

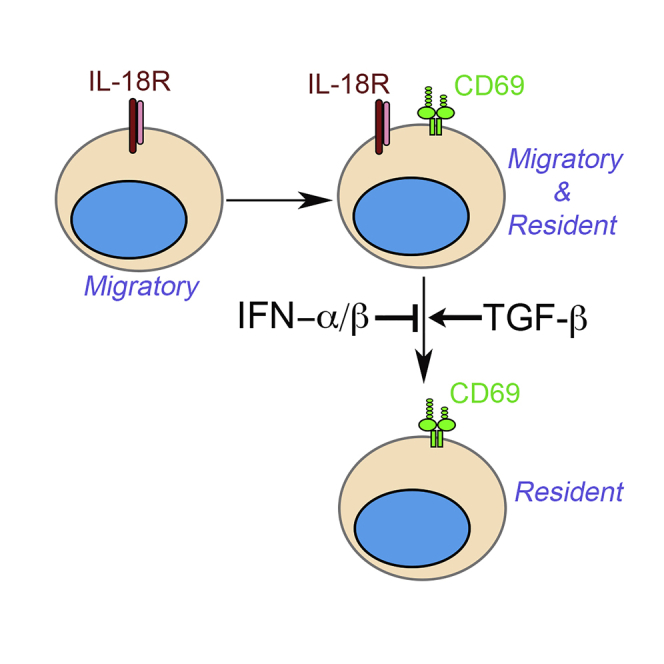
Summary
Stepwise induction of CD69 and CD103 marks distinct differentiation stages of mucosal Trms. But the majority of non-mucosal Trm lacks CD103 expression. The expression of CD69 alone cannot faithfully define Trm cells in heavily vascularized non-mucosal tissues, such as the kidney. Here, we found that a subset of kidney Trms downregulated IL-18 receptor during differentiation. Via global transcriptional analysis and parabiosis experiments, we have discovered that the downregulation of interleukin-18 receptor (IL-18R) is associated with the establishment of tissue residency. Together with the expression of CD69, IL-18Rlo exclusively identify tissue-resident cells whereas IL-18Rhi population contains both tissue-resident and migratory ones. Local cytokines including transforming growth factor β (TGF-β) and interferon α (IFN-α)/β as well as TGF-β-dependent suppression of transcription factor Tcf-1 are essential for IL-18R downregulation during kidney Trm differentiation. Together, we identified a convenient surface marker to distinguish bona fide kidney-resident CD8+ T cells as well as underlying molecular mechanisms controlling this differentiation process.
Subject areas: Immunology, Cell Biology, Transcriptomics
Graphical abstract
Highlights
-
•
CD8+ Trm cells downregulate IL-18 receptor during differentiation
-
•
IL-18Rhi population is composed of both migratory and resident subsets
-
•
IL-18Rlo population is exclusively tissue-resident
-
•
TGF-β promotes, whereas IFN-α/β inhibits, IL-18R downregulation
Immunology; Cell Biology; Transcriptomics
Introduction
Tissue-resident memory T (Trm) cells represent a distinct memory T cell population that is separated from the circulation and provides immediate protection against local reinfection (Cauley and Lefrancois, 2013; Clark, 2015; Iijima and Iwasaki, 2015; Mueller et al., 2013; Mueller and Mackay, 2016; Park and Kupper, 2015; Schenkel and Masopust, 2014; Thome and Farber, 2015). With variable accuracy, only a handful of surface markers, including CD69 and CD103, have been established to distinguish Trms from circulating T cells. The two-step differentiation model has been proposed for skin Trms (Mackay et al., 2013). After exiting bloodstream, effector CD8+ T cells induce CD69 expression as the first step followed by the acquisition of CD103 as the second step. It is generally accepted that CD103hi phenotype is tightly linked with the establishment of tissue residency and the loss of migratory capacity. However, the expression of CD103 is largely restricted to mucosal or barrier tissues, such as the intestines, skin, salivary glands, and lung. The vast majority of non-mucosal Trms do not express CD103 (Steinert et al., 2015). Whether CD69+ Trms within a non-mucosal tissue represent a homogeneous or heterogeneous cell population remains unknown. Because the upregulation of CD69 on Trm precursors is a very early event, whether additional differentiation steps exist after the induction of CD69 remains unknown.
Because the expression of CD69 alone cannot accurately identify tissue resident T cells, intravascular labeling technique is widely used in Trm field to distinguish blood borne versus tissue-resident cells (Anderson et al., 2014). However, intravascular labeling can only provide localization information of a cell population at a given time (i.e., when intravascular labeling is performed). The results from intravascular labeling cannot tell the migratory history or migratory potential of a cell. Indeed, it has been reported that a blood-borne CD8+ T cell can be a tissue-resident cell, especially in densely vascularized organs, such as the kidney (Steinert et al., 2015). It is unknown whether a cell surface marker exists to accurately identify bona fide tissue-resident T cells in the kidney.
Transforming growth factor β (TGF-β) is mainly involved in the acquisition of CD103 expression while dispensable for the initial induction of CD69 during Trm differentiation (Bergsbaken and Bevan, 2015; Thom et al., 2015). Even though TGF-β-independent induction of CD103 has been reported (Pizzolla et al., 2017), most previous research on TGF-β have been focused on CD103hi eTrm cells (i.e., Trm cells reside in the epithelial layers of barrier tissues) (Hu et al., 2015; Mackay et al., 2013; Mani et al., 2019; Sheridan et al., 2014; Zhang and Bevan, 2013). The function of TGF-β in non-barrier Trm differentiation remains less well defined.
Here, we discovered that kidney-resident CD8+ T cells downregulated IL-18 receptor (IL-18R) after induction of CD69 during differentiation. Compared with IL-18Rhi counterparts, IL-18Rlo cells carried more common Trm signatures at transcription level. Via parabiosis experiments, we discovered that the downregulation of IL-18R was tightly associated with the establishment of kidney residency regardless of vascular location. TGF-β and suppression of transcription factor Tcf-1 (T cell factor-1) were required for IL-18R downregulation, whereas type I interferon (IFN) signals inhibited this process. Together, we have established IL-18Rlo as a convenient surface marker to identify kidney-resident CD8+ T cells as well as the molecular mechanisms controlling the downregulation of IL-18R.
Results
Kidney Trm downregulates IL-18 receptor
Mucosal Trm cells induce the expression of CD69 and CD103 in a progressive order, i.e., the induction of CD69 occurs usually before turning on CD103 (Mackay et al., 2013). The expression of CD103 is often associated with limited migratory capacity and tissue-resident phenotype. However, the expression of CD103 is usually not detectable or substantially delayed in non-mucosal and non-barrier tissues, such as the kidney (Casey et al., 2012). To investigate the differentiation of kidney-resident T cells, we employed the well-established LCMV (lymphocytic choriomeningitis virus) infection model. Briefly, congenically marked P14 TCR transgenic CD8+ T cells specific for an LCMV epitope were adoptively transferred into unmanipulated C57BL/6 (B6) mice followed by acute LCMV Armstrong (LCMV Arm) infection (illustrated in Figure 1A). To distinguish blood-borne versus tissue-resident CD8+ T cells, intra-vascular labeling of CD8+ T cells was performed before euthanasia (Anderson et al., 2014). In both CD8α staining positive (intravascular, i.v.) and CD8α staining negative (extravascular, e.v.) compartments, a donor-derived P14 T cell population was clearly identified by congenic marker CD45.1 (Figure 1B). A subset of e.v. P14 T cells downregulated IL-18R after the induction of CD69. CD69+ Trm cells could be further divided into two differentiation stages, namely IL-18Rhi and IL-18Rlo subsets (Figures 1B and 1C). There was a slight increase in the percentage of IL-18Rlo subset at memory time points (e.g., day 42–62 in Figure 1D). Intriguingly, at a later memory time point (e.g., day 45), even though Trm cells were highly enriched in the e.v. compartment, we could consistently detect Trm-like cells (CD69+IL-18Rhi and CD69+IL-18Rlo) in the i.v. compartment (Figures 1C and 1E). To be noted, compared with mucosal Trms (small intestine intraepithelial lymphocyte, SI-IEL) isolated from the same animals, we could not detect CD103 expression on kidney Trms (Figure 1F). Together, kidney Trm cells downregulate IL-18R during differentiation. Both IL-18Rhi and IL-18Rlo cells co-exist for a prolonged period of time in the kidney. Intravascular labeling cannot fully distinguish Trm versus non-Trm in the kidney, especially at later time points.
Figure 1.

Downregulation of IL-18R during kidney Trm differentiation
(A) Experimental design. 104 congenically marked naive P14 T cells were transferred into B6 recipients followed by LCMV Arm infection on the next day.
(B and C) (B) Fourteen days after infection and (C) 45 days after infection, representative FACS profiles of kidney CD8+ T cells are shown. (i.v., intravascular and e.v., extravascular).
(D) The percentage of IL-18Rlo cells in e.v. CD69+ P14 T cells.
(E) The composition of i.v. (left) versus e.v. (right) P14 T cells at day 45 post-infection.
(F) The percentage of CD103+ cells in kidney P14s versus SI-IEL P14s at day 45. Pooled results from five independent experiments are shown in (D).
Representative results from two to three independent experiments are shown in (B), (C), (E), and (F). Each symbol in (D) and (F) represents the results from an individual mouse. ∗∗, p < 0.01; ∗∗∗, p < 0.001; and ∗∗∗∗, p < 0.0001 by one-way ANOVA with Tukey multi-comparison post-test. See also Figure S1. Bar graphs indicate the mean (±S.D.).
Downregulation of IL-18R is a common feature of Trms
Next, we investigated whether this phenomenon was kidney specific or not. To this end, we examined Trm cells isolated from salivary glands (SG) and SI-IEL of LCMV-infected animals. Both SG and SI-IEL Trms induce the expression of CD103 and downregulate circulating memory T cell marker Ly6C during differentiation. In both SG and SI-IEL, by first gating Trm cells based on their expression of IL-18R and CD69, it was clear that IL-18Rlo Trms carried higher levels of CD103 and lower levels of Ly6C compared with their IL-18Rhi counterparts (Figures 2A and 2B). Together, Trm cells downregulate IL-18R during differentiation, and IL-18Rlo cells may represent a mature subset in both mucosal and non-mucosal tissues.
Figure 2.

Downregulation of IL-18R during SG and SI-IEL Trm differentiation
Similar experimental setup to Figure 1. Day 25 (A) or day 45 (B) post-infection, SG P14 (upper row) and SI-IEL P14 (lower row) were isolated and subjected to flow cytometry analysis. Donor P14 T cells were first gated based on the expression of CD69 and IL-18R. The expression of Ly6C (left) and CD103 (right) on each subset were shown. Each group of connected symbols in (A) represents the results from an individual animal. ∗, p < 0.05; ∗∗∗, p < 0.001; and ∗∗∗∗, p < 0.0001 by one-way ANOVA with Tukey multi-comparison post-test. See also Figures S1 and S2.
IL-18R is not required for Trm differentiation
To determine whether IL-18R signaling is involved in Trm differentiation or maturation, we generated congenically marked Il18r1−/− P14 TCR transgenic mice. As illustrated in Figure S1A, naive P14 T cells were isolated from both control and Il18r1−/− mice, mixed at a 1:1 ratio and adoptively co-transferred into B6 recipients followed by LCMV Arm infection. We examined Trm differentiation in both mucosal (SI-IEL) and non-mucosal tissues (kidney). In SI-IEL, we could not detect any significant changes in the differentiation of CD103+ Trms in Il18r1−/− cells, including the upregulation of CD73 and downregulation of Cmah (cytidine monophospho-N-acetylneuraminic acid hydroxylase) activities (Figures S1B and S1C). When focused on the kidney, we did not detect any significant defects in CD69 induction, Ly6C downregulation, or CD38 induction in both intravascular and extravascular compartments (Figures S1D–S1F). Therefore, we concluded that the lack of IL-18R did not impact Trm differentiation and maintenance, which was consistent with a previous report focused on intestinal Trm (Bergsbaken et al., 2017). In addition, we confirmed that in a different systemic viral infection model (Vesicular Stomatitis Virus), similar subsets of CD69+IL-18Rhi and CD69+IL-18Rlo kidney Trm cell can be identified (not depicted). Thus, even though IL-18 signaling is not required for the formation of Trms, the expression of IL-18R can be used as a biomarker to separate Trm cells into various differentiation stages.
Memory CD8+ T cells exert effector functions in response to both antigen-specific and non-specific bystander stimulations. Cognate antigen induces robust activation of memory T cells, which represents the hallmark of immunological memory (Williams and Bevan, 2007). In the absence of cognate antigen, memory T cells rapidly produce inflammatory cytokines (e.g. IFN-γ) in response to type I IFN, IL-15, IL-12, and IL-18 (Berg et al., 2003; Berg and Forman, 2006; Kohlmeier et al., 2010; Kupz et al., 2012; Lauvau et al., 2016; Raue et al., 2013; Richer et al., 2015; Ruiz et al., 2014; Soudja et al., 2012). In contrast to type I IFN, IL-12, and IL-15, IL-18 is unique, as it is only required for bystander inflammatory responses without apparent involvement in antigen-elicited CD8+ T cell responses (Haring and Harty, 2009). However, this conclusion about bystander response of memory CD8+ T cells is almost exclusively based on the results from circulating cells residing in the secondary lymphoid organs. Whether Trm cells from non-lymphoid tissues respond to bystander inflammation in a similar manner as circulating memory T cells remains unclear. To probe the functional consequences of IL-18R downregulation during Trm differentiation, we examined their response to bystander inflammation. Because the large majority of SI-IEL Trms downregulate IL-18R at later time points (Figure 2B), we focused on SI-IEL Trms for this purpose. In contrast to splenic counterparts who responded to both cognate peptide (i.e., GP33-41 for P14 TCR) and bystander inflammation (i.e., IL-12+IL-18), SI-IEL Trm cells completely lost the response to bystander inflammation while maintaining the capacity to elicit a robust response to cognate antigen during ex vivo stimulation (Figure S2A). Using Il18r1−/− cells, we were able to demonstrate that the bystander response of splenic memory CD8+ T cells was largely IL-18R dependent. In contrast, both WT and Il18r1−/− SI-IEL Trms could not respond to bystander stimuli (Figure S2B). Interestingly, TCR signal induced both IFN-γ and tumor necrosis factor (TNF) production, whereas bystander inflammatory cytokine only induced IFN-γ (Figure S2A), which is likely due to the fact that TNF production requires Ca2+ signaling (Falvo et al., 2010). Together, we propose that IL-18R downregulation in Trms may be related to the loss of response to IL-18-mediated bystander inflammation, which warrants future investigation.
Transcriptional and functional distinction of IL-18Rhi versus IL-18Rlo Trms
To further characterize IL-18Rhi and IL-18Rlo kidney T cells, we FACS sorted both populations as well as CD69− kidney P14 T cells and performed RNA-seq analysis. Taking advantage of published transcriptional signatures of both tissue-resident and circulating memory T cells (Mackay et al., 2013, 2016; Milner et al., 2017), we focused our analysis on these sets of resident and circulating signature genes. As shown in Figure 3A, for most circulating signature genes, their expression exhibited a steady decrease from CD69− and CD69+IL-18Rhi to CD69+IL-18Rlo populations. Conversely, a dramatic induction of the majority of resident signature genes occurred in CD69+IL-18Rlo cells (Figure 3B).
Figure 3.

Transcriptional and functional distinction between IL-18Rhi and IL-18Rlo kidney Trms
Similar to Figure 1A, 12 days later, CD69−, CD69+IL-18Rhi, and CD69+IL-18Rlo P14 T cells were FACS sorted from the kidney and subjected to RNA-seq analysis. The expression of (A) circulating signature genes and (B) resident signature genes are shown. Frequently identified Trm-associated genes are marked by arrows.
(C) Day 12–14 post-infection, kidney CD8+ T cells are pre-gated on CD69−, CD69+IL-18Rhi, and CD69+IL-18Rlo cells, and the histograms of representative surface markers are shown. Each column in (A) and (B) represents a biologically independent replicate. Representative results from at least two independent experiments are shown in (C).
(D) Experimental design. Briefly, similar to Figure 1A, day 30 post-LCMV infection, the mice received GP33-41 peptide via an intravenous route together with Brefeldin A. Mice were euthanized 4 h later.
(E) MFI of IFN-γ (left) and MFI of granzyme B (right) on pre-gated P14 subsets isolated from the spleen and kidney.
(F) Representative FACS profiles to show IFN-γ and granzyme B expression in kidney P14 subsets. Dotted lines, without GP33-41; Solid lines, with GP33-41.
Each symbol in (E) represents the results from an individual mouse. Representative results from two independent experiments are shown in (E). N.S., not significant, ∗, p < 0.05; ∗∗, p < 0.01; and ∗∗∗, p < 0.001 by one-way ANOVA with Tukey multi-comparison post-test. See also Figure S3. Bar graphs indicate the mean (±S.D.).
In addition to IL-18Rα, several Trm-associated genes were differentially expressed at the protein level in CD69+IL-18Rhi versus CD69+IL-18Rlo kidney CD8+ T cells (Figure 3C). The downregulation of Ly6C and Cmah activity were largely restricted to CD69+IL-18Rlo cells. In contrast, the induction of CD38 and CXCR4 exhibited a stepwise increase during kidney Trm differentiation. Together, IL-18Rhi versus IL-18Rlo cells represent distinct kidney-resident T cell subsets. Compared with IL-18Rhi ones, IL-18Rlo cells carry more common Trm signatures.
To probe whether Trm differentiation stages impact their effector function during recall responses, we stimulated P14 memory T cells with their cognate peptide in vivo and measured their effector molecule production 4 h later before massive proliferation or before recruitment of circulating memory T cells occurred (illustrated in Figure 3D) (Beura et al., 2018; Park et al., 2018). To be noted, to avoid ex vivo incubation-induced confounding factors, cognate peptide and Golgi inhibitor were delivered in vivo and the production of effector molecules were measured on freshly isolated cells without further in vitro manipulation. As shown in Figure 3E left and Figure 3F, both IL-18Rhi and IL-18Rlo kidney Trm cells produced similar levels of IFN-γ upon re-stimulation. Consistent with recent findings that non-lymphoid tissue Trm cells are reactivated less efficiently than their lymphoid tissue counterparts (Low et al., 2020), both IL-18Rhi and IL-18Rlo Trms produced less IFN-γ than splenic memory T cells. Alternatively, this finding may be explained by inefficient access to cognate peptide for Trms in vivo. Indeed, when kidney Trms were isolated and stimulated ex vivo with peptide, we did not detect significant reduction of IFN-γ production (Figure S3). Different from IFN-γ, IL-18Rlo Trms produced significantly increased amount of granzyme B both at baseline and after re-stimulation (Figure 3E right and Figure 3F). In summary, compared with IL-18Rhi cells, IL-18Rlo Trms produce similar levels of IFN-γ and increased amounts of granzyme B upon reactivation in vivo.
IL-18Rlo Trm cells represent the bona fide tissue-resident population
To directly test the tissue residency of IL-18Rhi versus IL-18Rlo Trms, we performed parabiosis experiments. Briefly, B6 mice carrying P14 T cells with distinct congenic markers were surgically connected 30 days post-LCMV infection and examined 19–21 days later (Figure 4A). In the kidney, CD69+IL-18Rlo subset was almost exclusively derived from the hosts. Interestingly, in both i.v. and e.v. compartments, CD69+IL-18Rlo cells were similarly host-derived (Figures 4B and 4C). As expected, the vast majority of splenic P14 T cells were migratory and exhibited a roughly 50:50 ratio between host- and partner-derived cells, which was similar to CD69−IL-18Rhi cells in the kidney. In contrast, SI-IEL compartment was mostly occupied by host-derived Trm cells (Figure 4C). For both i.v. and e.v. compartments, the transition from CD69+IL-18Rhi to CD69+IL-18Rlo stages was associated with significantly improved tissue-residency (Figure 4C). Importantly, using mucosal SI-IEL as a well-established Trm reference, the percentage of tissue residency was comparable among SI-IEL, kidney i.v. CD69+IL-18Rlo, and e.v. CD69+IL-18Rlo subsets (Figure 4C). These findings clearly demonstrate that the results from intravascular labeling does not tell the migratory history of a T cell. In stark contrast, the downregulation of IL-18R can be used as a convenient marker to faithfully define kidney-resident CD8+ T cells.
Figure 4.

IL-18Rlo T cells represent a bona fide kidney-resident population
(A) Parabiosis experimental design for (B) to (D). Naive P14 T cells with distinct congenic markers were separately transferred into B6 mice followed by LCMV Arm infection. D30 p.i., pairs of mice carrying congenically different P14s were surgically connected and examined 19–21 days later.
(B) Representative FACS profiles of kidney CD8+ T cells (n = 10).
(C) The composition (host- versus partner-derived) of each indicated P14 subset.
(D) Parabiosis experimental design for (E). D12 p.i., pairs of mice carrying congenically different P14s were surgically connected and examined 15 days later.
(E) Percentage of host-derived versus partner-derived cells within different P14 subsets are shown (n = 8).
Pooled results from two independent experiments are shown in (C) and (E). N.S., not significant, ∗∗, p < 0.01; ∗∗∗, p < 0.001 by paired Student t test or one-way ANOVA with Tukey multi-comparison post-test. Bar graphs indicate the mean (±S.D.).
Because we have demonstrated that IL-18Rlo kidney-resident CD8+ T cells are formed at early stages after acute viral infection (i.e., day 12–14 post-infection, see Figures 1B and 1D), to further prove that IL-18R downregulation defines tissue residency even at these early stages, we performed another set of parabiosis experiments at an early time point (illustrated in Figure 4D). Briefly, B6 mice carrying P14 T cells with distinct congenic markers were surgically connected 12 days post-LCMV infection and examined 15 days later. As shown in Figure 4E, CD69+IL-18Rlo kidney CD8+ T cells were exclusively derived from the hosts, indistinguishable from SI-IEL counterparts. In contrast, CD69+IL-18Rhi cells contained a mixed population of both migratory and resident T cells.
Local cytokines control the downregulation of IL-18R in kidney Trms
Cytokine signals (e.g., IL-33, TNF and TGF-β) control the differentiation and maintenance of mucosal Trm cells (Bergsbaken et al., 2017; Hu et al., 2015; Mani et al., 2019; Skon et al., 2013; Slutter et al., 2017). To pinpoint the signals controlling IL-18R downregulation, we first used an ex vivo culture system. Day 4.5 post-infection when effector CD8+ T cells started to migrate from the secondary lymphoid organs to the periphery (Masopust et al., 2010), splenic P14 T cells were cultured overnight in the presence of a panel of different stimuli and the expression of IL-18R was measured. Both TCR and TGF-β significantly reduced the expression of IL-18R (Figure S4). IFN-β dramatically enhanced the expression of IL-18R, whereas IL-18, IL-33, and TNF did not yield significant changes (Figure S4). Based on this observation, we set up in vivo systems to investigate the involvement of TGF-β and type I IFN in the downregulation of IL-18R in kidney-resident CD8+ T cells.
TGF-β is specifically required for the downregulation of IL-18R during kidney Trm differentiation
TGF-β provides an essential signal for the induction of mucosal or epithelial Trm marker CD103 (El-Asady et al., 2005). Further, we have demonstrated that TGF-β promotes the formation of kidney Trms via enhancing effector CD8+ T cell extravasation at early stages (Ma et al., 2017). To investigate the possible roles of TGF-β in the downregulation of IL-18R during kidney Trm differentiation, we employed TGF-β receptor conditional knockout (i.e., Tgfbr2f/fdLck-cre (Zhang and Bevan, 2012), hereafter referred to as Tgfbr2−/−) P14 T cells. Similar to the experimental design illustrated in Figure S1A, congenically marked naive control and Tgfbr2−/− P14 T cells were purified, mixed, and adoptively co-transferred into B6 recipients followed by LCMV infection. At day 14 post-infection, the percentage of CD69+IL-18Rhi subset was comparable between control and Tgfbr2−/− P14 T cells (see blue numbers in Figure 5A), consistent with the results from the gut and salivary glands (Bergsbaken and Bevan, 2015; Thom et al., 2015). We did detect a slight but significant decrease of total number of CD69+IL-18Rhi Tgfbr2−/− cells in the e.v. compartment (see blue symbols in Figure 5B), most likely due to defective extravasation of Tgfbr2−/− cells (Ma et al., 2017). However, the downregulation of IL-18R was significantly impaired in Tgfbr2−/− P14 T cells (see red numbers and symbols in Figures 5A and 5B). Further, this defect was not due to delayed downregulation of IL-18R in Tgfbr2−/− cells (Figure 5C). Importantly, at later time points (day 42–45) when there was a significant population of CD69+ cells in the i.v. compartment of the kidney, we could clearly detect defective downregulation of IL-18R in Tgfbr2−/− P14 T cells isolated from both i.v. and e.v. compartments (Figure 5C), suggesting that TGF-β signal can be delivered to induce tissue-resident differentiation at both extravascular and intravascular sites. Furthermore, we performed another parabiosis experiment using WT and Tgfbr2−/− P14 T cells. Briefly, congenically distinct WT and Tgfbr2−/− P14 T cells were adoptively transferred into different B6 recipients followed by LCMV infection. Thirty days later, pairs of mice received WT, and Tgfbr2−/− P14 T cells were surgically connected. The distribution and differentiation of P14 T cells were examined 15 days after parabiosis surgery (Figure S5A). As shown in Figure S5B, only host-derived WT control P14 T cells differentiated into IL-18Rlo kidney Trms, whereas Tgfbr2−/− P14 T cells exhibited indistinguishable differentiation patterns in host versus partner mice.
Figure 5.

TGF-β and type I IFN control the downregulation of IL-18R during kidney Trm differentiation
Congenically marked control and Tgfbr2−/− P14 T cells were co-transferred into B6 mice followed by LCMV infection. Fourteen days post-infection, representative FACS profiles of pre-gated P14 T cells in both i.v. and e.v. compartments are shown in (A). (B) Day 14 and (C) day 42–45 post-infection, the ratio of (Tgfbr2−/− P14/Control P14) in each pre-gated kidney P14 subset. (D) Experimental design for (E) to (G). At day 4 and day 7 post-infection, 1 mg anti-IFNAR-1 or isotype control antibody was given i.p. Kidney P14 cells were examined later. (E) Day 30 post-infection, representative FACS profiles of kidney CD8+ T cells. (F) Day 12 and (G) day 30 post-infection, the percentage of IL-18Rlo cells in donor P14 (left) or host CD8+ T cells (right) in the kidney. Each symbol in (B), (C), (F), and (G) represents the results from an individual mouse. Pooled results from two to five independent experiments are shown in (B), (C), (F), and (G). N.S., not significant, ∗∗, p < 0.01, ∗∗∗; p < 0.001; and ∗∗∗∗, p < 0.0001 by Student t test or one-way ANOVA with Tukey multi-comparison post-test. See also Figures S4–S6. Bar graphs indicate the mean (±S.D.).
Here, we demonstrated that using the expression of IL-18R as a marker, CD69+ Trm cells could be further divided into two differentiation steps. Interestingly, TGF-β signaling is specifically required for the second step of Trm differentiation in the kidney, i.e., the downregulation of IL-18R after the induction of CD69. A similar scenario exists in mucosal or epithelial Trms, where TGF-β is only required for the induction of CD103 after acquiring CD69 (Bergsbaken and Bevan, 2015; Thom et al., 2015). Thus, TGF-β may represent the essential signal to Trm differentiation in both mucosal and non-mucosal tissues after establishing early residency.
Type I IFN inhibits IL-18R downregulation in kidney Trms
Next, we focused on type I IFNs, which provide the major inflammatory signal in our LCMV infection system. Interfering IFNα/β during the early stages of LCMV Arm infection (i.e., within the first 2–3 days) significantly impacted viral clearance and CD8+ T cell priming/expansion (not depicted (Cousens et al., 1999; Kolumam et al., 2005)). Further, effector CD8+ T cells usually leave secondary lymphoid organs and seed peripheral tissues around day 4 after LCMV Arm infection (Masopust et al., 2010). Thus, to avoid complications associated with early IFNα/β blocking and to specifically target Trm forming stages, we undertook a strategy illustrated in Figure 5D. Briefly, naive P14 T cells with congenic markers were adoptively transferred into B6 mice followed by LCMV infection. At day 4 and day 7 after infection, recipient mice were treated by anti-IFNAR1 blocking antibody or isotype control antibody. Trm cells were examined later. This type I IFN blocking strategy did not significantly alter the expansion or accumulation of P14 T cells in both secondary lymphoid and non-lymphoid tissues, including the kidney (Figure S6A) or effector CD8+ T cell differentiation in the spleen (Figure S6B). Importantly, we did not detect significant changes in viral clearance (Figure S6C). Consistent with previous findings that type I IFN was not required for the induction of CD69 on Trms in vivo (Mackay et al., 2015a), the expression of CD69 was largely intact (Figure 5E). In contrast, as shown in Figures 5E–5G, type I IFN blockade significantly enhanced the downregulation of IL-18R during kidney Trm differentiation. The enhanced downregulation of IL-18R can be detected as early as day 12 post-infection (Figure 5F). Interestingly, transient type I IFN blockade had a long-lasting effect on kidney-resident CD8+ T cells, as we could detect significant changes in kidney Trms at day 30 post-infection (Figures 5E and 5G). This result further supports the model that shortly after arrival at the kidney, CD8+ T cells acquire tissue residency under the influence of local signals. In addition, the effects of type I IFN were not restricted to monoclonal TCR transgenic P14 T cells; endogenous polyclonal kidney CD8+ T cells were also impacted in a similar manner (Figures 5F and 5G, right panels). Thus, inflammatory signal type I IFN negatively regulates the downregulation of IL-18R during kidney Trm differentiation.
Repression of transcription factor Tcf-1 is involved in the downregulation of IL-18R during Trm maturation
During Trm differentiation, TGF-β-mediated downregulation of T-box transcription factors T-bet and Eomes plays an essential role (Mackay et al., 2015b). To determine the transcription factors underlying TGF-β controlled Trm differentiation, we first focused on T-box transcription factors. However, neither Tbx21 nor Eomes deficiency could rescue defective IL-18R downregulation in Tgfbr2−/− cells in the kidney (unpublished observations). Instead, consistent with our own RNA-seq results (Figure 3) and published RNA-seq results (Mackay et al., 2016; Milner et al., 2017), the expression of Tcf-1 (T cell factor-1, encoded by Tcf7) was suppressed during kidney Trm differentiation at the protein level (Figure 6A). Interestingly, compared with their WT control counterparts, both Tgfbr2−/− CD69+IL-18Rhi and residual Tgfbr2−/− CD69+IL-18Rlo cells expressed significantly higher levels of Tcf-1. To elucidate the function of Tcf-1 downregulation during Trm differentiation, we used a retroviral system to force express Tcf7 in P14 T cells (Figure 6B). Consistent with published results of Tcf-1 in circulating effector and memory CD8+ T cells (Zhou et al., 2010), overexpression of Tcf7 did not significantly impact the expansion of P14 T cells in the spleen, whereas the differentiation of KLRG-1- memory precursors was enhanced and KLRG-1+ effector cells was inhibited (Figure S7). Compared with empty GFP control retrovirus, Tcf7 overexpression significantly dampened the downregulation of IL-18R during kidney Trm differentiation at both early (day 14) and later (day 30) stages (Figures 6C–6E). At transcription level, suppression of Tcf-1 has been associated with Trm differentiation (Mackay et al., 2016; Milner et al., 2017). Recently, it has been reported that TGF-β-dependent downregulation of Tcf-1 is essential for lung-resident CD8+ T cell differentiation at functional level (Wu et al., 2020). Here, we showed that TGF-β suppressed the expression of Tcf-1, which may be essential for the downregulation of IL-18R associated with kidney Trm differentiation.
Figure 6.

The downregulation of transcription factor Tcf-1 is required for IL-18R downregulation in the kidney
(A) Similar to the experimental setup in Figure 5A, day 14 post-LCMV infection, the expression of Tcf-1 was determined in pre-gated CD69+IL-18Rhi and CD69+IL-18Rlo kidney P14 T cells by flow cytometry.
(B) Experimental design. Activated P14 T cells were spin-infected by control retrovirus (GFP only) or retrovirus carrying Tcf7 cDNA. One hour after spin infected, GFP-P14 and Tcf7-GFP-P14 were mixed and 2x105 cells co-transferred into recipient B6 mice followed by LCMV infection. P14 T cells were examined later.
(C) Day 14 post-infection, representative FACS profiles of pre-gated kidney P14 T cells are shown.
(D and E) (D) Day 14 and (E) day 30 post-infection, the percentage of IL-18Rlo cells in GFP+CD69+ P14 kidney T cells is shown. Each pair of symbols in (A), (D), and (E) represents the results from an individual recipient. Pooled results from two independent experiments are shown in (A), (D), and (E). ∗, p < 0.05 and ∗∗, p < 0.01 by paired Student t test. See also Figure S7.
Discussion
Together, we have analyzed Trm differentiation in the kidney and established that the downregulation of IL-18R is a distinct marker associated with the establishment of tissue residency. Further, this finding can be extended to other tissues, including SG and SI-IEL. As a convenient biomarker, IL-18Rlo subset precisely indicates kidney residency regardless of its intra- or extra-vascular location. In contrast, IL-18Rhi subset carries a mixture of tissue-resident and migratory T cells. TGF-β promotes, whereas type I IFN inhibits, the downregulation of IL-18R during kidney Trm differentiation. Further, the downregulation of transcription factor Tcf-1 is associated with this differentiation step of kidney Trms. Consistent with recent findings that reveal the heterogeneity of mucosal Trms (Kurd et al., 2020; Milner et al., 2020), our findings demonstrate that kidney CD8+ T cells contain heterogeneous populations of cells with different levels of tissue residency.
Limitations of the study
In the current project, we focused on a systemic acute viral infection model and defined a differentiation process associated with CD8+ tissue residency. Whether this finding could be extended to other infection models was not determined.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Nu Zhang (zhangn3@uthscsa.edu)
Materials availability
This study did not generate new unique reagents.
Data and code availability
Original RNA-seq results can be accessed by GSE111801. The raw data supporting the current study are available from the Lead Contact upon request. All software is commercially available.
Methods
All methods can be found in the accompanying Transparent methods supplemental file.
Acknowledgments
We thank Dr. Tessa Bergsbaken (Rutgers New Jersey Medical School) and Dr. Linda Wakim (University of Melbourne, Australia) for critical reading of the manuscript, Dr. Haihui Xue (Hackensack University Medical Center) for providing Tcf7-GFP retroviral vector, and Dr. Linxi Li (University of Arkansas for Medical Sciences) for help with parabiosis experiments. This work is supported by NIH grants AI125701 and AI139721, Cancer Research Institute CLIP program, and American Cancer Society grant RSG-18-222-01-LIB to N.Z. and National Natural Science Foundation of China (No. 81522038, No. 81270024, and No. 81220108017) to Q.L. We thank Dr. Ben Daniel and Karla Gorena for FACS sorting. Flow Cytometry data was generated in the UT Health San Antonio Flow Cytometry Shared Resource Facility, which is supported in part by UT Health San Antonio, the Mays Cancer Center P30 Cancer Center Support Grant (NIH-NCI P30 CA054174) and the NIH National Center for Advancing Translational Sciences Clinical Translational Science Award (NIH-NCATS UL1 TR002645). We thank Drs. Zhao Lai, Yi Zou and Yidong Chen for RNA-seq analysis and informatics assistance. RNA-seq analysis was performed by the Genome Sequencing Core Facility at UTHSCSA, which is supported by NIH/NCI Cancer Center Support Grant P30 CA054174, NIH Shared Instrument grant 1S10OD021805-01, and Cancer Prevention and Research Institute of Texas (CPRIT) Core Facility grant RP160732. Special thanks to Dr. Rizi Ai (UCSD) for suggestions and help on bioinformatics.
Author contributions
Conceptualization, W.L., Y.L., Q.L., and N.Z.; Methodology, C.M. and X. L.; Investigation, W.L., Y.L., C.M., L.W., G.L., S.M., S.S., K.K-H.F., H.W., Q.L., M.Z., and E.L.D.; Writing–Original Draft, W.L., Q.L., X.Z., Y.Q., and N.Z.; Writing–Review & Editing, N.Z.; Supervision, Q.L. and N.Z.; Funding Acquisition, Q.L. and N.Z.
Declaration of interests
The authors declare no competing interests.
Published: January 22, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2020.101975.
Contributor Information
Qianjin Lu, Email: qianlu5860@gmail.com.
Nu Zhang, Email: zhangn3@uthscsa.edu.
Supplemental information
References
- Anderson K.G., Mayer-Barber K., Sung H., Beura L., James B.R., Taylor J.J., Qunaj L., Griffith T.S., Vezys V., Barber D.L., Masopust D. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat. Protoc. 2014;9:209–222. doi: 10.1038/nprot.2014.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg R.E., Crossley E., Murray S., Forman J. Memory CD8+ T cells provide innate immune protection against Listeria monocytogenes in the absence of cognate antigen. J. Exp. Med. 2003;198:1583–1593. doi: 10.1084/jem.20031051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg R.E., Forman J. The role of CD8 T cells in innate immunity and in antigen non-specific protection. Curr. Opin. Immunol. 2006;18:338–343. doi: 10.1016/j.coi.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Bergsbaken T., Bevan M.J. Proinflammatory microenvironments within the intestine regulate the differentiation of tissue-resident CD8(+) T cells responding to infection. Nat. Immunol. 2015;16:406–414. doi: 10.1038/ni.3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsbaken T., Bevan M.J., Fink P.J. Local inflammatory cues regulate differentiation and persistence of CD8(+) tissue-resident memory T cells. Cell Rep. 2017;19:114–124. doi: 10.1016/j.celrep.2017.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beura L.K., Mitchell J.S., Thompson E.A., Schenkel J.M., Mohammed J., Wijeyesinghe S., Fonseca R., Burbach B.J., Hickman H.D., Vezys V. Intravital mucosal imaging of CD8(+) resident memory T cells shows tissue-autonomous recall responses that amplify secondary memory. Nat. Immunol. 2018;19:173–182. doi: 10.1038/s41590-017-0029-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey K.A., Fraser K.A., Schenkel J.M., Moran A., Abt M.C., Beura L.K., Lucas P.J., Artis D., Wherry E.J., Hogquist K. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J. Immunol. 2012;188:4866–4875. doi: 10.4049/jimmunol.1200402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauley L.S., Lefrancois L. Guarding the perimeter: protection of the mucosa by tissue-resident memory T cells. Mucosal Immunol. 2013;6:14–23. doi: 10.1038/mi.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark R.A. Resident memory T cells in human health and disease. Sci. Transl. Med. 2015;7:269rv261. doi: 10.1126/scitranslmed.3010641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousens L.P., Peterson R., Hsu S., Dorner A., Altman J.D., Ahmed R., Biron C.A. Two roads diverged: interferon alpha/beta- and interleukin 12-mediated pathways in promoting T cell interferon gamma responses during viral infection. J. Exp. Med. 1999;189:1315–1328. doi: 10.1084/jem.189.8.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Asady R., Yuan R., Liu K., Wang D., Gress R.E., Lucas P.J., Drachenberg C.B., Hadley G.A. TGF-{beta}-dependent CD103 expression by CD8(+) T cells promotes selective destruction of the host intestinal epithelium during graft-versus-host disease. J. Exp. Med. 2005;201:1647–1657. doi: 10.1084/jem.20041044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falvo J.V., Tsytsykova A.V., Goldfeld A.E. Transcriptional control of the TNF gene. Curr. Dir. Autoimmun. 2010;11:27–60. doi: 10.1159/000289196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haring J.S., Harty J.T. Interleukin-18-related genes are induced during the contraction phase but do not play major roles in regulating the dynamics or function of the T-cell response to Listeria monocytogenes infection. Infect. Immun. 2009;77:1894–1903. doi: 10.1128/IAI.01315-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y., Lee Y.T., Kaech S.M., Garvy B., Cauley L.S. Smad4 promotes differentiation of effector and circulating memory CD8 T cells but is dispensable for tissue-resident memory CD8 T cells. J. Immunol. 2015;194:2407–2414. doi: 10.4049/jimmunol.1402369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima N., Iwasaki A. Tissue instruction for migration and retention of TRM cells. Trends Immunol. 2015;36:556–564. doi: 10.1016/j.it.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmeier J.E., Cookenham T., Roberts A.D., Miller S.C., Woodland D.L. Type I interferons regulate cytolytic activity of memory CD8(+) T cells in the lung airways during respiratory virus challenge. Immunity. 2010;33:96–105. doi: 10.1016/j.immuni.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolumam G.A., Thomas S., Thompson L.J., Sprent J., Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med. 2005;202:637–650. doi: 10.1084/jem.20050821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupz A., Guarda G., Gebhardt T., Sander L.E., Short K.R., Diavatopoulos D.A., Wijburg O.L., Cao H., Waithman J.C., Chen W. NLRC4 inflammasomes in dendritic cells regulate noncognate effector function by memory CD8(+) T cells. Nat. Immunol. 2012;13:162–169. doi: 10.1038/ni.2195. [DOI] [PubMed] [Google Scholar]
- Kurd N.S., He Z., Louis T.L., Milner J.J., Omilusik K.D., Jin W., Tsai M.S., Widjaja C.E., Kanbar J.N., Olvera J.G. Early precursors and molecular determinants of tissue-resident memory CD8(+) T lymphocytes revealed by single-cell RNA sequencing. Sci. Immunol. 2020;5:eaaz6894. doi: 10.1126/sciimmunol.aaz6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauvau G., Boutet M., Williams T.M., Chin S.S., Chorro L. Memory CD8(+) T cells: innate-like sensors and orchestrators of protection. Trends Immunol. 2016;37:375–385. doi: 10.1016/j.it.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low J.S., Farsakoglu Y., Amezcua Vesely M.C., Sefik E., Kelly J.B., Harman C.C.D., Jackson R., Shyer J.A., Jiang X., Cauley L.S. Tissue-resident memory T cell reactivation by diverse antigen-presenting cells imparts distinct functional responses. J. Exp. Med. 2020;217:e20192291. doi: 10.1084/jem.20192291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C., Mishra S., Demel E.L., Liu Y., Zhang N. TGF-beta controls the formation of kidney-resident T cells via promoting effector T cell extravasation. J. Immunol. 2017;198:749–756. doi: 10.4049/jimmunol.1601500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay L.K., Braun A., Macleod B.L., Collins N., Tebartz C., Bedoui S., Carbone F.R., Gebhardt T. Cutting edge: CD69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J. Immunol. 2015;194:2059–2063. doi: 10.4049/jimmunol.1402256. [DOI] [PubMed] [Google Scholar]
- Mackay L.K., Minnich M., Kragten N.A., Liao Y., Nota B., Seillet C., Zaid A., Man K., Preston S., Freestone D. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science. 2016;352:459–463. doi: 10.1126/science.aad2035. [DOI] [PubMed] [Google Scholar]
- Mackay L.K., Rahimpour A., Ma J.Z., Collins N., Stock A.T., Hafon M.L., Vega-Ramos J., Lauzurica P., Mueller S.N., Stefanovic T. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 2013;14:1294–1301. doi: 10.1038/ni.2744. [DOI] [PubMed] [Google Scholar]
- Mackay L.K., Wynne-Jones E., Freestone D., Pellicci D.G., Mielke L.A., Newman D.M., Braun A., Masson F., Kallies A., Belz G.T., Carbone F.R. T-box transcription factors combine with the cytokines TGF-beta and IL-15 to control tissue-resident memory T cell fate. Immunity. 2015;43:1101–1111. doi: 10.1016/j.immuni.2015.11.008. [DOI] [PubMed] [Google Scholar]
- Mani V., Bromley S.K., Aijo T., Mora-Buch R., Carrizosa E., Warner R.D., Hamze M., Sen D.R., Chasse A.Y., Lorant A. Migratory DCs activate TGF-beta to precondition naive CD8(+) T cells for tissue-resident memory fate. Science. 2019;366:eaav5728. doi: 10.1126/science.aav5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masopust D., Choo D., Vezys V., Wherry E.J., Duraiswamy J., Akondy R., Wang J., Casey K.A., Barber D.L., Kawamura K.S. Dynamic T cell migration program provides resident memory within intestinal epithelium. J. Exp. Med. 2010;207:553–564. doi: 10.1084/jem.20090858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner J.J., Toma C., He Z., Kurd N.S., Nguyen Q.P., McDonald B., Quezada L., Widjaja C.E., Witherden D.A., Crowl J.T. Heterogenous populations of tissue-resident CD8(+) T cells are generated in response to infection and malignancy. Immunity. 2020;52:808–824.e7. doi: 10.1016/j.immuni.2020.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner J.J., Toma C., Yu B., Zhang K., Omilusik K., Phan A.T., Wang D., Getzler A.J., Nguyen T., Crotty S. Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature. 2017;552:253–257. doi: 10.1038/nature24993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller S.N., Gebhardt T., Carbone F.R., Heath W.R. Memory T cell subsets, migration patterns, and tissue residence. Annu. Rev. Immunol. 2013;31:137–161. doi: 10.1146/annurev-immunol-032712-095954. [DOI] [PubMed] [Google Scholar]
- Mueller S.N., Mackay L.K. Tissue-resident memory T cells: local specialists in immune defence. Nat. Rev. Immunol. 2016;16:79–89. doi: 10.1038/nri.2015.3. [DOI] [PubMed] [Google Scholar]
- Park C.O., Kupper T.S. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat. Med. 2015;21:688–697. doi: 10.1038/nm.3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.L., Zaid A., Hor J.L., Christo S.N., Prier J.E., Davies B., Alexandre Y.O., Gregory J.L., Russell T.A., Gebhardt T. Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat. Immunol. 2018;19:183–191. doi: 10.1038/s41590-017-0027-5. [DOI] [PubMed] [Google Scholar]
- Pizzolla A., Nguyen T.H.O., Smith J.M., Brooks A.G., Kedzieska K., Heath W.R., Reading P.C., Wakim L.M. Resident memory CD8(+) T cells in the upper respiratory tract prevent pulmonary influenza virus infection. Sci. Immunol. 2017;2:eaam6970. doi: 10.1126/sciimmunol.aam6970. [DOI] [PubMed] [Google Scholar]
- Raue H.P., Beadling C., Haun J., Slifka M.K. Cytokine-mediated programmed proliferation of virus-specific CD8(+) memory T cells. Immunity. 2013;38:131–139. doi: 10.1016/j.immuni.2012.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richer M.J., Pewe L.L., Hancox L.S., Hartwig S.M., Varga S.M., Harty J.T. Inflammatory IL-15 is required for optimal memory T cell responses. J. Clin. Invest. 2015;125:3477–3490. doi: 10.1172/JCI81261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz A.L., Soudja S.M., Deceneux C., Lauvau G., Marie J.C. NK1.1+ CD8+ T cells escape TGF-beta control and contribute to early microbial pathogen response. Nat. Commun. 2014;5:5150. doi: 10.1038/ncomms6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenkel J.M., Masopust D. Tissue-resident memory T cells. Immunity. 2014;41:886–897. doi: 10.1016/j.immuni.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan B.S., Pham Q.M., Lee Y.T., Cauley L.S., Puddington L., Lefrancois L. Oral infection drives a distinct population of intestinal resident memory CD8(+) T cells with enhanced protective function. Immunity. 2014;40:747–757. doi: 10.1016/j.immuni.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skon C.N., Lee J.Y., Anderson K.G., Masopust D., Hogquist K.A., Jameson S.C. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol. 2013;14:1285–1293. doi: 10.1038/ni.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slutter B., Van Braeckel-Budimir N., Abboud G., Varga S.M., Salek-Ardakani S., Harty J.T. Dynamics of influenza-induced lung-resident memory T cells underlie waning heterosubtypic immunity. Sci. Immunol. 2017;2:eaag2031. doi: 10.1126/sciimmunol.aag2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soudja S.M., Ruiz A.L., Marie J.C., Lauvau G. Inflammatory monocytes activate memory CD8(+) T and innate NK lymphocytes independent of cognate antigen during microbial pathogen invasion. Immunity. 2012;37:549–562. doi: 10.1016/j.immuni.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinert E.M., Schenkel J.M., Fraser K.A., Beura L.K., Manlove L.S., Igyarto B.Z., Southern P.J., Masopust D. Quantifying memory CD8 T cells reveals regionalization of immunosurveillance. Cell. 2015;161:737–749. doi: 10.1016/j.cell.2015.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thom J.T., Weber T.C., Walton S.M., Torti N., Oxenius A. The salivary gland acts as a sink for tissue-resident memory CD8(+) T cells, facilitating protection from local cytomegalovirus infection. Cell Rep. 2015;13:1125–1136. doi: 10.1016/j.celrep.2015.09.082. [DOI] [PubMed] [Google Scholar]
- Thome J.J., Farber D.L. Emerging concepts in tissue-resident T cells: lessons from humans. Trends Immunol. 2015;36:428–435. doi: 10.1016/j.it.2015.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams M.A., Bevan M.J. Effector and memory CTL differentiation. Annu. Rev. Immunol. 2007;25:171–192. doi: 10.1146/annurev.immunol.25.022106.141548. [DOI] [PubMed] [Google Scholar]
- Wu J., Madi A., Mieg A., Hotz-Wagenblatt A., Weisshaar N., Ma S., Mohr K., Schlimbach T., Hering M., Borgers H., Cui G. T cell factor 1 suppresses CD103+ lung tissue-resident memory T cell Development. Cell Rep. 2020;31:107484. doi: 10.1016/j.celrep.2020.03.048. [DOI] [PubMed] [Google Scholar]
- Zhang N., Bevan M.J. TGF-beta signaling to T cells inhibits autoimmunity during lymphopenia-driven proliferation. Nat. Immunol. 2012;13:667–673. doi: 10.1038/ni.2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N., Bevan M.J. Transforming growth factor-beta signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity. 2013;39:687–696. doi: 10.1016/j.immuni.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Yu S., Zhao D.M., Harty J.T., Badovinac V.P., Xue H.H. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity. 2010;33:229–240. doi: 10.1016/j.immuni.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original RNA-seq results can be accessed by GSE111801. The raw data supporting the current study are available from the Lead Contact upon request. All software is commercially available.