranges from −8.892 kcal/mol to −7.257 kcal/mol.
Table 1.
Structure-based virtual screening of UNPD, NPACT and SANDB compound database against Estrogen protein.
| S. No | Compound ID | Glide XP score (kcal/mol) | Glide energy (kcal/mol) | MM-GBSA (kcal/mol) | Interaction Residues |
|---|---|---|---|---|---|
| 1 | SANDB_11243993 | −14.253 | −47.537 | −64.232 | THR_347,GLU_353,ARG_394,LEU_387 |
| 2 | SANDB_11336474 | −12.925 | −48.619 | −57.102 | THR_347,GLU_353,ARG_394 |
| 3 | SANDB_9841162 | −11.833 | −37.264 | −48.684 | GLU_353,THR_347 |
| 4 | NPACT_01454 | −13.852 | −45.98 | −64.36 | ARG_394,LEU_387,GLU_353 |
| 5 | NPACT_01463 | −13.116 | −43.029 | −65.012 | THR_347,GLY_420 |
| 6 | UNPD_481 | −8.65 | −35.75 | −72.133 | GLU_353,THR_347,ARG_394, GLY_420 |
| 7 | UNPD_179 | −8.35 | −59.396 | −63.34 | GLU_353,THR_347,ARG_394 |
Fig. 2.

Two dimensional interaction diagrams of the top hit compounds identified in structure based virtual screening.
3.3. Pharmacophore model
Four point pharmacophore model AHRR with one hydrophobic (H2), one acceptor (A1), and two aromatic rings (R4, R6) was developed using set compounds. The four point pharmacophore models are shown in Fig. 3. The top five ligands selected based on docking score are the diversity in structure and it is shown in Fig. 4. The best five ligands were superimposed on pharmacophore model to show the site points of the pharmacophore are matching with functional groups which will highlight the electronic features playing an important role in the interaction of receptor ligand. The displaying Pharmacophore model of most of active compound is shown in Table 2.
Fig. 3.
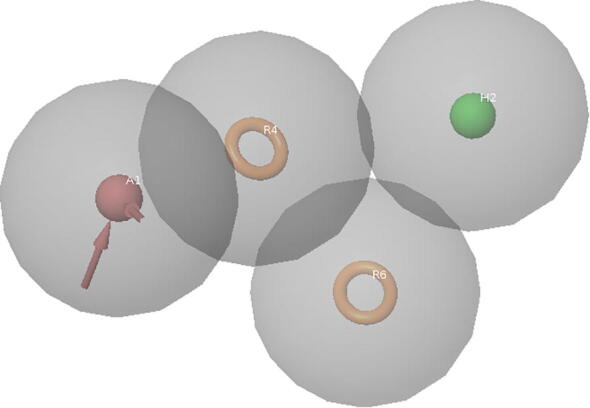
Diagram displaying Pharmacophore model of most of active compound.
Fig. 4.

Mapping of most active from the data on designed pharmacophore model showing the matching site points of ligand structure and pharmacophore.
Table 2.
Summary of Fitness score data set compounds.
| S.no | Name of Compound | Fitness |
|---|---|---|
| 1 | 11243993 | 1.787 |
| 2 | 11336474 | 1.822 |
| 3 | NPACT01463 | 2.018 |
| 4 | NPACT01454 | 1.867 |
| 5 | 9841162 | 2.005 |
3.4. Pharmacophore based virtual screening
Virtual screening was helpful to screen potential compound of interest from inactive molecules. The fitness of molecules with pharmacophore model was measured by fitness score and the map helped to locate the important structural characters required for 1LDG protein inhibition. Therefore, the leads with high fitness score were identified for DFT and molecular dynamics studies. Pharmacophore development observations were carried out to identify the drug of interest. Also phase based screening was carried out against various libraries and based on the hits active compounds were detected. The obtained hits were subjected to high throughput virtual screening to find potential lead molecules. The high throughput virtual screening (HTVS) was done using Glide and rest of the 10% of top scored ligands were analyzed to Glide SP (Standard Precision) dock. Glide was used to analyze all the ligands. The docking score for legands along with the residues forming interaction with the receptor is shown in Table 3, Table 4. Among the analyzed ligands, the score of the ligand IBS2016 was 9.825 k/cal.
Table 3.
Summary of HOMO, LUMO Solv.Energy data set compounds.
| Compound | HOMO (eV) | LUMO (eV) | Solv.Energy (Kcal/mol) |
|---|---|---|---|
| 11243993 | −0.21 | 0 | −24.78 |
| NPACT01454 | −0.21 | 0 | −20.09 |
| NPACT01463 | −0.2 | 0 | −17.09 |
Table 4.
The ADME properties for the top eight compounds.
| Compound | qPlogPw | %Human oral absorption | Rule of five | Donor H | Acceptor H |
|---|---|---|---|---|---|
| 11243993 | 13.195 | 74.104 | 0 | 5 | 4.7 |
| 11336474 | 11.377 | 89.908 | 2 | 2 | 2.2 |
| NPACT01463 | 10.273 | 88.665 | 0 | 4 | 4.7 |
| NPACT01454 | 11.106 | 86.32 | 0 | 5 | 5.3 |
| 9841162 | 15.563 | 62.888 | 1 | 3 | 3.9 |
3.5. DFT analysis
The properties related to donating and accepting electron of the all the 3 screened compounds were analysed. Also, high energy gap between HOMO and LUMO produce unfavourable state of electron transition and decreases the reactivity with protein. In the current study, low energy gap (HOMO-LUMO) between the 3 screened compounds shown that all the compounds are more reactive towards proteins (Table 3).
3.6. Binding free energy
Binding free energy was calculated based on docking complex analysis using Prime/MM–GBSA technique. PRIME MM/GBSA solvation energy (ΔGbind) of compounds 11243993, NPACT01463, NPACT01454 ranged −74.651, −54.312, and −62.743 kcal/mol (Table 2). In our study, binding energy was obtained and this binding energy was similar with the results of Glide XP.
3.7. ADME properties predictions
ADME properties of target compound were analyzed. The processes such as, absorption, distribution, metabolism and excretion were the important process in the development of drugs. In this study, eight compounds were analyzed using various databases to check pharmaceutical relevant properties and drug likeliness. Schrodinger software suite was used to analyze the ADME properties using Lipinski’s rule of five.
3.8. Molecular dynamics analysis
3.8.1. Analysis of structure flexibility
The root-mean square deviation (RMSD) of backbone atoms was evaluated and also conformation drift of the protein was analyzed. At the initial 5000 ps, stimulation raise was observed because of severe relaxation of the model in the water environment. In the overall experiment, RMSD level did not vary significantly and the average stimulation period was <0.03 nm for about 20 ns. These analysis revealed that the identified compounds showed high stability in 3ERT protein. Also structural flexibility was analyzed. The protein structure was maintained in stable form and the size was about 1.8 nm. The Rg of the backbone of the protein is described in Fig. 5.
Fig. 5.
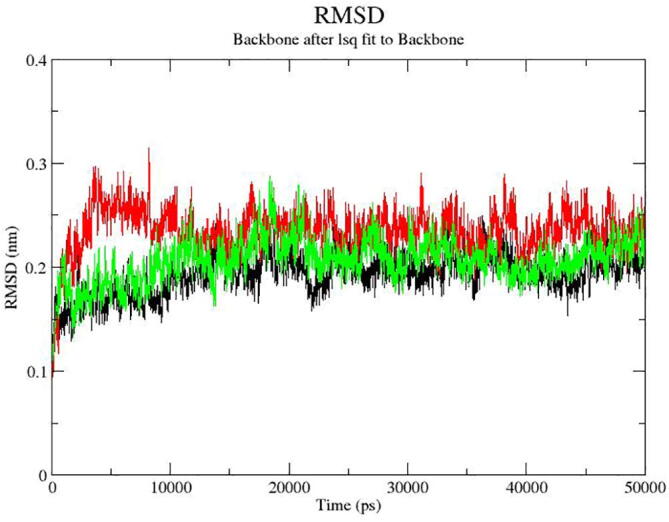
The RMSD plot of the screened compounds (11243993, NPACT01463, NPACT01454) docked in the 3ERT protein.
4. Discussion
In this investigation, the in silico analysis was carried out to analyze docking against the Estrogen protein of Breast Cancer utilizing virtual screening methods. Furthermore, considering various databases were attempted to use estrogen utilizing pharmacopore in curing breast cancer. Pharmacophore modeling and 3ERT protein were taken out utilizing PHASE module of the Schrödinger suite implemented in molecular modeling package, Maestro (Maestro 9.3.5, 2012) [Protein preparation wizard, Epik version 2.2]. PHASE recognize the spatial alignment of various functional groups that are general and necessary for potential biological activity of the lead molecules [Dixon et al., 2006]. The structure of the lead molecules were made by using LigPrep and the structure was made.
Pharmacophore property of the drug molecule is a key in the process of drug discovery. The molecular structure is very important in pharmacological and biological functions. The quality of pharmacophore is evaluated by different methods. RSMD method is more frequent in determining the quality of pharmacophore [Shubina et al., 2015]. Also, vector score and volume of the score were useful for the determination of Van der Waals interaction of the molecule [Gaulton et al., 2012]. Identification of active principles based on pharmacophore process shows information about various interactions. In the case of ligand-based pharmacophore identification there is the lack of receptor structure [Krovat, and Langer, 2003]. Alignment based on pharmacophore stress to locate all unique groups together while in natural the highly complex receptor exists [Verma et al., 2010]. Generally, the ligands interacted with the receptor and adjust their geometrics. Hence, analyzing the structure of the receptor is useful to identify the interactions between receptor ligand [Sastry et al., 2010]. In this study, molecular docking was performed and the top scores were analyzed. Molecular docking is useful to the identification of suitable site of receptor protein. The interaction energy of ligand – receptor was obtained after docking the ligands with active site (3ERT).
VS or virtual screening method is based on computational analysis widely used in the field of drug research. This method is useful to identify the structure, drug target and the analysis of protein receptor complex. The main focus of the study is to screen the drug of target using VS. VS revealed anticancer activity of various natural products and conformational changes of ER –α over expressed on most of the protein [Marquette, and Nabell, 2012]. Analysis of active conformation of ER-α was performed as the target protein for VS. Two factors were considered to screen the new chemically and clinically ER positive anticancer drug molecule [Samanta et al., 2012]. The first approach is based on chemical modification of available chemical substances, which lead to novel molecule with potential therapeutic property also retaining initial action of drug. The approach is to study various pharmacological pathways and analyzing various molecular mechanisms, signaling pathway such as, sulphatase and aromatase [Bouras et al., 2002]. Novel drug is always welcome to overcome the existing antihormonal drugs in new directions. Drug of interest targeting ER membrane alpha and beta nuclear transcription factors would be interesting methods to develop lead molecules [Angelis et al., 2005, Cao et al., 2013].
5. Conclusion
In this analysis, 3ERT was targeted utilizing rational approach accorded with in silico predictive capacity for inhibiting the development of cancer cells. The investigation harnessed the potential of pharmacophore modeling in exploring the molecules with strong binding affinity and biologically relevant interactions. Docking studies were acted in the wake of signifying the screened compounds over the recorded compounds as well as data set utilized to build pharmacophore sample. Prime MMGBSA scores were also well corraletd with the Glide docking scores. Molecular dynamics simulation is acted to verify the constancy of the protein complex structures. The RMSD, RMSF and hydrogen bond interaction outcome denotes that the complex is highly constant through the simulation period.
Declaration of Competing Interest
None declared.
Acknowledgement
JJSR thank UGC Inno/ASIST (F. 14-13/2013); DST- Fund for Improvement of S&T Infrastructure in Universities & Higher Educational Institutions (FIST) (SR/FST/LSI-667/2016) (C); DST-Promotion of University Research and Scientific Excellence (PURSE) (No. SR/PURSE Phase 2/38 (G), 2017 and MHRD-RUSA 2.0, New Delhi (F.24-51/2014-U, Policy (TNMulti-Gen), Dept. of Edn. Govt. of India, Dt.09.10.2018). The authors extend their appreciation to the Researchers supporting project number (RSP-2020/7) King Saud University, Riyadh, Saudi Arabia.
Footnotes
Peer review under responsibility of King Saud University.
References
- Angelis M.D., Stossi F., Carlson K.A., Katzenellenbogen B.S., Katzenellenbogen J.A. Indazole estrogens: highly selective ligands for the estrogen receptor b. J. Med. Chem. 2005;48:1132–1144. doi: 10.1021/jm049223g. [DOI] [PubMed] [Google Scholar]
- Bai Z., Gust R. Breast Cancer, Estrogen Receptor and Ligands. Arch. Pharm. Chem. 2009;342:133–149. doi: 10.1002/ardp.200800174. [DOI] [PubMed] [Google Scholar]
- Bouras T., Southey M.C., Chang A.C., Reddel R.R., Willhite D., Glynne R., Henderson M.A., Armes J.E., Venter D.J. Stanniocalcin 2 is an estrogen-responsive gene coexpressed with the estrogen receptor in human breast cancer. Cancer Res. 2002;62:1289–1295. [PubMed] [Google Scholar]
- Cao X., Jiang J., Zhang S., Zhu L., Zou J., Diao Y., Xiao W., Shan L., Sun H., Zhang W., Huang J., Li H. Discovery of natural estrogen receptor modulators with structure-based virtual screening. Bioorg. Med. Chem. Lett. 2013;23:3329–3333. doi: 10.1016/j.bmcl.2013.03.105. [DOI] [PubMed] [Google Scholar]
- Dixon S.L., Smondyrev A.M., Knoll E.H., Rao S.N., Shaw D.E., Friesner R.A. J. Comput. Aided Mol. Des. 2006;20:647e671. doi: 10.1007/s10822-006-9087-6. [DOI] [PubMed] [Google Scholar]
- Gaulton A., Bellis L.J., Bento A.P., Chambers J., Davies M., Hersey A., Light Y., McGlinchey S., Michalovich D., Al-Lazikani B., Overington J.P. ChEMBL: alarge-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40:D1100–D1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano S.H., Buzdar A.U., Hortobagyi G.N. Breast cancer in men. Ann. Intern. Med. 2002;137:678–687. doi: 10.7326/0003-4819-137-8-200210150-00013. [DOI] [PubMed] [Google Scholar]
- Jemal A., Bray F., Center M.M., Ferlay J., Ward E., Forman D. Global cancer statistics. CA Cancer J. Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Jiang X., Orr B.A., Kranz D.M., Shapiro D.J. Estrogen induction of the granzyme B inhibitor, proteinase inhibitor 9, protects cells against apoptosis mediated by cytotoxic T lymphocytes and natural killer cells. Endocrinology. 2006;147:1419–1426. doi: 10.1210/en.2005-0996. [DOI] [PubMed] [Google Scholar]
- Krovat E.M., Langer T. Nonpeptide angiotensin II receptor antagonists: chemical feature based pharmacophore identification. J. Med. Chem. 2003;46:716–726. doi: 10.1021/jm021032v. [DOI] [PubMed] [Google Scholar]
- Maria M.N., Fernando P., Florencia P., Nestor L., Hugo K., Silvana N. Immunohistochemical characterization of neoplastic cells of breast origin. Diagn. Pathol. 2012;7:73. doi: 10.1186/1746-1596-7-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquette C., Nabell L. Chemotherapy-Resistant Metastatic Breast Cancer. Curr. Treat. Options Oncol. 2012;13:263–275. doi: 10.1007/s11864-012-0184-6. [DOI] [PubMed] [Google Scholar]
- McDonnell D.P., Wardell S.E., Norris J.D. Oral selective estrogen receptor downregulators (SERDs) a break through endocrine therapy for breast cancer. J. Med. Chem. 2015;58:4883–4887. doi: 10.1021/acs.jmedchem.5b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandakumar A. Indian Council of Medical Research; New Delhi Google Scholar: 2001. National Cancer Registry Programme (2001) Consolidated report of the population based cancer registries 1990–1996. [Google Scholar]
- Niinivehmasa S.P., Manivannana E., Rauhamäkia S., Huuskonenc J., Pentikäinena O.T. Identification of estrogen receptor α ligands with virtual screening techniques. J Mol Graph Model. 2016;64:30–39. doi: 10.1016/j.jmgm.2015.12.006. [DOI] [PubMed] [Google Scholar]
- Nilsson S., Makela S., Treuter E., Tujague M., Thomsen J., Andersson G.R., Enmark E., Pettersson K., Warner M., Gustafsson J.A. Mechanisms of estrogen action. Physiol. Rev. 2001;81:1535–1565. doi: 10.1152/physrev.2001.81.4.1535. [DOI] [PubMed] [Google Scholar]
- Park M.T., Kim M.J., Kang Y.H., Choi S.Y., Lee J.H., Choi J.A. Phytosphingosine in combination with ionizing radiation enhances apoptotic cell death in radiation-resistant cancer cells though ROS-dependent and -independent AIF release. Blood. 2005;10(5):1724–1733. doi: 10.1182/blood-2004-07-2938. [DOI] [PubMed] [Google Scholar]
- Peng J., Sengupta S., Jordan V.C. Potential of Selective Estrogen Receptor Modulators as Treatments and Preventives of Breast Cancer. Anticancer Agents Med Chem. 2009;9:481–499. doi: 10.2174/187152009788451833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samanta S., Sharma V.M., Khan A., Mercurio A.M. Regulation of IMP3 by EGFR signaling and repression by ERbeta: implications for triple-negative breast cancer. Oncogene. 2012 doi: 10.1038/onc.2011.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry M., Lowrie J.F., Dixon S.L., Sherman W. Large-scale systematic analysis of 2D fingerprint methods and parameters to improve virtual screening enrichments. J. Chem. Inf. Model. 2010;50(5):771–784. doi: 10.1021/ci100062n. [DOI] [PubMed] [Google Scholar]
- Schrödinger Release, Protein preparation wizard, Epik version 2.2, Impact version 5.7, Prime version 3.0, Schrödinger, LLC, New York, NY, 2011.
- Shubina V., Niinivehmas S., Pentikainen O.T. Reliability of virtual screeningmethods in prediction of PDE4B inhibitor activity. Curr. Drug Discov. Technol. 2015;12(2):117–126. doi: 10.2174/1570163812666150702123018. [DOI] [PubMed] [Google Scholar]
- Suganya J., Radha M., Naorem D.L., Nishandhini M. In Silico Docking Studies of Selected Flavonoids - Natural Healing Agents against Breast Cancer. Asian Pac. J. Cancer Prev. 2014;15:8155–8159. doi: 10.7314/apjcp.2014.15.19.8155. [DOI] [PubMed] [Google Scholar]
- Tan H., Zhong Y., Pan Z. Autocrine regulation of cell proliferation by estrogen receptor-alpha in estrogen receptor-alpha-positive breast cancer cell lines. BMC Cancer. 2009;9:31. doi: 10.1186/1471-2407-9-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torre L.A., Bray F., Siegel R.L., Ferlay J., Lortet Tieulent J., Jemal A. Global cancer statistics, 2012. CA: Cancer J. Clinicians. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- Verma J., Khedkar V.M., Coutinho E.C. 3D-QSAR in drug design—a review. Curr. Top. Med. Chem. 2010;10(1):95–115. doi: 10.2174/156802610790232260. [DOI] [PubMed] [Google Scholar]