
Figure 2. Overview of different mass spectrometry techniques.
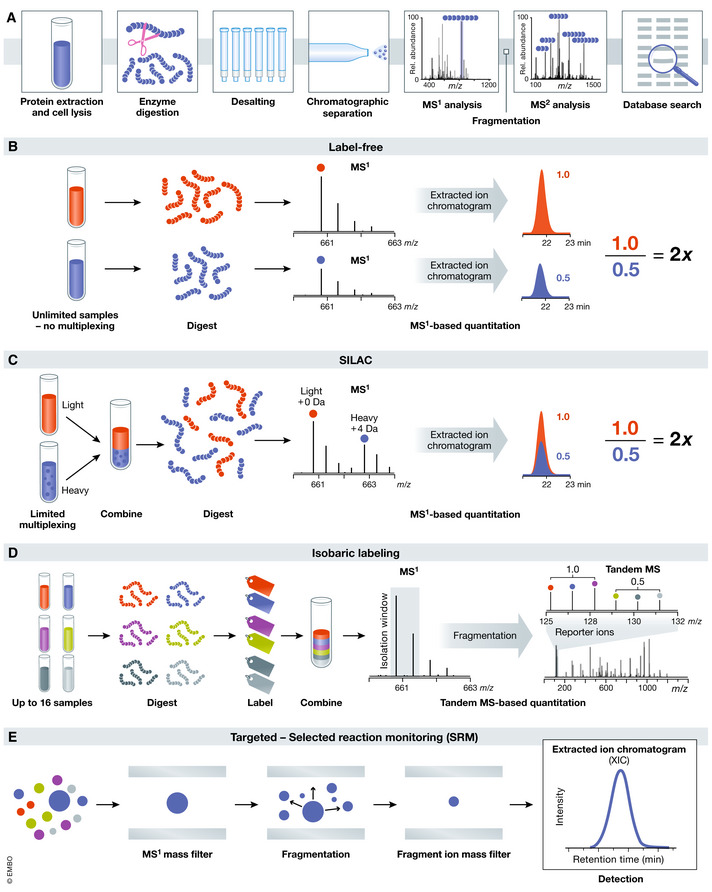
(A) Workflow for bottom‐up proteomics. Preparing proteomic samples for LC‐MS/MS analysis requires protein extraction, proteolysis, and, optionally, peptide‐level fractionation. Online LC separation of complex peptide mixtures introduces analytes into the mass spectrometer for precursor and fragment ion mass analysis. Tandem mass spectra are matched to theoretical spectra generated in silico to garner peptide sequences that are used for protein inference. (B) Label‐free quantitation. Following protein digestion, for each sample, an equal amount of peptides is separately loaded on the column. Relative quantitation is performed by comparing the extracted peak intensity of a given peptide across runs in the dataset. (C) SILAC. During cell culture, “light” or “heavy” versions of specific amino acids are metabolically incorporated into samples. Following sample preparation, cell lysates are mixed in equal total protein ratios and digested into peptides. Intensities of peptide extracted ion chromatograms from the MS1 scan can be used to quantify relative protein abundances between samples. (D) Isobaric labeling. Each sample is digested into peptides, labeled with a unique isobaric label, and mixed in equal ratios. During MS/MS analysis, each tag yields a fragment with a unique mass that can be used for relative quantitation. (E) Targeted MS. In SRM, each fragment of a protein of interest is individually monitored and quantified. The peptide of interest is first isolated, and its characteristic fragments can be monitored for quantitation. Only the specific peptide and fragment masses selected by the user are monitored over the analysis.