

Abstract
Alzheimer’s disease (AD) is the most common neurodegenerative disease affecting more than 50 million people worldwide. The pathology of this multifactorial disease is primarily characterized by the formation of amyloid-β (Aβ) aggregates; however, other etiological factors including metal dyshomeostasis, specifically copper (Cu), zinc (Zn), and iron (Fe), play critical role in disease progression. Because these transition metal ions are important for cellular function, their imbalance can cause oxidative stress that leads to cellular death and eventual cognitive decay. Importantly, these transition metal ions can interact with the amyloid-β protein precursor (AβPP) and Aβ42 peptide, affecting Aβ aggregation and increasing its neurotoxicity. Considering how metal dyshomeostasis may substantially contribute to AD, this review discusses polyphenols and the underlying chemical principles that may enable them to act as natural chelators. Furthermore, polyphenols have various therapeutic effects, including antioxidant activity, metal chelation, mitochondrial function, and anti-amyloidogenic activity. These combined therapeutic effects of polyphenols make them strong candidates for a moderate chelation-based therapy for AD.
Keywords: Alzheimer’s disease, amyloid-β, metalloproteins, copper, zinc, iron, polyphenols, metal chelation therapy
INTRODUCTION
Neurodegenerative diseases (NDDs) are a consequence of progressive neuronal death which are typically associated with the accumulation of protein aggregates in the central nervous system (CNS), and eventually leading to loss of cognitive or motor function [1–3]. Although most NDDs are sporadic, approximately 10–20% of cases are caused by inherited mutations that sensitize carriers exposed to environmental factors [4, 5]. Alzheimer’s disease (AD) is the most common NDD, followed by Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD), multiple sclerosis (MS), and prion diseases [6–13]. The etiology of NDDs is closely related to aging, but other factors are also involved, such as genomic instability, de novo and somatic mutations, epigenetic alterations, and environmental factors that include stressful situations, infections, and poor nutrition [4, 14]. NDDs may occur at specific sites or simultaneously in multiple regions of the brain [15, 16]. For example, the cerebral cortex and hippocampus are the two primary brain regions affected by neuronal loss [17]. In AD, neuronal loss mainly occurs in the nucleus basalis; in PD, the substantia nigra and striatum are the most affected by neuronal loss [15]. ALS usually affect the upper brain and lower spinal cord, whereas HD severely affects the striatal medium spiny neurons [15]. Although MS is generally associated with cortical demyelination, neuronal loss is reported in the basal ganglia, cerebellar cortex, spinal gray matter, and thalamus. In contrast, prion diseases can affect large areas of the brain, regardless of the origin of the prion [12–16].
AD is a devastating NDD that affects more than 50 million people worldwide, and its prevalence among the population between 65 and 85 years of age is estimated between 0.6% to 8.4% [18]. Furthermore, the 2018 World Alzheimer Report estimates that the number of cases will triple by 2050 [19, 20]. The main histological features that confirm AD are extracellular deposits of Aβ fibrils and intracellular neurofibrillary tangles (NFTs) [21]. The extracellular fibrils are composed of Aβ42 peptides with a characteristic β-sheet structure, whereas NFTs are composed of phosphorylated tau protein [21, 22]. Other characteristic features of AD are the synaptic damage, mitochondrial dysfunction [23, 24], inflammation [25], and oxidation of biomolecules, and metal dyshomeostasis of calcium (Ca), copper (Cu), zinc (Zn), and iron (Fe) [22]. AD is identified by progressive memory loss, cognitive impairment, early neurovascular changes, accumulation of Aβ42 peptide, tau protein tangles, and neuronal death [26, 27]. The etiology and pathology of AD is complex and multifactorial, where exogenous and endogenous factors can trigger the disorder [19]. Exogenous factors that may contribute to AD include lifestyle factors, including smoking, dietary deficiencies, and mental or physical inactivity. Chronic diseases, such as diabetes, obesity, and cardiovascular diseases are also linked to AD [28, 29]. Endogenous factors such as oxidative stress, metal ion dyshomeostasis, extra- and intracellular Aβ42 peptide concentration, and intracellular tau protein phosphorylation are related to AD progression [19].
There is evidence that essential metals, such as Cu, Zn, and Fe, are dysregulated in AD [30]. Of these biologically relevant transition metals, Cu receives the most attention because it interacts with both amyloid-β protein precursor (AβPP) and Aβ42 peptide [31]. Furthermore, non-essential neurotoxic metals, such as aluminum (Al), lead (Pb), cadmium (Cd), and mercury (Hg), may have a role in triggering AD [31–33]. Cu and Fe both increase the production of reactive oxygen species (ROS), causing neuronal death and subsequent cognitive defects [4]. Zn modulates Cu binding to Aβ42 peptide, thus indirectly affecting ROS production. Additionally, AβPP and Aβ42 peptide are metalloproteins that have the ability to sequester metal ions, and the resulting complex can increase the redox toxicity and induce Aβ aggregation [30–33]. Unbound Cu and Fe ions can also react with hydrogen peroxide (H2O2) and generate hydroxyl radicals (HO•) through a Fenton reaction mechanism, thus causing oxidative stress to cells [4, 34–39]. For these reasons, we speculate that metal imbalance is closely linked to AD pathogenesis. A major challenge to discovering new drugs against AD is the lack of an animal model that reflects the entirety of AD pathology, as well as the lack of biomarker specificity [40, 41]. There is an urgent need to discover new drugs that can prevent or halt AD progression.
Investigations of AD etiology have uncovered many factors that contribute to AD progression. Due to the complexity of AD contributing factors, there are many promising multifunctional compounds for targeting these different pathological components in a parallel fashion [42]. Thus, the interest in natural products, specifically the large family of polyphenols, is based on current investigations that demonstrate therapeutic properties such as antioxidant, anti-amyloidogenic activity, cell signaling modulation, and metal chelation activity (Fig. 1) [43–45]. Polyphenols have also been demonstrated to have neuroprotective activity against AD [1, 46], PD [24, 47–50], HD [51, 52], and ALS [53, 54]. Metal chelation is a promising avenue for research due to increasing evidence of metal dyshomeostasis in AD; however, massive chelation as an approach may not be favorable because it could disrupt normal metalloproteins activity [45, 55]. Alternatively, chelation could be mediated by molecules with moderate yet specific affinities for targeted transition metals, thus reducing the neurotoxic effects caused by interaction between the metals and Aβ42 peptide [56, 57]. Such classes of emerging compounds are referred to as metal-protein attenuating compounds (MPACs) [55–58]. This review will focus on the chelating properties of polyphenols that are natural sources of potential MPACs and discuss their potential therapeutic effects in mitigating metal ion dyshomeostasis in a moderate chelation treatment for AD. We are focused on Cu, Zn, and Fe because these d-block transition metals are essential to humans, relatively abundant in the CNS, and involved in AD triggering.
Fig. 1.
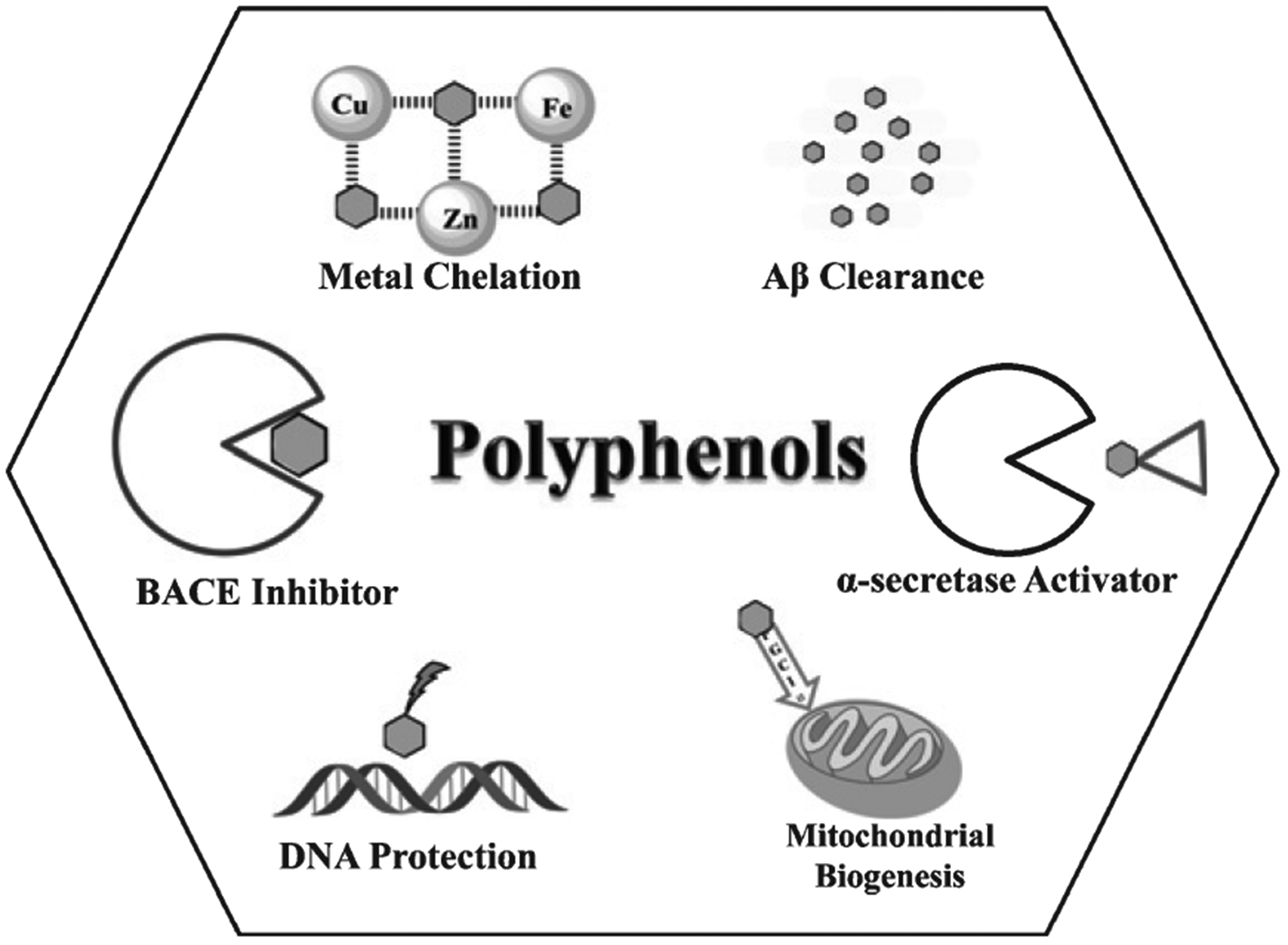
Potential therapeutic effects of polyphenols. Metal chelation occurs via coordination with oxophilic transition metals, such as Cu, Zn, and Fe, which are abundant in the CNS. Transition metals can induce ROS, which damage the genome. Therefore, polyphenols have DNA protection activity. Polyphenols modulate the AβPP pathway in two different ways. One pathway activates α-secretase by removing the pro-domain, which induces the non-amyloidogenic pathway. A second pathway favors neuroprotection, wherein polyphenols inhibit the β-site amyloid protein precursor cleaving enzyme 1 (BACE1), leading to decreased Aβ concentration. Polyphenols also promote Aβ clearance through non-covalent interactions with amino acid residues that disrupt Aβ structure stability, thus leading to fibril disaggregation. Polyphenols can also induce mitochondrial biogenesis by stimulating PGC-1α.
LEVELS OF TRANSITION METAL IONS IN THE BRAIN
Transition metal ions with accessible d orbitals are necessary for critical biological processes, including oxygen transport, oxygen activation, neurotransmitter synthesis, DNA replication and transcription, cell-cell interactions, and extracellular matrix construction and demolition [59–62]. In these processes, metal ions are structural and metabolic cofactors, as well as electron transporters. Metalloproteins such as ceruloplasmin, superoxide dismutase, hemoglobin, and cobalamin contain the transition metals Cu, Zn, Fe, cobalt (Co), and manganese (Mn) [30, 62, 63]. Exposure to environmental metals such as Mn, Cu, Zn, Fe, Pb, Hg, and Al, is a risk factor for NDDs [64, 65]. For example, excess Fe and inhalation of Mn dust are linked to PD [66]. Also, essential metals (Cu, Zn, and Fe) and non-essential metals (Al) are major contributors to AD and PD pathophysiology [37, 66]. In AD brains, Cu concentrations are as high as 400 μM and Zn concentrations reach 1 mM, which are much higher than the acknowledged healthy concentrations of Cu (70 μM) and Zn (350 μM) [37, 67]. Both Cu and Zn metabolism are regulated by metallothioneins and an antagonistic relationship may exist between Cu and Zn [17, 33]. In AD, metallothionein I and II are upregulated, further suggesting an inverse relationship between Cu and Zn. This phenomenon is most notable in the metal-rich hippocampus and amygdala. In AD, transition metal ions, notably Cu, Zn, and Fe, first accumulate on Aβ fibrils. Moreover, these metals have roles in both Aβ fibril and tautangles aggregation that may be attributed to their oxophilic character. Because d-block transition metal ions have neurodegenerative effects on the brain, their role in AD is of high interest [17, 68–70].
Copper levels
Cu(II) has a d9 electron configuration, which distorts the octahedral orientation in a Jahn-Teller manner and stabilizes square planar formation [17, 71]. Protein binding distorts the configuration of Cu(II) complexes in a way that is important for regulating Cu catalysis both in cells and in the extracellular space. In the body, extracellular Cu(II) is attached to cysteine (Cys) and histidine (His) sidechains. Inside cells under normal conditions, Cu is almost in the reduced form Cu(I) and tightly bound to proteins [72]. Because Cu is a redox active transition metal ion involved in dioxygen and peroxide activation, its role in producing and clearing ROS in the CNS is not surprising [73, 74]. Indeed, in AD pathogenesis, light metal ions, such as Fe and Zn, compete for the active and transporter sites of concentrated Cu ions [75, 76].
Cu transporter 1 (CTR1) and Cu ATPases (ATP7A and ATP7B) are responsible for intracellular Cu(I) regulation, whereas the divalent metal transporter 1 (DMT1) is involved in regulating Cu(II) [77]. Healthy serum levels of Cu range between 1020–1050 μg/L [70], whereas serum levels of unbound Cu in AD patients tend to be higher; these higher Cu serum levels are attributed to disrupted ion transport [78]. Excess Cu produces abnormal behavior involved in the development of NDDs. Menkes and Wilson’s diseases are examples of how disruption of metal ion homeostasis can be detrimental [66]. In Menkes disease, a mutation in ATP7A causes Cu deficiency in the cerebellum region [79]. In contrast, Wilson’s disease occurs when an ATP7B mutation causes excess Cu in the basal ganglia area, leading to Cu toxicity [79–81]. Nonetheless, both disorders are congenital and of particular research interest because these were the first NDDs linked to Cu metabolism.
Cu is frequently studied because AD manifests in both disruption regions of the metal ion, but primarily in cortical gray matter where Aβ fibrils and neurofibrillary tangles are found [69]. During short-term alterations of Cu levels, metal-binding proteins, such as metallothionein and ceruloplasmin, act as buffers. However, during a long-term increase of Cu concentration, metal binding proteins are unregulated to prevent further toxicity. After a critical concentration is reached, free circulating Cu levels increase. Additionally, when Cu is bound to smaller proteins, it can cross the blood-brain barrier (BBB) and cause neurotoxic effects such as Aβ42 aggregation and also change DNA conformation. DNA conformation changes may be caused by interactions between the Aβ-Cu complex and trinucleotides CAG and CTG, which disrupt the B conformation of DNA and create cellular instability [82].
Zinc levels
Zn(II) has a closed shell d10 diamagnetic electron configuration, similar to Cu(I) [83, 84]. However, the ionization energy of Zn(II) is too large to reach oxidized states. Zn usually exists in tetrahedral or octahedral orientations, but also forms pentagonal complexes with bipyramidal trigonal and square pyramidal configurations. Because of its closed d shell, Zn(II) has a high positive charge density, even compared to Ca(II). This property allows Zn(II) to bind to proteins and nucleic acids, acting as an important structural component in many proteins and as a co-factor for more than 300 enzymes [85]. Zn(II) contributes to regulating cellular processes and signaling pathways because a high positive charge makes Zn(II) a strong Lewis acid, particularly in hydrolysis reactions. In the brain, Zn(II) is found within synaptic vesicles and glutamatergic terminals. Similar to Cu, Zn(II) dysregulation causes neurotoxic effects, including DNA nicking and affecting Aβ42 aggregation [40, 85]. Zn(II) homeostasis is regulated by three different groups of macromolecules: Zn transporters, Zn-regulated, and Fe-regulated transporter proteins, and metallothioneins [77, 85]. Zn metallothioneins have four isoforms; of these, metallothionein III is brain specific and is found with Zn in astrocytes, neurons, and synaptic vesicles. Healthy serum levels of Zn range between 720–899 μg/L [70, 86]. However, a meta-analysis of Zn serum levels in 1027 AD patients and 1949 healthy people as controls found discordant results. Nonetheless, the analysis concluded that Zn levels are lower in patients with AD [69] and other meta-analyses have reached the same conclusion [31, 86, 87].
Iron levels
Like Cu, Fe has two major oxidation states in the body, Fe(II) and Fe(III). Cycling between the two oxidation states occurs easily because they differ by a single electron. Fe and Cu are Fenton catalysts affecting ROS levels [88–91]. In cells, the reduced form of Fe is usually stabilized by heme group binding and formation of a distorted octahedron by two axial ligands [59, 60, 92]. Fe has many electron spin states and several different configurations, including tetrahedral; however, an octahedral shape is common for both oxidation states [91]. An oxidation-reduction state can cause neurotoxic effects, such as affecting the production, folding, and aggregation of proteins, but, when this property holds, transition metal ions have essential roles in DNA synthesis, neurotransmission, and oxygen transport. Transition metal ions also act as cofactors for key enzymes in the biosynthesis of neurotransmitters, such as dopamine and noradrenaline [39, 88]. The controlled transport of Fe is mediated by proteins that undergo a series of oxidations and reductions. For instance, ceruloplasmin oxidation is required for Fe transport by transferrin. Other proteins, such as DMT1 and ferroportin, are involved in Fe transport, whereas ferritin controls Fe storage. The accumulation of Fe in neurodegenerative sites may be attributed to altered levels of ferritin and transferrin that decrease export of metal ions from cells [86, 93]. Ferroptosis, an apoptotic pathway related to increased Fe concentration in cells, is triggered regardless of ROS production and is linked to neurodegeneration [58, 65]. Additionally, meta-analyses have shown lower Fe serum levels in AD patients than in healthy counterparts, which range between 633–2444.4 μg/L [69, 86].
ALZHEIMER’S DISEASE AND TRANSITION METAL IONS IN THE BRAIN
AβPP is a multidomain transmembrane glycoprotein and the most common isoforms have 695, 751, and 770 amino acid (aa) residues [89, 94]. AβPP has two Cu binding sites, one between residues 124 and 189, and another at the E2 domain between residues 376 and 554. The first domain is formed by two histidine residues (His147 and His151), one tyrosine residue (Tyr168), and two water molecules. Together these form a pentagonal complex with Cu(II) in a non-planar orientation, favoring reduction to Cu(I) (Fig. 2a)[95]. After Cu reduction, a disulfide bridge forms between neighboring cysteine residues (Cys144 and Cys158) [36]. The second domain is formed by four histidine residues (His388, His457, His507, and His511). The tetra-coordinated complex with Cu(II) distorts the square planar geometry, leading to a structural change in AβPP (Fig. 2b)[96]. AβPP can be cleaved by α-, β-, and γ-secretases, and cleaving leads to two pathways: non-amyloidogenic and amyloidogenic [97]. In the first pathway, AβPP is cleaved in the middle of the Aβ sequence region by α-secretase (a disintegrin and metalloproteinase) at Lys687, resulting in a secreted AβPPα fragment (sAβPPα) and a C-terminal fragment (CTFα) [98]. Further cleaving of CTFα by γ-secretase leads to formation of protein 3 (p3), a soluble and neuroprotective peptide [99, 100]. In the second pathway, AβPP is cleaved past Met671 by β-secretase, forming a secreted AβPPβ fragment (sAβPPβ) and a C-terminal fragment (CTFβ) [101]. CTFβ is then cleaved by γ-secretase producing the Aβ42 peptide in one of two isoforms, Aβ40 or Aβ42, and Aβ42 aggregates at a higher rate [102]. Recently, the composition of Aβ fibrils has been revised, so that the recognized percentage of N-truncated Aβ42 peptides is increased [103, 104].
Fig. 2.
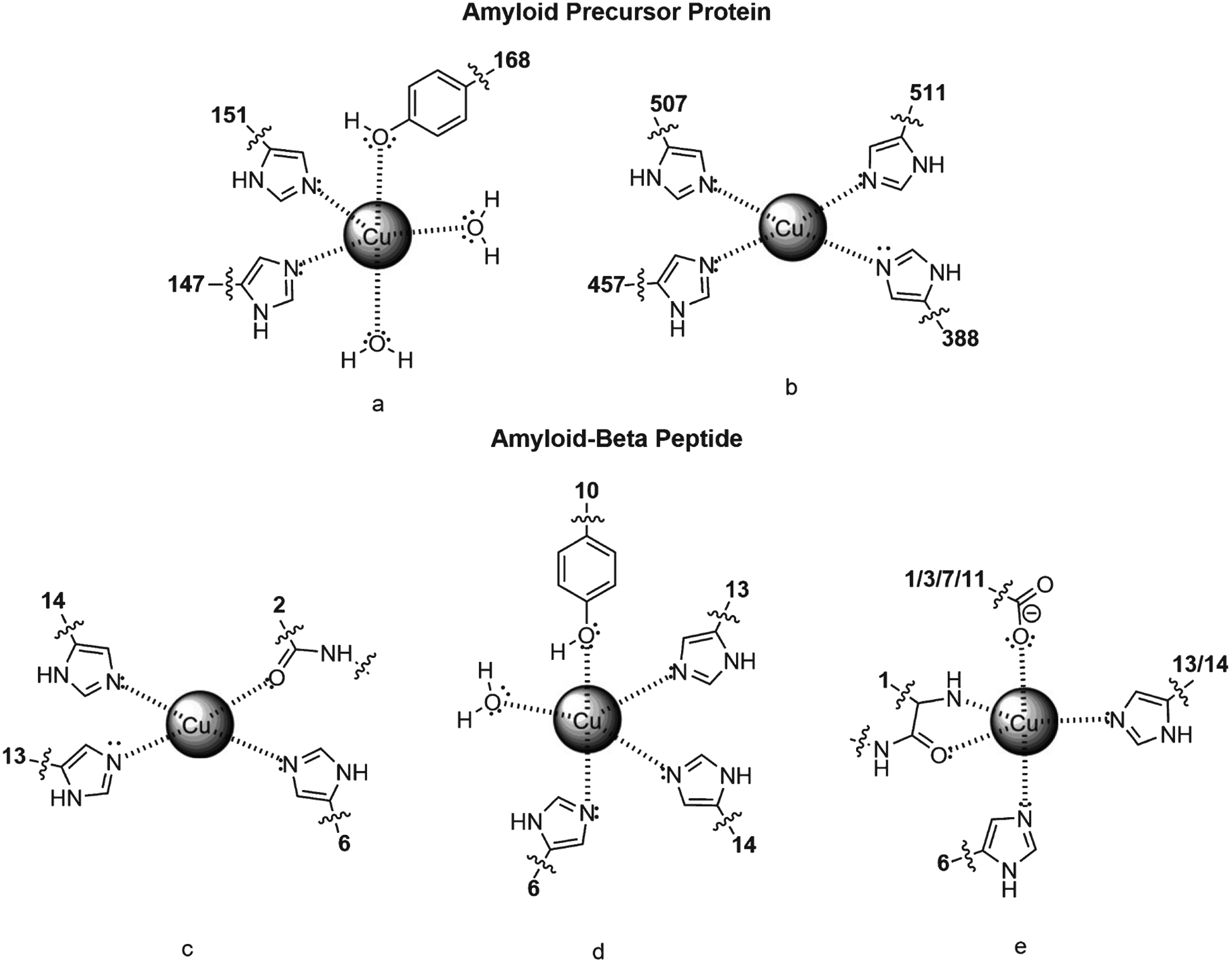
Cu binding interactions with AβPP and Aβ42. a) Configuration of Cu with residues His147, His151, Tyr168 in APP’s copper binding region and two water molecules in a trigonal bipyramidal geometry. b) Configuration of Cu with residues His388, His457, His507, and His511 in APP E2 domain. c) Cu binding with Aβ42 residues Ala2, His6, His13, and His14 in a tetrahedral square planar configuration. d) Cu binding to Aβ42 residues His6, Tyr10, His13, His14, and a water molecule in a trigonal bipyramidal configuration. e) Square-base pyramidal arrangement of Cu with Aβ42 residues Asp1, His6, His 13 or 14, and the carboxylate of Asp1, Glu3, Asp7, or Glu11.
AβPP and Aβ42 peptide could be considered metalloproteins because they sequester transition metal ions, as shown by nuclear magnetic resonance (NMR), electron spin resonance (ESR), and crystallography results [36, 105, 106]. When Aβ42 peptide is formed, a new Cu binding site is created with His6, His13, and His14 residues and the controversial keto-moieties of Ala2. At this binding site, Cu binding has a square planar orientation (Fig. 2c) [107, 108]. Another proposed binding orientation based on X-ray absorption spectroscopy (XAS) indicated the existence of a five-coordinated complex formed by three histidine residues (His6, His13, His14), one tyrosine (Tyr10), and one water molecule (Fig. 2d) [109–111]. Finally, IR spectroscopy suggests that carboxylate groups of aspartate (Asp1 or Asp7) or glutamate (Glu3 or Glu11) interact with transition metals instead of Tyr10 (Fig. 2e) [112, 113]. Because the Aβ42 ligand is disordered, the high valence Cu(II) orientation is highly dynamic, which has important implications for the mechanism of redox cycling and Cu reactivity [114, 115].
Aβ42 peptides have four basic interactions, three of which correspond to the imidazol ring of His6, His13, and His14 [64]. The fourth interaction is based on the nucleophilic (or Lewis base) character and size of the residue, which increases the possibility of interacting with a metal. The phenol group of Tyr10 is nucleophilic and larger than the carboxylate groups of either aspartate (Asp1 or Asp7) or glutamate (Glu3 or Glu11) residues, allowing it to interact with the metal. The hydroxyl group of Tyr10 (pKa~10) could become deprotonated by water due to interaction with the metal ion or from hydrogen bonds with neighboring residues. For these reasons, Tyr10 has a greater probability of interacting with the metal than as part ate (Asp1 or Asp7) and glutamate (Glu3 or Glu11) residues (Fig. 2d). However, interaction with metal ions increases the acidity of amide groups, which compete as ligands for the metal ion [92]. Although more complex stoichiometry is reported, it is generally accepted that the stoichiometry of Aβ40/42–Cu2+ complexes is generally a 1 : 1 molar ratio [30, 116].
According to the amyloid-β hypothesis of AD, the Aβ42 peptide is implicated in AD progression, such that an imbalance between Aβ42 production and clearance triggers AD pathogenesis in the human brain [21, 117]. This hypothesis is supported by experimental results showing that mutations in genes encoding for AβPP, presenilin 1 or 2 (PS1 or PS2), or Apolipoprotein E allele 4 (APOEε4), lead to accumulation of Aβ42 in the brain [118–120]. Aβ42 aggregation in the brain is considered the hallmark of AD [119, 121]. Aβ42 fibril formation is a multistep process that starts with monomers, followed by oligomers, protofibrils, and eventually mature fibrils [21, 122, 123]. Furthermore, Aβ42 aggregation induced by transition metal ions produce ROS such as H2O2 [124]. Soluble oligomers are considered neurotoxic because they can induce dyshomeostasis, oxidative injury, inflammation, and the development of tau protein tangles [15, 40, 125].
It is important to understand the close relationship between oxidation-reduction cycles and transition metal ion activity in Aβ42 aggregation. Here, we focus on how transition metal ions affect Aβ42 aggregation and ROS production by forming an Aβ-metal complex.
Effects of transition metal ions on Aβ42 aggregation
Transition metal ions may promote aggregation of Aβ42 peptide [126]. Surface plasmon resonance experiments show that the aggregation constant ka increases when Cu(II), Zn(II), and Fe(III) are added to growth media [127, 128]. Of the three transition metal ions, Cu(II) addition resulted in the largest ka increase(13.6-fold) compared to Aβ42 aggregation alone. Moreover, Cu(II) may preferentially bind monomers and small oligomers of the peptide, whereas Zn(II) promotes formation of larger aggregates. Thus, Cu(II) and Zn(II) may have different mechanisms to induce aggregation. Zn(II) also induces aggregation of Aβ42 peptides by promoting a conformation change from the α-helix intermediate to a larger β-sheet structure [129]. In contrast, Cu(II) may increase toxicity by stabilizing intermediate aggregates [116].
Aβ42 monomers have a mostly random-coil structure that evolves into β-sheet structure as the fibrillization progresses [130]. However, transition metal ions affect peptide aggregation by changing Aβ42 conformation and interpeptide connectivity. For example, Cu catalyzes dimer formation via a dityrosine product of radical chemistry [111, 129, 131, 132]. Moreover, NMR spectroscopy and surface plasmon resonance experiments have shown that intermolecular complexes form via Zn(II)-induced oligomerization [133, 134]. These studies indicate that His6, His13, and aa residues 11–14 are the crucial residues involved in forming the seeding dimer. Furthermore, both Zn and Cu induce amorphous aggregation while inhibiting formation of β-sheets [116, 129, 135]. This amorphous quality is confirmed by chromatographic and NMR studies, which also suggest that the Aβ-Cu/Zn building block is monomeric during oligomerization and subsequent metal ion reshuffling [129]. In addition to amorphous aggregates, a granular has been identified only in the presence of metal ions [136]. Because Aβ42 is a disordered protein, it is affected by changes in the population of thermally accessible Aβ42 conformers in disordered chains. These population changes are induced by interactions with transition metal ions or by the chain environment [137, 138]. This issue has been investigated for Aβ42 using MS and solid state NMR [139, 140].
ROS production by Aβ-metal complex interaction
Oxidative stress caused by Fenton and Haber-Weiss reactions produces ROS and reactive nitrogen species that play a key role in degenerative diseases [141]. Transition metal ions catalyze reactions that produce radicals that damage DNA, proteins, and lipids. In AD, hydroxyl radicals produced by Fe(III) cause Aβ42 neurotoxicity and turn soluble fibrinogen into insoluble aggregates [141]. There are higher concentrations of Fe in AD plaques (1 mM) than in healthy brain tissue (350 μM). Also, Fe affects Aβ42 aggregation and tau phosphorylation [141]. In ROS chemistry, the hydroxyl radical is the most aggressive [142–144]. Hydroxyl radicals are formed when each one-electron transfer step is facilitated by a single electron reductant like ascorbate, and an ion like Cu or Femediates one-electron transfer, with the Fenton reaction as the last step (Fig. 3).
Fig. 3.
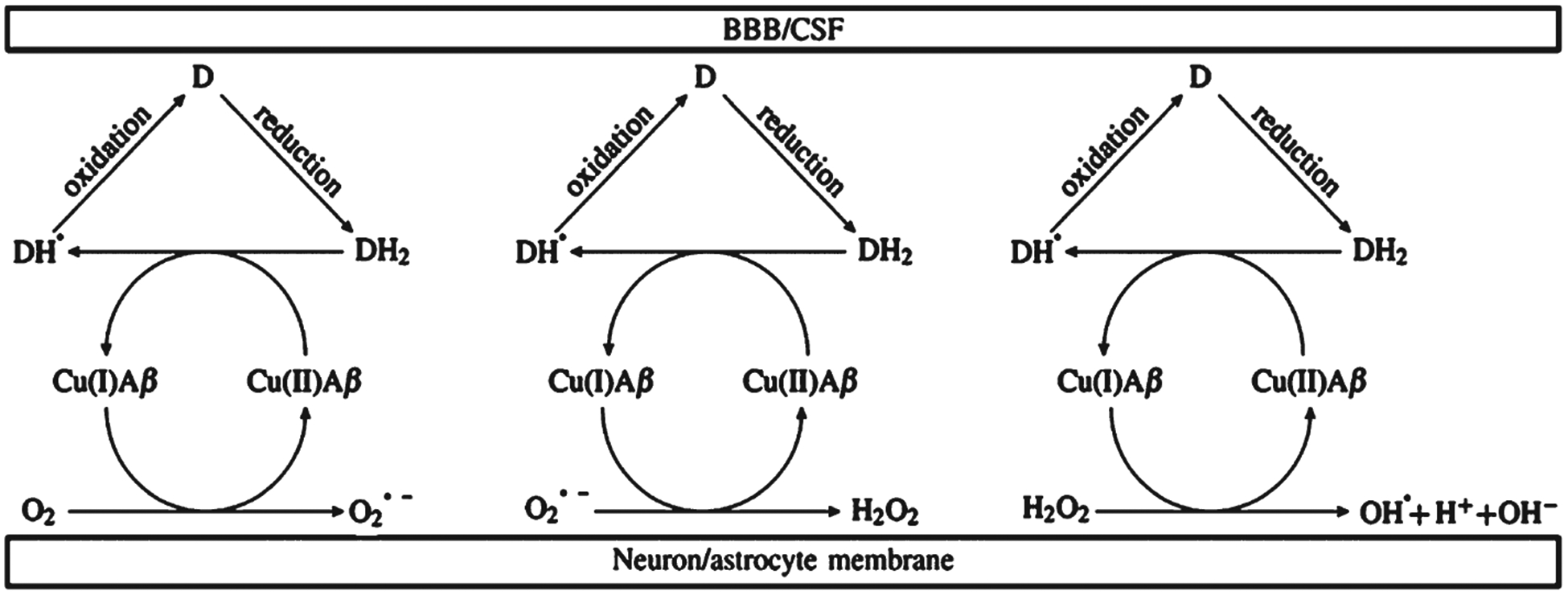
One-electron transfer cycle during ROS production within the synaptic cleft. DH2 is ascorbic acid and D is dehydroascorbic acid. The blood-brain barrier (BBB) is in contact with cerebrospinal fluid (CSF).
Dioxygen is formed by splitting the triplet state of dioxygen into two radicals (two doublet species) [115]. This transient species is a Cu(I) complex that is produced in small amounts by reducing molecules, much like Fe(II) species in Fenton chemistry [143]. Splitting produces Cu(II) and superoxide, both in doublet spin states. Reducing agents are abundant in cells and synapses [145]. The most representative reducing agent is ascorbic acid, which is chemically similar to polyphenols, but characterized by greater solubility in water and a lower first dissociation pKa constant. Ascorbic acid (DH2) at pH~7 is almost completely dissociated into ascorbate (DH−) and one-electron oxidation produces an ascorbyl radical (DH•). DH is easily oxidized into dehydroascorbic acid, the 2-electron oxidized form of ascorbic acid (D), which is actively transported through BBB [145].
The first step in Fig. 3 is the inverted initial part of a Haber-Weiss reaction, where oxidation of Fe(II) to Fe(III) is mediated by an initial amount of superoxide [141]. The direction of this step depends on the amount of reducing agent (ascorbate is more abundant in the synapse than superoxide) and on binding between the ligand and transition metal ion. Notably, high valence transition metal ions, Cu(II) or Fe(III), can be destabilized by geometrical distortions when the ligand binding is not rigid. Indeed, Aβ is an intrinsically disordered peptide. The pro-oxidant role of Aβ-Cu(II) is based on Cu binding to Aβ because this phenomenon catalyzes ROS production [131]. Aβ-Cu(II) intermediates produce more hydroxyl radicals (OH•) and H2O2 than monomeric Aβ-Cu(II), supporting the hypothesis that aggregation intermediates are more toxic than fibrils [126, 129]. Comparison of ROS production kinetics of the Aβ-Cu(II) complex in the presence of ascorbate with those of physiologically normal Cu complexes, such as those formed with aa, albumin, and metallothionenin III, showed that Aβ-Cu(II) produced more hydroxyl radicals than the other complexes [146, 147]. Similarly, studies comparing the production of H2O2 by Aβ42 monomers, oligomers, and fibrils, in the presence of Cu(II), showed that oligomers produced more ROS than the other two forms, supporting the hypothesis that oligomers are the main source of ROS generation [148]. The first step in ROS production catalyzed by Aβ-Cu is the in situ reduction of dioxygen to super-oxide (O2•−) [115, 149–151]. Superoxide rapidly attacks other sites in the Aβ42 peptide and the environment, including sites that stabilize pre-organized oligomers by forming covalent bonds [132, 147, 152]. Thus, the oxidative damage in AD may be due to prooxidant activity of Aβ-Cu(II) complexes. ROS effects include the oxidation of the Aβ42 peptide, including decarboxylation and deamination of Asp1, oxidation of histidine and methionine, and cleavage of the main Aβ42 chain [111, 131, 153].
Characterization of the Aβ-Fe(II/III) complex is more complicated. In vitro experiments show that in the reduced form, Fe binds the Aβ42 monomer in a 1 : 1 ratio, but oxidized Fe becomes intercalated with monomers, indicating an extended intermolecular Fe-mediated interaction network [141, 153, 154]. Indeed, these results are consistent with in vivo observations of nanostructured FeO particles that co-localized with Aβ42 aggregates [155]. Zn(II) is assumed to be neuroprotective, compared to Cu(II) and Fe(III), due to its displacement effect on redox-active metal ions. Indeed, in contrast to redox active metal ions, Zn(II) decreases neurotoxic effects in cell culture [111]. A proposed mechanism for the neuroprotective effects of Zn(II) is a metal swap between a Zn(II)-loaded metallothionein III and the Aβ-Cu(II) complex, thus reducing ROS production. However, a reported down-regulation of metallothionein III in AD would prevent the metal swap and increase neurotoxic effects of the Aβ-Cu(II) complex [129].
METAL CHELATION THERAPY
Metal chelation is a prominent therapy for dyshomeostasis of transition metal ions in the brain and could be used to prevent production of toxic Aβ42 species and ROS [156]. Transition metal ions in free form are highly toxic and usually tightly bound to proteins; therefore, chelation is an essential therapy to control metal ion levels during conditions where metals are more likely to be released from proteins [33, 157]. Chelators have a high binding affinity for metals, and metal chelation therapy is used for heavy metal poisoning and diseases caused by an excess of metal ions [158, 159]. However, because chelation therapy has low specificity and many side effects, few chelating compounds are approved by the FDA (Fig. 4), [160, 161]. Dithiol BAL (British Anti-Lewisite) was developed during World War II and is used to treat arsenic, mercury, and lead poisoning [162]. Similarly, calcium-EDTA is only used for heavy metal poisoning, and is used in conjunction with BAL for acute lead poisoning [160]. However, because BAL has hypertension, nausea, and vomiting as side effects, synthetic derivatives with less severe side effects have been developed [163]. DMSA, better known as succimer, is a BAL-derivative approved for treating lead poisoning in children [160]. The main advantage of succimeris its reduced toxicity, which makes it suitable for long-term administration and able to decrease lead and methylmercury deposits in the brain [162]. Another thiol-containing chelating agent is d-penicillamine (DPA), which is approved for treating Wilson’s disease and available in oral form [160]. DPA is effective for rapid elimination of Cu through urine, and is given in combination with Zn salts to promote metallothionein synthesis [81]. Similarly, clinically licensed Fe chelators are used to treat Fe overload caused by frequent blood transfusions in patients with genetic blood diseases. The standard Fe chelator is deferoxamine (DFOA), which is highly efficient due to its shielding effect on metal ions that prevents it from being reduced [162]. However, DFOA is inconvenient to administer, so a combined therapy of deferiprone (DFP) and deferasirox (DFX) is also available [164]. Metal chelation therapy is used in very specific circumstances, and chelation in AD must be applied differently than the classic approach of massive chelation [165]. A major challenge for metal chelation in AD arises from the need for the chelation compound to cross the BBB, which requires a hydrophobic and low molecular weight chelator [165, 166]. Clinically available chelators are hydrophilic and intended to act on tissues; therefore, these are not suitable therapeutic molecules for AD [158, 159, 167]. Another challenge is that chelation for AD treatment must avoid complete depletion of biometals, because biometals have essential neurological roles in metalloproteins activity [157, 158, 165–168].
Fig. 4.
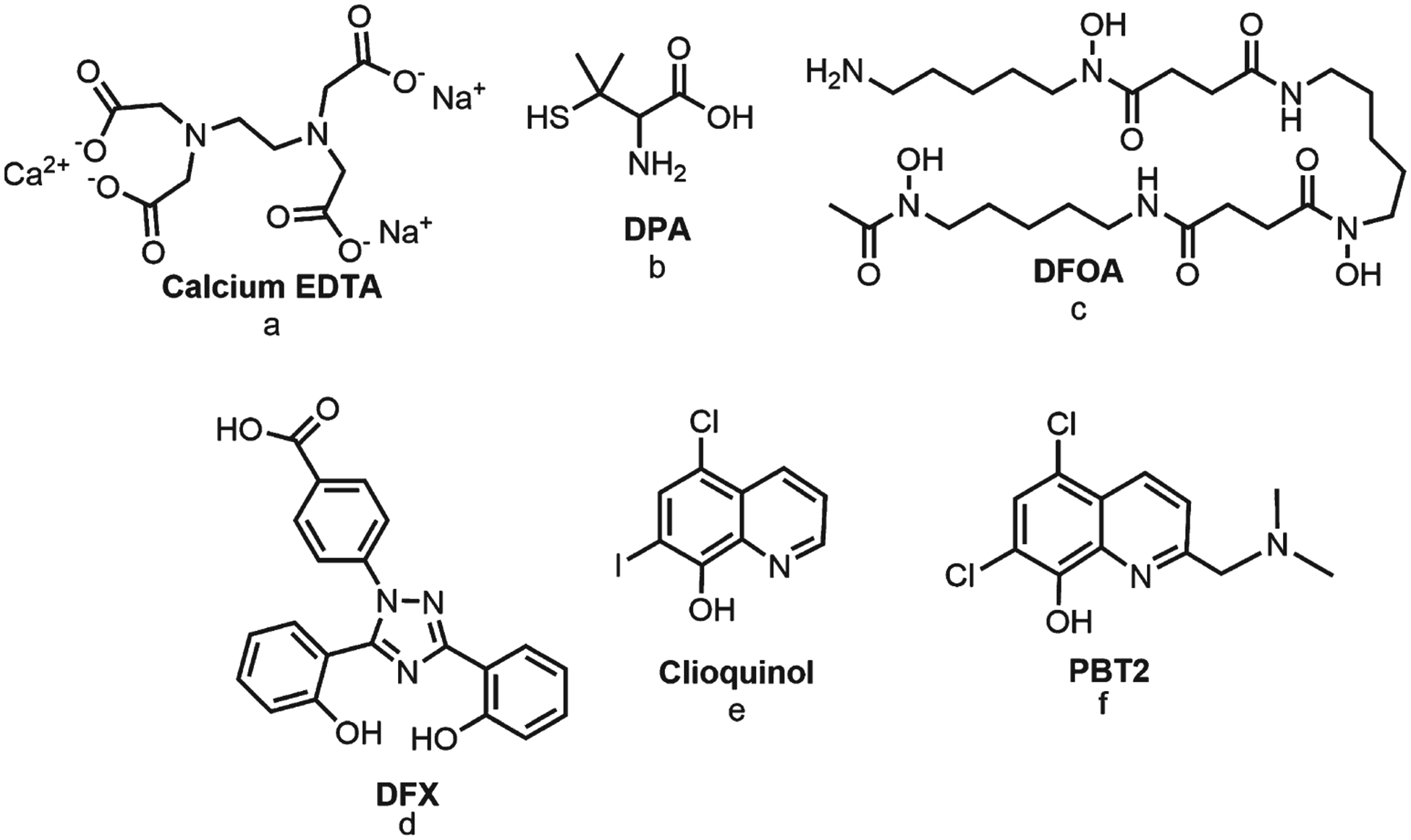
Types of metal chelation compounds. Compounds a–d are approved for treating metal ion excess. Compounds e–f are considered metal-protein attenuating compounds (MPAC).
Potential chelators of interest for AD therapy have been called metal-protein attenuating compounds (MPACs). MPACs should have specific and moderate chelation activity to prevent metal depletion [56, 165, 166, 168–172]. The main difference between metal chelators and MPACs is the binding constant, which should be weaker in MPACs than in traditional chelators [173]. Also, metal chelation therapy for AD should focus on redistributing metals to interrupt abnormal metal-protein interactions, rather than excreting metals from the brain [165, 169]. Additional challenges to metal chelation therapy for AD include understanding potential metal-chelator complexes and anticipating how disaggregated Aβ42 peptides will be diverted from pathological pathways [165, 174]. Considering these requirements, hydroxyquinolines are potential MPACs [173]. Clioquinol, an 8-hydroxyquinoline derivative, is a hydrophobic molecule with therapeutic activity in AD, including solubilizing the Cu(II)/Zn(II)-Aβ complex, inhibiting Aβ42 aggregation, and decreasing redox toxicity [164, 168, 175, 176]. However, the safety of clioquinol as an AD treatment is still undetermined [172, 174, 177–179]. Clioquinol has a hydroxyl and amine moieties which coordinate with Cu(II) and Zn(II) to form a stable five-membered ring [168, 180]. PBT2 is another hydroxyquinoline derivative that has undergone phase II clinical trials for AD treatment [181, 182]. PBT2 chelates Cu(II) and Zn(II) ions trapped between Aβ fibrils, and facilitates re-uptake of ions into cells [182]. Despite these favorable results, PBT2 is no longer being as an AD therapy; however, it is currently researched as a drug for HD [183].
Polyphenols as metal chelators in Alzheimer’s disease
Polyphenols are natural compounds that are abundant in fruits, vegetables, and seeds [1, 184]. These compounds possess several nutrition benefits to protect against heart disease, diabetes, and brain disorders [3, 185, 186]. Polyphenols from dietary origin are considered safe for most consumers but the consumption of fortified food or supplements need to be carefully regulated. The fortified food or supplements containing the high quantity of polyphenols can trigger undesirable effects such as adverse interaction with medications, kidney damage, increase stroke, mortality, etc. [187, 188]. As nutraceutical compounds polyphenols can be used as preventive strategy or to mitigate decrease progression [189–191]. Naturally occurring polyphenols, such as curcumin, epigallocatechin-3-gallate (EGCG), quercetin, myricetin, and others, are considered candidate therapeutics for AD and other diseases [3, 156]. Curcumin, which is found in Turmeric longa, appears to have therapeutic activity, including anti-inflammatory, neurotoxic metal chelating, neuroprotective, anti-amyloidogenic, and reducing oxidative damage to mitochondria and DNA [192–197]. Epidemiological studies demonstrate that the curcumin consumption can decrease the incidence of AD 4.4 times [198]. Significantly reduced AD incidence rate in the south asian and indian people is thought to be attributed to the abundant use of turmeric as an important spice in indian diet, which supports this hypothesis [198]. Curcumin chelates Cu and Fe transition ions, which play roles in AD pathology [156, 196, 199]. The keto-enol moiety is crucial for metal coordination [200, 201]. Additionally, the hydroxyl groups of curcumin interact with ROS to form a stable species that does not damage DNA [202]. EGCG is the most abundant catechin found in the green tea plant Camellia sinensis. The green tea plant has antioxidant, metal chelating, anti-inflammatory, anti-carcinogenic, and anti-apoptotic properties [46, 203, 204]. EGCG sequesters Fe and Cu transition ions, which increases its potential as a neuroprotective treatment [205, 206]. EGCG forms two five-membered stable rings with transition metals found at hydroxyl groups in ring B and at the gallic acid moiety, thus preventing ROS production [203, 206, 207]. The effects of EGCG on Aβ aggregation in the presence of Cu(II) have been explored with electrochemical and optical techniques, and the results were concordant with thioflavin-T assays [208]. A decrease in the electrochemical signal in a square wave voltammetry experiment indicated chelation of Cu(II), whereas TEM imaging showed unstructured aggregates, unlike the structured aggregates that formed in the absence of polyphenol [208]. These results suggest that the anti-aggregation potential of EGCG is due to Cu(II) chelation, which prevents the Cu(II) ion from interacting with His residues in the peptide [208]. Quercetin and myricetin are promising therapeutic candidates due to their bifunctionality as antioxidants and chelating molecules [209, 210]. Structures of quercetin and myricetin indicate three potential metal binding sites. A six-membered aromatic ring forms at the β-ketophenolate (5-hydroxy-4-keto site), whereas a five-membered ring forms at the α-ketoenolate (3-hydroxy-4-keto site). The third metal binding site involves the catechol moieties, which exhibit antioxidant and chelating activities [211]. However, there is controversy surrounding the β-ketophenolate and α-ketoenolate binding sites of flavonoids. There is evidence of interactions between the β-ketophenolate moiety of flaviolin and the active-site metal in cytochrome P450 [212]. In contrast, there is evidence of preferred binding between the α-ketoenolate moiety of quercetin and Cu(II)-Aβ, indicating different modes of metal binding in the flavonoids group [211]. Additionally, computer models suggest that Cu(II) binds quercetin, indicating that the α-ketoenolate site is the most probable chelation site [210]. However, the catechol binding site is considered to have the highest metal binding affinity [213].
Chelators characteristic of polyphenols
Polyphenols are metal chelators with high therapeutic potential, suggested by their characteristic coordination with oxophilic transition metals that forms stable five- or six-membered rings [156, 196, 199, 214–216]. The metal chelation potential of polyphenols strongly depends on the catechol moieties and combinations of hydroxyl and carbonyl groups [217, 218]. Combinations of these groups and moieties define metal binding sites. Therefore, we propose that polyphenols be classified into three groups (Fig. 5). Most polyphenols would be included in a “one-metal binding site” group, because they have only one potential chelator site. This group includes curcuminoids, lignans, still-benes, isoflavonoids, flavanols, and anthocyanins. A “two-metal binding sites” group includes flavones and flavanones. A “three-metal binding sites” group includes flavonols, flavanols, and tannins. This classification also strongly depends on the type of substitutions, as previously reviewed [1, 213]. Investigations suggest that the therapeutic efficacy of polyphenols in preventing metal-free and metal-induced Aβ42 aggregation is proportional to the number of catechol moieties present in the therapeutic compound [219]. However, in polyphenols with overlapping metal binding sites, such as the pyrogallol group in delphinidin (Fig. 5), only one of the potential binding sites may chelate, due to disruption of an adjacent site [213].
Fig. 5.
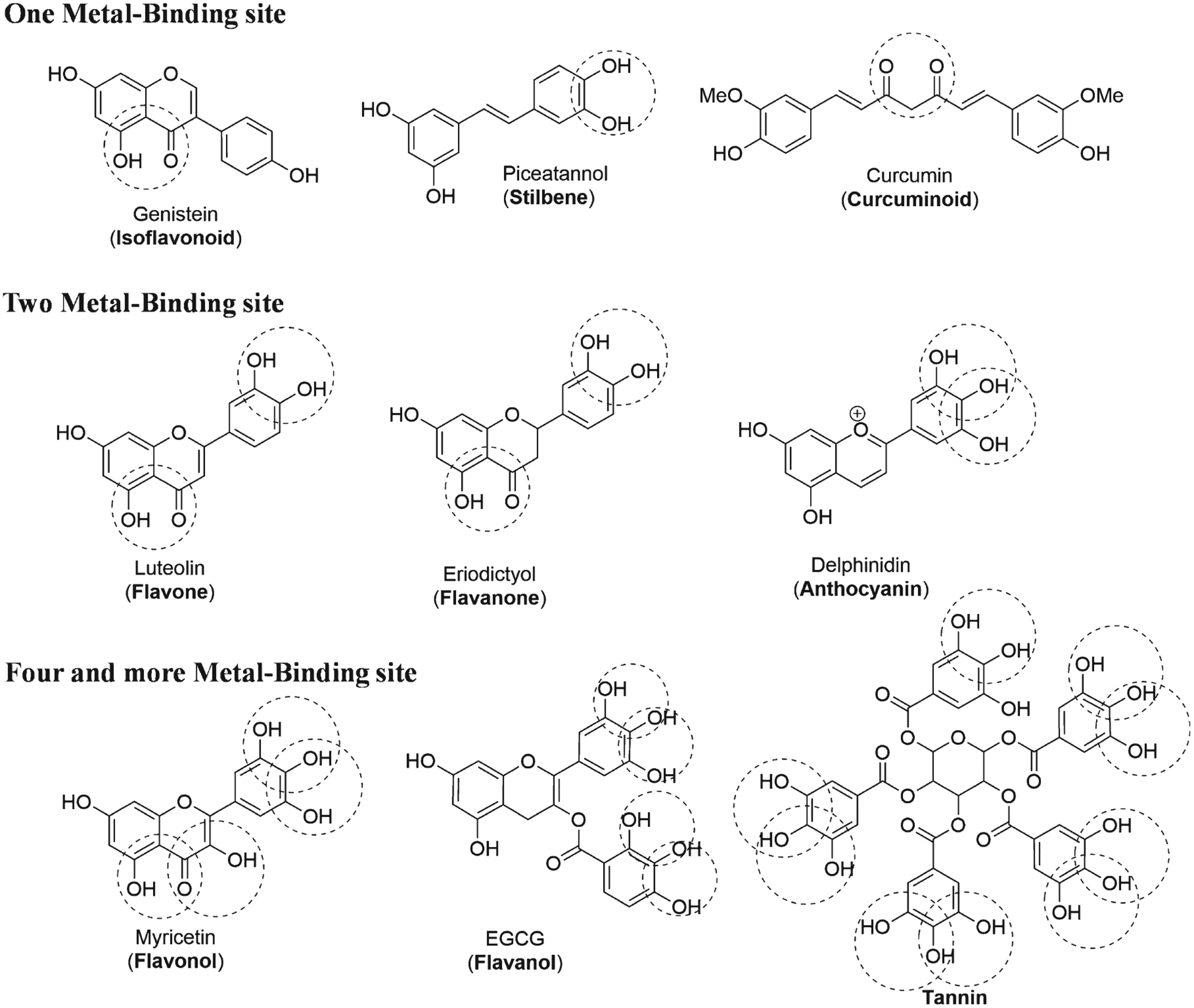
Classification of polyphenols according to the number of metal binding sites (common names in parenthesis).
Chelation therapy exploits physical and chemical properties of metals, such as the principles of Pearson, ionic radius, solvation, chemical stability, bioavailability, metabolism, and natural excretion [162, 174]. Among these, the principles of Pearson explain the driving forces behind chelation therapy. The principles of Pearson or Hard-Soft Acid-Base (HSAB) Theory state that Lewis acids and bases can be considered “hard” or “soft” based on their ion size and polarizability [220]. A “hard” molecule is any species that is small and slightly polarized, whereas a “soft” molecule is larger in size and more polarized. Moreover, Pearson states that hard species tend to couple with hard species electrostatically, with a mostly ionic bond character. Soft species couple with soft species, with electron exchange and/or electron density changes dominated by covalent and/or dispersive nonbonding interactions [220]. Thus, the stability of metal chelate complexes is determined by the hardness/softness of ligands and ions [163]. Generally, ligands are hard bases containing electron donors that are oxygen or nitrogen groups, such as hydroxyl, carboxylate or amine moieties [163, 221]. However, this classification does not clearly divide “hard” and “soft” ligands and ions, because there are also “intermediate” species. For example, transition metal ions involved in AD pathology are intermediate acids with a hardness that decreases with size [Fe(II) > Cu(II) > Zn(II)] [222]. This intermediate nature of transition metals ions is important because it allows them to interact with both “hard” and “soft” bases. Unoccupied d orbitals of transition metal ions can be filled by a basic ligand to form a highly stable and soluble metal complex, thus introducing metal binding chemistry into biological processes [83, 84, 91]. In addition to the formation of coordination bonds, chelation involves formation of stable rings because fewer geometrical constraints arise from the metal-ligand bond (due to the involvement of d orbitals) in contrast to C-C or C-N bonds [223].
The principles of Pearson can be used to describe polyphenols as chelating molecule. Any deprotonated phenolic group will reveal an oxygen with a high charge density, thus resulting in a “hard” ligand [213, 216]. The relatively high pKa of phenols (~9–10) decreases (to ~5–8) because the proton is displaced by metals, such as Fe(III) and Cu(II) [92]. Thus, metal chelation by polyphenols occurs at pH 7.4 [200, 213]. The phenolic pKa value is important for metal chelation, because metal chelation is more efficient at lower pKa values [224]. We also consider that the binding of a metal ion to a polyphenol is favored by oxophilic attraction between a metal and “hard” deprotonated oxygen atoms, creating a stable five- or six-membered ring (Fig. 6) [216]. For example, curcuminoids form a stable six-membered ring with transition metals due to curcuminoid keto groups, which have enolic tautomer of acetylacetone properties [225, 226]. Similarly, genistein and luteolin form six-membered rings with transition metals via their adjacent 5-hydroxyl and keto groups. Five-membered rings are formed by the catechol moieties in piceatannol, delphinidin, and EGCG. Flavanones, flavones, and flavonols, such as eriodictyol, baicilein, and myricetin, also form stable five- and six-member heterocycle rings with transition metals [63, 174, 227]. An electrospray mass spectrometry study showed that Cu and Fe are chelated by some flavonoids, including kaempferol, quercetin, myricetin, luteolin, naringenin, and catechin. The stoichiometry for Cu(II)/flavonoid ratios is usually 1 : 1 and 1 : 2, whereas those for Fe(II) vary among studied polyphenols. In Fe(II) chelation stoichiometry, the reducing ability of the chelation compound increases with the number of hydroxyl groups (myricetin > quercetin > catechin > luteolin > kaempferol > naringenin). The study concluded that the 4-oxo, 3- and 5-hydroxyl groups, and ortho-catechol groups are the metal chelating sites in these molecules [228]. Additionally, Cu and Fe metal chelation assays have shown that polyphenols have moderate to strong chelating activity [224, 229, 230].
Fig. 6.
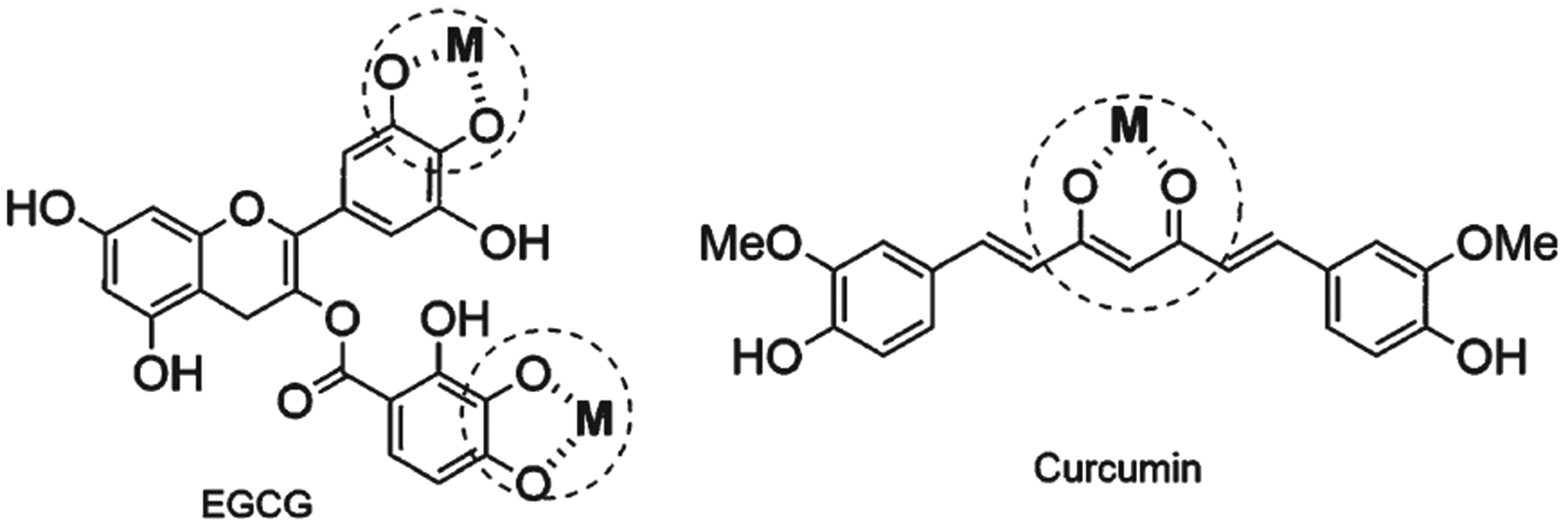
Polyphenols interact with transition metal ions to form a stable 5- membered ring as EGCG or 6-membered ring as curcumin.
Polyphenols interact with transition metal ions
During ROS production in the synaptic cleft, antioxidants disrupt the initiating reaction and scavenge the free radicals created by propagation reactions, such as Fenton and Haber-Weiss reactions (Fig. 7) [144, 218]. Polyphenols have antioxidant effects due to phenolic hydroxyl groups, which reduce free radicals more efficiently than ascorbate. Phenolic hydroxyl groups are more stable because of the resonance of aromatic radical structures [231]. Additionally, polyphenols have low reduction potentials, such that the oxidized form of a given target molecule is more likely to be reduced by receiving an electron. In other words, polyphenols tend to donate electrons to biologically relevant radicals, which have higher standard reduction potentials. The stability of the newly formed radical polyphenol determines its structural requirements. One requirement is that moieties that maximize flavonoid antioxidant potential include a catechol group, a 2,3-double bond conjugation with a 4-oxogroup, and 3- and 5-hydroxyl groups. These required groups all mediate stable electron delocalization [216, 218, 231]. Moreover, there is a positive correlation between the number of hydroxyl groups and the antioxidant potential of a polyphenol.
Fig. 7.
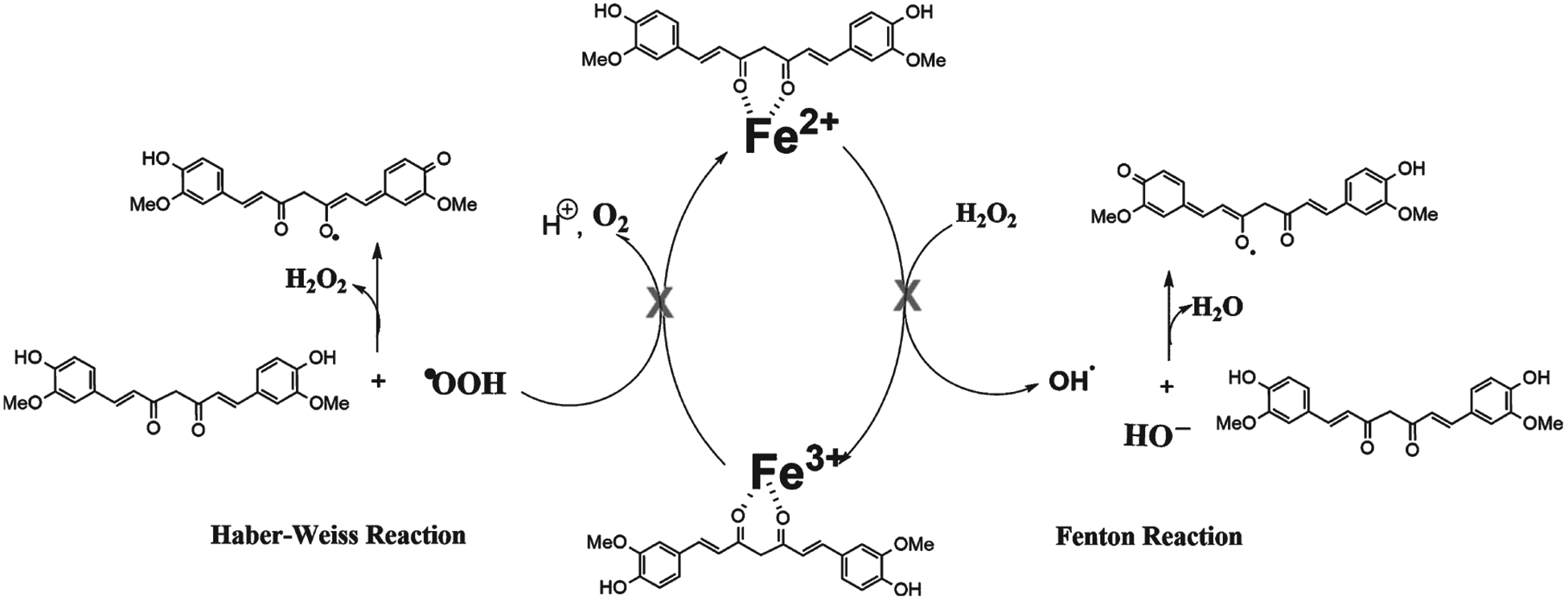
Polyphenols interact with transition metal ions and avoid Haber-Weiss and Fenton reactions.
Polyphenols interact with the Aβ-metal complex
The well-known therapeutic effect of polyphenols in fibril disaggregation may stem from polyphenol hydrophobic nature and metal chelation activity. The bifunctionality of polyphenols, specifically metal chelation activity and Aβ interaction, forms a ternary complex between the Aβ42 peptide, metal ion, and polyphenol, which further decreases the toxicity of the metal-Aβ species, in addition to antioxidant effects (Fig. 8) [42, 207, 211]. Characterization of the complex formed by curcumin and two truncated Aβ42 segments using mass spectrometry and spectroscopic techniques showed that curcumin partially chelates Cu(II) from the Aβ6–14 binding site, despite of the strong His interactions [232]. This result suggests that curcumin is a potential metal redistributor. Moreover, the interactions between Cu(II), curcumin, and Aβ14–23 peptide show a stable ternary complex, indicating that the polyphenol simultaneously acts as a chelator and direct interactor with the Aβ42 peptide. Picciano and Vaden demonstrated the bifunctionality of curcumin. Further, molecular models show that Aβ42 peptide conformational changes are essential for oligomerization and these changes are affected by Cu(II) and curcumin [139, 152, 233, 234]. When curcumin is included in these models, curcumin reduces β-sheet conformation and increases stability of an α-helical structure, and the α-helical structure stabilizes monomeric forms [234]. These models indicate the possibility of an Aβ-Cu(II)-curcumin ternary complex, making curcumin a double-action Aβ42 aggregation inhibitor, via metal chelation activity and direct interaction with Aβ42 peptide. With EGCG, nuclear magnetic resonance (NMR) and mass spectrometry studies of Aβ-Cu/Zn(II)-EGCG ternary complexes suggested that metal chelation, Aβ42 interactions, and ternary complex formation occur in parallel [207]. There is also evidence that flavonols, such as quercetin and myricetin, form Aβ-metal-flavonol ternary complexes via hydrophilic interactions between the hydroxyl groups and the Aβ42 peptide [211].
Fig. 8.
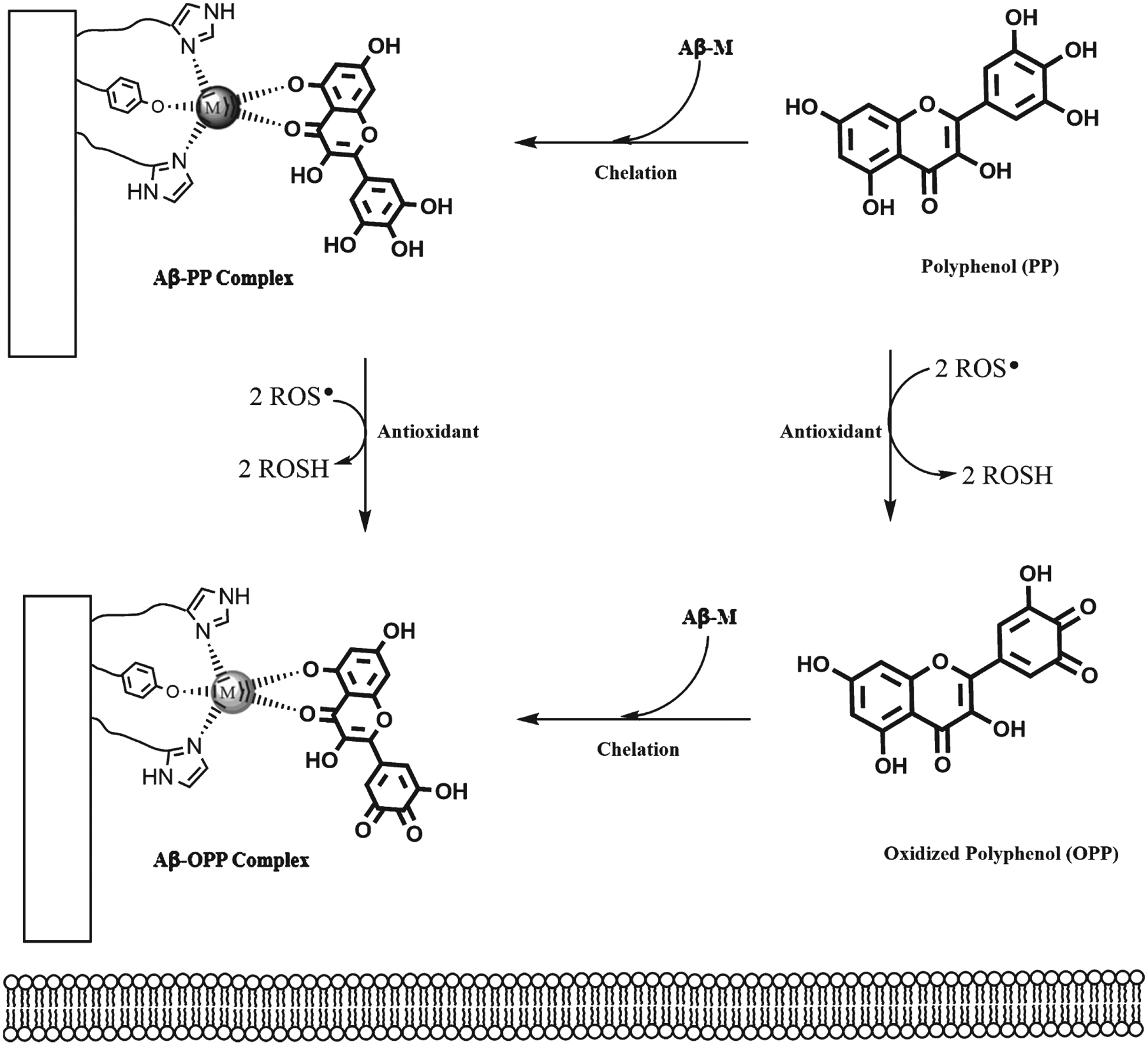
Bifunctionality of polyphenols, which form an Aβ-M-PP ternary complex and also interact with ROS.
Based on the existing body of knowledge, we propose that the stability of Aβ-metal-polyphenolternary complex arises from metal oxophilicity and the “hardness” of the groups that determinethe potential metal binding in the polyphenol. Additionally, stereoelectronic effects and resulting five- or six-membered rings increase the stability of the Aβ-metal-polyphenol ternary complex. Moreover, π − π stacking between polyphenol phenolic rings and Aβ42 peptide aromatic aa residues destabilize β-sheet structures [235]. Overall, the therapeutic effects of polyphenols decrease neurotoxicity caused by Aβ42 peptide and dysregulated metals.
In Fig. 9, we propose a mechanism by which polyphenols act as chelator molecules. AβPP and Aβ42 are considered metalloproteins because both have a metal binding site. Polyphenols can interact directly with metal transition ions and also interact with the Aβ-M complex. We suggest that polyphenols are neuroprotective, and should be studied as natural chelator molecules for regulating metal transition ion imbalance in the brain.
Fig. 9.
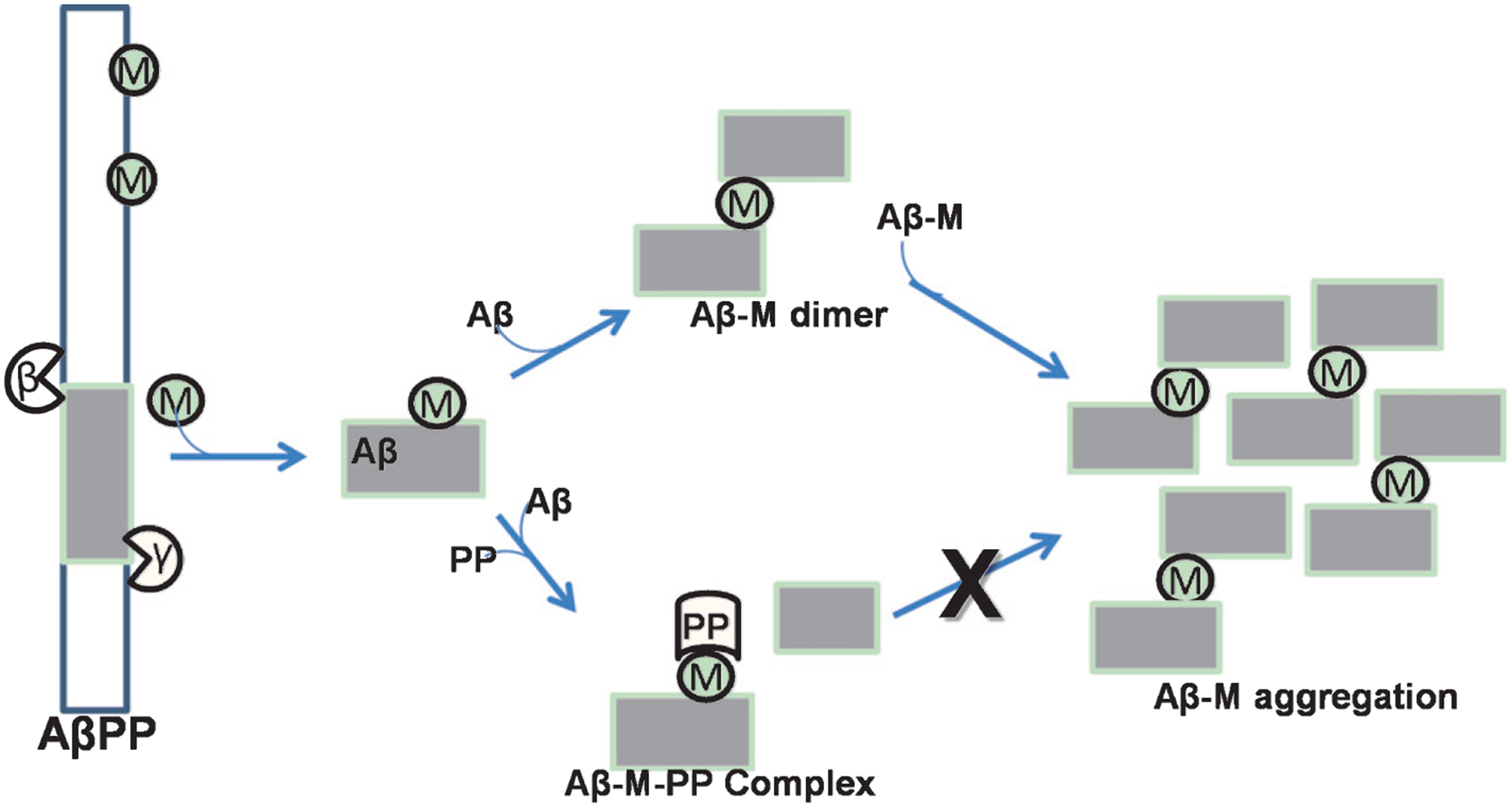
Potential mechanism of polyphenol neuroprotection caused by the ternary complex Aβ-M-PP, which may prevent Aβ-M aggregation.
Polyphenols as potential drug for the treatment of Alzheimer’s disease
We strongly advocate that polyphenols have potential to be a new drug against AD and others neurodegenerative disorders. Polyphenols have also been considering nutraceutical compounds with the advantage to be taken as dietary supplement in any stage of illness. Notably, five drugs have been approved by Food and Drug Administration (FDA) for treatment of AD: these are donepezil, galantamine, rivastigmine, memantine, and the combination of donepezil with memantine, all with limited short term benefits [236, 237]. We consider that polyphenols have the potential to be converted in a new drug in the fighting against AD. Curcumin, a well-known polyphenol, has similarity with donepezil (Fig. 10) where both have two aromatic rings and they are connected with a chain that contain keto group. There are synthetic compounds that took the half part of each compound and created a novel molecule with major potential than their precursor such as donepezil and curcumin [238]. The disadvantage of curcumin is the low bioavailability and its degradation in the organism. If we improve the bioavailability and avoid the degradation of this polyphenol, it can be converted to a new drug against AD.
Fig. 10.
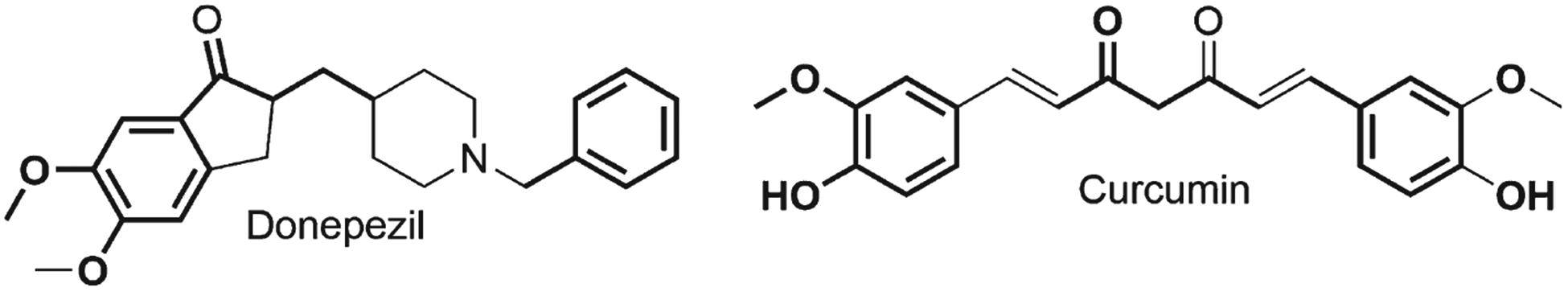
Structural comparison between donepezil and curcumin.
CONCLUSIONS
Polyphenols have high therapeutic potential because they are natural chelators that can cross the BBB. These biomolecules reduce metal accumulation by forming one or more stable 5- or 6-membered rings. The chelation activity of polyphenols avoids metal imbalance, prevents ROS production, and disturbs Aβ fibril patterns. The chelation potential of polyphenols may be related to their anti-amyloidogenic properties that allow for the formation of ternary complexes between Aβ, metal ions, and the polyphenol. We propose that the chelation potential and antioxidant properties of polyphenols make these biomolecules strong pleiotropic candidates for chelation therapy compounds. We also consider that polyphenols have therapeutic potential for AD. Polyphenols are a natural product that can interact with transition metal ions and the Aβ-metal complex.
ACKNOWLEDGMENTS
The authors acknowledge research fundings by: National Secretariat for Science, Technology, and Innovation of Panama (SENACYT) grant number [FID17-002], INDICASAT Internal Grant [JR04-2020] to KSR and National Institute of Neurological Disorders and Stroke (NINDS) of the National Institute of Health (NIH) grant R01NS088645 to MLH. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
J.L-B., and K.S.R. are grateful to Melo Brain’s Grant, MEF Nutritional Grant for support and INDICASAT Internal Grant. JL-B. and K.S.R. also thank the National Science System (SNI) of National Secretariat for Science, Technology, and Innovation of Panama (SENACYT) for support. GLP is grateful to INDICASAT for financial support of the collaboration.
Footnotes
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-0185r1).
REFERENCES
- [1].Lakey-Beitia J, Berrocal R, Rao KS, Durant AA (2015) Polyphenols as therapeutic molecules in Alzheimer’s disease through modulating amyloid pathways. Mol Neurobiol 51, 466–479. [DOI] [PubMed] [Google Scholar]
- [2].Cho KS, Shin M, Kim S, Lee SB (2018) Recent advances in studies on the therapeutic potential of dietary carotenoids in neurodegenerative diseases. Oxid Med Cell Longev 2018, 4120458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lakey-Beitia J, JK D, Hegde ML (2019) Carotenoids as novel therapeutic molecules against neurodegenerative disorders: Chemistry and molecular docking analysis. Int J Mol Sci 20, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wang H, Dharmalingam P, Vasquez V, Mitra J, Boldogh I, Rao KS, Kent TA, Mitra S, Hegde ML (2017) Chronic oxidative damage together with genome repair deficiency in the neurons is a double whammy for neurodegeneration: Is damage response signaling a potential therapeutic target? Mech Ageing Dev 161, 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Shaw BF, Valentine JS (2007) How do ALS-associated mutations in superoxide dismutase 1 promote aggregation of the protein? Trends Biochem Sci 32, 78–85. [DOI] [PubMed] [Google Scholar]
- [6].Reddy PH, Shirendeb UP (2012) Mutant huntingtin, abnormal mitochondrial dynamics, defective axonal transport of mitochondria, and selective synaptic degeneration in Huntington’s disease. Biochim Biophys Acta 1822, 101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Reddy PH, Mao P, Manczak M (2009) Mitochondrial structural and functional dynamics in Huntington’s disease. Brain Res Rev 61, 33–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mao P, Manczak M, Calkins MJ, Truong Q, Reddy TP, Reddy AP, Shirendeb U, Lo HH, Rabinovitch PS, Reddy PH (2012) Mitochondria-targeted catalase reduces abnormal APP processing, amyloid β production and BACE1 in a mouse model of Alzheimer’s disease: Implications for neuroprotection and lifespan extension. Hum Mol Genet 21, 2973–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gitler AD, Dhillon P, Shorter J (2017) Neurodegenerative disease: Models, mechanisms, and a new hope. Dis Model Mech 10, 499–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. [DOI] [PubMed] [Google Scholar]
- [11].Riva N, Agosta F, Lunetta C, Filippi M, Quattrini A (2016) Recent advances in amyotrophic lateral sclerosis. J Neurol 263, 1241–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dutta R, Trapp BD (2011) Mechanisms of neuronal dys-function and degeneration in multiple sclerosis. Prog Neurobiol 93, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Peggion C, Sorgato MC, Bertoli A (2014) Prions and prion-like pathogens in neurodegenerative disorders. Pathogens 3, 149–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mass E, Jacome-Galarza CE, Blank T, Lazarov T, Durham BH, Ozkaya N, Pastore A, Schwabenland M, Chung YR, Rosenblum MK, Prinz M, Abdel-Wahab O, Geissmann F (2017) A somatic mutation in erythro-myeloid progenitors causes neurodegenerative disease. Nature 549, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Farooqui T, Farooqui AA (2009) Aging: An important factor for the pathogenesis of neurodegenerative diseases. Mech Ageing Dev 130, 203–215. [DOI] [PubMed] [Google Scholar]
- [16].Bleiholder C, Bowers MT (2017) The solution assembly of biological molecules using ion mobility methods: From amino acids to amyloid β-protein. Annu Rev Anal Chem 10, 365–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kepp KP (2017) Alzheimer’s disease: How metal ions define β-amyloid function. Coord Chem Rev 351, 127–159. [Google Scholar]
- [18].Lakey-Beitia J, Doens D, Kumar DJ, Murillo E, Fernández PL, Rao K, Durant-Archibold AA (2017) Anti-amyloid aggregation activity of novel carotenoids: Implications for Alzheimer’s drug discovery. Clin Interv Aging 12, 815–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Crous-Bou M, Minguillón C, Gramunt N, Molinuevo JL (2017) Alzheimer’s disease prevention: From risk factors to early intervention. Alzheimers Res Ther 9, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Patterson C (2018) World Alzheimer Report 2018. The state of the art of dementia research: New frontiers. Alzheimer’s Disease International, London, UK. [Google Scholar]
- [21].Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297, 353–356. [DOI] [PubMed] [Google Scholar]
- [22].Kawahara M, Kato-Negishi M, Tanaka K (2017) Cross talk between neurometals and amyloidogenic proteins at the synapse and the pathogenesis of neurodegenerative diseases. Metallomics 9, 619–633. [DOI] [PubMed] [Google Scholar]
- [23].Dos Santos TW, Pereira QC, Teixeira L, Gambero A, Villena JA, Ribeiro ML (2018) Effects of polyphenols on thermogenesis and mitochondrial biogenesis. Int J Mol Sci 19, 2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].van der Merwe C, van Dyk HC, Engelbrecht L, van der Westhuizen FH, Kinnear C, Loos B, Bardien S (2017) Curcumin rescues a PINK1 knock down SH-SY5Y cellular model of Parkinson’s disease from mitochondrial dysfunction and cell death. Mol Neurobiol 54, 2752–2762. [DOI] [PubMed] [Google Scholar]
- [25].Hashimoto M, Rockenstein E, Crews L, Masliah E (2003) Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer’s and Parkinson’s diseases. Neuromolecular Med 4, 21–35. [DOI] [PubMed] [Google Scholar]
- [26].Bäckman L, Jones S, Berger A-K, Laukka EJ, Small BJ (2004) Multiple cognitive deficits during the transition to Alzheimer’s disease. J Intern Med 256, 195–204. [DOI] [PubMed] [Google Scholar]
- [27].Blennow K, de Leon MJ, Zetterberg H (2006) Alzheimer’s disease. Lancet 368, 387–403. [DOI] [PubMed] [Google Scholar]
- [28].Oliver DMA, Reddy PH (2019) Molecular basis of Alzheimer’s disease: Focus on mitochondria. J Alzheimers Dis 72, S95–S116. [DOI] [PubMed] [Google Scholar]
- [29].Bhatti GK, Reddy AP, Reddy PH, Bhatti JS (2020) Lifestyle modifications and nutritional interventions in aging-associated cognitive decline and Alzheimer’s disease. Front Aging Neurosci 11, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Eskici G, Axelsen PH (2012) Copper and oxidative stress in the pathogenesis of Alzheimer’s disease. Biochemistry 51, 6289–6311. [DOI] [PubMed] [Google Scholar]
- [31].Bagheri S, Squitti R, Haertlé T, Siotto M, Saboury AA (2018) Role of copper in the onset of Alzheimer’s disease compared to other metals. Front Aging Neurosci 9, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Jan A, Azam M, Siddiqui K, Ali A, Choi I, Haq Q (2015) Heavy metals and human health: Mechanistic insight into toxicity and counter defense system of antioxidants. Int J Mol Sci 16, 29592–29630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chen P, Miah MR, Aschner M (2016) Metals and neurodegeneration. F1000Res 5, 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Mayes J, Tinker-Mill C, Kolosov O, Zhang H, Tabner BJ, Allsop D (2014) β-amyloid fibrils in Alzheimer disease are not inert when bound to copper ions but can degrade hydrogen peroxide and generate reactive oxygen species. J Biol Chem 289, 12052–12062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hamley IW (2012) The amyloid beta peptide: A chemist’s perspective role in Alzheimer’s and fibrillization. Chem Rev 112, 5147–5192. [DOI] [PubMed] [Google Scholar]
- [36].Kong GK-W, Adams JJ, Harris HH, Boas JF, Curtain CC, Galatis D, Masters CL, Barnham KJ, McKinstry WJ, Cappai R, Parker MW (2007) Structural studies of the Alzheimer’s amyloid precursor protein copper-binding domain reveal how it binds copper ions. J Mol Biol 367, 148–161. [DOI] [PubMed] [Google Scholar]
- [37].Hegde ML, Hegde PM, Rao KS, Mitra S (2011) Oxidative genome damage and its repair in neurodegenerative diseases: Function of transition metals as a double-edged sword. J Alzheimers Dis 24, 183–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Gerber H, Wu F, Dimitrov M, Garcia Osuna GM, Fraering PC (2017) Zinc and copper differentially modulate amyloid precursor protein processing by γ-secretase and amyloid-β peptide production. J Biol Chem 292, 3751–3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Weinreb O, Mandel S, Youdim MBH, Amit T (2013) Targeting dysregulation of brain iron homeostasis in Parkinson’s disease by iron chelators. Free Radic Biol Med 62, 52–64. [DOI] [PubMed] [Google Scholar]
- [40].Suram A, Hegde ML, Rao KSJ (2007) A new evidence for DNA nicking property of amyloid β-peptide (1–42): Relevance to Alzheimer’s disease. Arch Biochem Biophys 463, 245–252. [DOI] [PubMed] [Google Scholar]
- [41].Chiang K, Koo EH (2014) Emerging therapeutics for Alzheimer’s disease. Annu Rev Pharmacol Toxicol 54, 381–405. [DOI] [PubMed] [Google Scholar]
- [42].Savelieff MG, DeToma AS, Derrick JS, Lim MH (2014) The ongoing search for small molecules to study metal-associated amyloid-β species in Alzheimer’s disease. Acc Chem Res 47, 2475–2482. [DOI] [PubMed] [Google Scholar]
- [43].Li W, Wu M, Tang L, Pan Y, Liu Z, Zeng C, Wang J, Wei T, Liang G (2015) Novel curcumin analogue 14p protects against myocardial ischemia reperfusion injury through Nrf2-activating anti-oxidative activity. Toxicol Appl Pharmacol 282, 175–183. [DOI] [PubMed] [Google Scholar]
- [44].Murillo E, Britton GB, Durant AA (2012) Antioxidant activity and polyphenol content in cultivated and wild edible fruits grown in Panama. J Pharm Bioallied Sci 4, 313–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Khan AN, Hassan MN, Khan RH (2019) Gallic acid: A naturally occurring bifunctional inhibitor of amyloid and metal induced aggregation with possible implication in metal-based therapy. J Mol Liq 285, 27–37. [Google Scholar]
- [46].Cascella M, Bimonte S, Muzio MR, Schiavone V, Cuomo A (2017) The efficacy of epigallocatechin-3-gallate (green tea) in the treatment of Alzheimer’s disease: An overview of pre-clinical studies and translational perspectives in clinical practice. Infect Agent Cancer 12, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zhou ZD, Xie SP, Saw WT, Ho PGH, Wang HY, Zhou L, Zhao Y, Tan EK (2019) The therapeutic implications of tea polyphenols against dopamine (da) neuron degeneration in Parkinson’s disease (PD). Cells 8, 911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Jayaraj RL, Elangovan N, Manigandan K, Singh S, Shukla S (2014) CNB-001 a novel curcumin derivative, guards dopamine neurons in MPTP model of Parkinson’s disease. Biomed Res Int 2014, 236182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Shrikanth Gadad B, K. Subramanya P, Pullabhatla S, S. Shantharam I, K.S. R(2012) Curcumin-glucoside, a novel synthetic derivative of curcumin, inhibits alpha-synuclein oligomer formation: Relevance to Parkinson’s disease. Curr Pharm Des 18, 76–84. [DOI] [PubMed] [Google Scholar]
- [50].Hor SL, Teoh SL, Lim WL (2019) Plant polyphenols as neuroprotective agents in Parkinson’s disease targeting oxidative stress. Curr Drug Targets 20, 2174. [DOI] [PubMed] [Google Scholar]
- [51].Naia L, Rosenstock TR, Oliveira AM, Oliveira-Sousa SI, Caldeira GL, Carmo C, Laço MN, Hayden MR, Oliveira CR, Rego AC (2017) Comparative mitochondrial-based protective effects of resveratrol and nicotinamide in Huntington’s disease models. Mol Neurobiol 54, 5385–5399. [DOI] [PubMed] [Google Scholar]
- [52].Pasinetti GM, Wang J, Marambaud P, Ferruzzi M, Gregor P, Knable LA, Ho L (2011) Neuroprotective and metabolic effects of resveratrol: Therapeutic implications for Huntington’s disease and other neurodegenerative disorders. Exp Neurol 232, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Srinivasan E, Rajasekaran R (2018) Comparative binding of kaempferol and kaempferide on inhibiting the aggregate formation of mutant (G85R) SOD1 protein in familial amyotrophic lateral sclerosis: A quantum chemical and molecular mechanics study. Biofactors 44, 431–442. [DOI] [PubMed] [Google Scholar]
- [54].Sohail A, Bhat WF, Bhat SA, Furkan M, Shah A, Bano B (2018) Investigating the preventive effects of baicalin and gallocatechin against glyoxal-induced cystatin aggregation. J Biomol Struct 36, 3791–3802. [DOI] [PubMed] [Google Scholar]
- [55].Tosato M, Di Marco V (2019) Metal chelation therapy and Parkinson’s disease: A critical review on the thermodynamics of complex formation between relevant metal ions and promising or established drugs. Biomolecules 9, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Cukierman DS, Accardo E, Gomes RG, De Falco A, Miotto MC, Freitas MCR, Lanznaster M, Fernández CO, Rey NA (2018) Aroylhydrazones constitute a promising class of ‘metal-protein attenuating compounds’ for the treatment of Alzheimer’s disease: A proof-of-concept based on the study of the interactions between zinc (II) and pyridine-2-carboxaldehyde isonicotinoyl hydrazon. J Biol Inorg Chem 23, 1227–1241. [DOI] [PubMed] [Google Scholar]
- [57].Van Bulck M, Sierra-Magro A, Alarcon-Gil J, Perez-Castillo A, Morales-Garcia J (2019) Novel approaches for the treatment of Alzheimer’s and Parkinson’s disease. Int J Mol Sci 20, 719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Sensi SL, Granzotto A, Siotto M, Squitti R (2018) Copper and zinc dysregulation in Alzheimer’s disease. Trends Pharmacol Sci 39, 1049–1063. [DOI] [PubMed] [Google Scholar]
- [59].Bertini I, Gray HB, Steifel EI, Valentine JS (2007) Metal ions and proteins: Binding, stability and folding. In Biological Inorganic Chemistry: Structure and Reactivity. University Science Books, Sausalito, CA, pp. 31–41. [Google Scholar]
- [60].Crichton RR (2018) The role of metals in biology - an overview. In Biological Inorganic Chemistry: A New Introduction to Molecular Structure and Function. Academic Press, London, UK, pp. 1–13. [Google Scholar]
- [61].Ackerman CM, Chang CJ (2018) Copper signaling in the brain and beyond. J Biol Chem 293, 4628–4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Mitra J, Guerrero E, Hegde PM, Wang H, Boldogh I, Rao KS, Mitra S, Hegde ML (2014) New perspectives on oxidized genome damage and repair inhibition by pro-oxidant metals in neurological diseases. Biomolecules 4, 678–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kepp KP (2012) Bioinorganic chemistry of Alzheimer’s disease. Chem Rev 112, 5193–5239. [DOI] [PubMed] [Google Scholar]
- [64].Bharathi, Ravid R, Rao KSJ(2006) Role of metals in neuronal apoptosis: Challenges associated with neurode-generation. Curr Alzheimer Res 3, 311–26. [DOI] [PubMed] [Google Scholar]
- [65].Toni M, Massimino ML, De Mario A, Angiulli E, Spisni E (2017) Metal dyshomeostasis and their pathological role in prion and prion-like diseases: The basis for a nutritional approach. Front Neurosci 11, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Brown DR (2011) Metals in neurodegenerative disease. Metallomics 3, 226. [DOI] [PubMed] [Google Scholar]
- [67].Lovell M, Robertson J, Teesdale W, Campbell J, Markesbery W (1998) Copper, iron and zinc in Alzheimer’s disease senile plaques. J Neurol Sci 158, 47–52. [DOI] [PubMed] [Google Scholar]
- [68].Ayton S, Lei P, Bush AI (2015) Biometals and their therapeutic implications in Alzheimer’s disease. Neurotherapeutics 12, 109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Li DD, Zhang W, Wang ZY, Zhao P (2017) Serum copper, zinc, and iron levels in patients with Alzheimer’s disease: A meta-analysis of case-control studies. Front Aging Neurosci 9, 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].González-Domínguez R, García-Barrera T, Gómez-Ariza JL (2014) Characterization of metal profiles in serum during the progression of Alzheimer’s disease. Metallomics 6, 292–300. [DOI] [PubMed] [Google Scholar]
- [71].La Penna G, Minicozzi V, Morante S, Rossi GC, Stellato F (2015) A first-principle calculation of the XANES spectrum of Cu 2+ in water. J Chem Phys 143, 124508. [DOI] [PubMed] [Google Scholar]
- [72].Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O’Halloran TV(1999) Undetectable intracellular free copper: The requirement of a copper chaperone for superoxide dismutase. Science 284, 805–808. [DOI] [PubMed] [Google Scholar]
- [73].Sayre LM, Perry G, Smith MA (1999) Redox metals and neurodegenerative disease. Curr Opin Chem Biol 3, 220–225. [DOI] [PubMed] [Google Scholar]
- [74].Bush AI (2000) Metals and neuroscience. Curr Opin Chem Biol 4, 184–191. [DOI] [PubMed] [Google Scholar]
- [75].Maynard CJ, Bush AI, Masters CL, Cappai R, Li QX (2005) Metals and amyloid-β in Alzheimer’s disease. Int J Exp Pathol 86, 147–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Bush AI (2003) The metallobiology of Alzheimer’s disease. Trends Neurosci 26, 207–214. [DOI] [PubMed] [Google Scholar]
- [77].Li Y, Jiao Q, Xu H, Du X, Shi L, Jia F, Jiang H (2017) Biometal dyshomeostasis and toxic metal accumulations in the development of Alzheimer’s disease. Front Mol Neurosci 10, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Squitti R, Simonelli I, Ventriglia M, Siotto M, Pasqualetti P, Rembach A, Doecke J, Bush AI (2013) Meta-analysis of serum non-ceruloplasmin copper in Alzheimer’s disease. J Alzheimers Dis 38, 809–822. [DOI] [PubMed] [Google Scholar]
- [79].De Bie P, Muller P, Wijmenga C, Klomp LWJ (2007) Molecular pathogenesis of Wilson and Menkes disease: Correlation of mutations with molecular defects and disease phenotypes. J Med Genet 44, 673–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Lutsenko S, Gupta A, Burkhead JL, Zuzel V (2008) Cellular multitasking: The dual role of human Cu-ATPases in cofactor delivery and intracellular copper balance. Arch Biochem Biophys 476, 22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Horn N, Møller LB, Nurchi VM, Aaseth J (2019) Chelating principles in Menkes and Wilson diseases: Choosing the right compounds in the right combinations at the right time. J Inorg Biochem 190, 98–112. [DOI] [PubMed] [Google Scholar]
- [82].Govindaraju M, Rao Jayanth KS, Jagadeesh Kumar D, Prasada Rao UJS, Sambasiva Rao KRS, Rao KS (2017) Studies on copper and Aβ1–16-induced conformational changes in CAG/CTG trinucleotide repeats sequence. J Alzheimers Dis Rep 1, 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Cotton FA, Wilkinson G, Murillo CA, Bochmann M (1980) Zinc, cadmium, and mercury. In Advanced Inorganic Chemistry. John Wiley and Sons, New York, NY, pp. 590–591. [Google Scholar]
- [84].Huheey JE, Keiter EA, Keiter RL (1993) Some descriptive chemistry of the metals. In Inorganic Chemistry: Principles of Structure and Reactivity. HarperCollins College Publishers, New York, NY, pp. 586–587. [Google Scholar]
- [85].Szewczyk B (2013) Zinc homeostasis and neurodegenerative disorders. Front Aging Neurosci 5, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Wang ZX, Tan L, Wang HF, Ma J, Liu J, Tan MS, Sun JH, Zhu XC, Jiang T, Yu JT (2015) Serum iron, zinc, and copper levels in patients with Alzheimer’s disease: A replication study and meta-analyses. J Alzheimers Dis 47, 565–581. [DOI] [PubMed] [Google Scholar]
- [87].Ventriglia M, Brewer GJ, Simonelli I, Mariani S, Siotto M, Bucossi S, Squitti R (2015) Zinc in Alzheimer’s disease: A meta-analysis of serum, plasma, and cerebrospinal fluid studies. J Alzheimers Dis 46, 75–87. [DOI] [PubMed] [Google Scholar]
- [88].Singh YP, Pandey A, Vishwakarma S, Modi G (2019) A review on iron chelators as potential therapeutic agents for the treatment of Alzheimer’s and Parkinson’s diseases. Mol Divers 23, 509–526. [DOI] [PubMed] [Google Scholar]
- [89].Hureau C (2012) Coordination of redox active metal ions to the amyloid precursor protein and to amyloid-β peptides involved in Alzheimer disease. Part 1: An overview. Coord Chem Rev 256, 2164–2174. [Google Scholar]
- [90].Lane DJR, Ayton S, Bush AI (2018) Iron and Alzheimer’s disease: An update on emerging mechanisms. J Alzheimers Dis 64, S379–S395. [DOI] [PubMed] [Google Scholar]
- [91].Greenwood NN, Earnshaw A (1997) Iron, ruthenium and osmium. In Chemistry of the Elements. Butterworth-Heinemann, Oxford, UK, pp. 1070–1098. [Google Scholar]
- [92].Furlan S, La Penna G (2012) Metal ions and protons compete for ligand atoms in disordered peptides: Examples from computer simulations of copper binding to the prion tandem repeat. Coord Chem Rev 256, 2234–2244. [Google Scholar]
- [93].Crespo ÂC, Silva B, Marques L, Marcelino E, Maruta C, Costa S, Timóteo  Vilares A, Couto FS, Faustino P, Correia AP, Verdelho A, Porto G, Guerreiro M, Herrero A, Costa C, de Mendonça A, Costa L, Martins M (2014) Genetic and biochemical markers in patients with Alzheimer’s disease support a concerted systemic iron homeostasis dysregulation. Neurobiol Aging 35, 777–785. [DOI] [PubMed] [Google Scholar]
- [94].Reddy PH, Beal MF (2008) Amyloid beta, mitochondrial dysfunction and synaptic damage: Implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol Med 14, 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Hesse L, Beher D, Masters CL, Multhaup G (1994) The βA4 amyloid precursor protein binding to copper. FEBS Lett 349, 109–116. [DOI] [PubMed] [Google Scholar]
- [96].Multhaup G, Schlicksupp A, Hesse L, Beher D, Ruppert T, Masters CL, Beyreuther K (1996) The amyloid precursor protein of Alzheimer’s disease in the reduction of copper(II) to Copper(I). Science 271, 1406–1409. [DOI] [PubMed] [Google Scholar]
- [97].Kummer MP, Heneka MT (2014) Truncated and modified amyloid-beta species. Alzheimers Res Ther 6, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Narasingapa RB, Jargaval MR, Pullabhatla S, Htoo HH, Rao JKS, Hernandez JF, Govitrapong P, Vincent B (2012) Activation of α-secretase by curcumin-aminoacid conjugates. Biochem Biophys Res Commun 424, 691–696. [DOI] [PubMed] [Google Scholar]
- [99].Grimm MOW, Mett J, Stahlmann CP, Grösgen S, Haupenthal VJ, Blümel T, Hundsdörfer B, Zimmer VC, Mylonas NT, Tanila H, Muller U, Grimm HS, Hartmann T (2015) APP intracellular domain derived from amyloidogenic β- and γ-secretase cleavage regulates neprilysin expression. Front Aging Neurosci 7, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Zhou Y, Suram A, Venugopal C, Prakasam A, Lin S, Su Y, Li B, Paul SM, Sambamurti K (2008) Geranylgeranyl pyrophosphate stimulates γ-secretase to increase the generation of Aβ and APP-CTFγ. FASEB J 22, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Rajendran L, Schneider A, Schlechtingen G, Weidlich S, Ries J, Braxmeier T, Schwille P, Schulz JB, Schroeder C, Simons M, Jennings G, Knolker H-J, Simons K (2008) Efficient inhibition of the Alzheimer’s disease-secretase by membrane targeting. Science 320, 520–523. [DOI] [PubMed] [Google Scholar]
- [102].Teplow DB, Lazo ND, Bitan G, Bernstein S, Wyttenbach T, Bowers MT, Baumketner A, Shea JE, Urbanc B, Cruz L, Borreguero J, Stanley HE (2006) Elucidating amyloid β-protein folding and assembly: A multidisciplinary approach. Acc Chem Res 39, 635–645. [DOI] [PubMed] [Google Scholar]
- [103].Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schiossmacher M, Whaley J, Swindlehurst C, McCormack R, Wolfert R, Selkoe D, Lieberburg I, Schenk D (1992) Isolation and quantification of soluble Alzheimer’s β-peptide from biological fluids. Nature 359, 325–327. [DOI] [PubMed] [Google Scholar]
- [104].Barritt JD, Viles JH (2015) Truncated amyloid-β (11–40/42) from Alzheimer disease binds Cu 2+ with a femtomolar affinity and influences fiber assembly. J Biol Chem 290, 27791–27802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Cherny RA, Legg JT, McLean CA, Fairlie DP, Huang X, Atwood CS, Beyreuther K, Tanzi RE, Masters CL, Bush AI (1999) Aqueous dissolution of Alzheimer’s disease Aβ amyloid deposits by biometal depletion. J Biol Chem 274, 23223–23228. [DOI] [PubMed] [Google Scholar]
- [106].Barnham KJ, McKinstry WJ, Multhaup G, Galatis D, Morton CJ, Curtain CC, Williamson NA, White AR, Hinds MG, Norton RS, Beyreuther K, Masters CL, Parker MW, Cappai R (2003) Structure of the Alzheimer’s disease amyloid precursor protein copper binding domain. J Biol Chem 278, 17401–17407. [DOI] [PubMed] [Google Scholar]
- [107].Dorlet P, Gambarelli S, Faller P, Hureau C (2009) Pulse EPR spectroscopy reveals the coordination sphere of copper(II) ions in the 1–16 amyloid-β Peptide: A key role of the first two N-terminus residues. Angew Chem Int Ed Engl 48, 9273–9276. [DOI] [PubMed] [Google Scholar]
- [108].Drew SC, Masters CL, Barnham KJ (2009) Alanine-2 carbonyl is an oxygen ligand in Cu2+ coordination of Alzheimer’s disease amyloid-β peptide - relevance to N-terminally truncated forms. J Am Chem Soc 131, 8760–8761. [DOI] [PubMed] [Google Scholar]
- [109].Stellato F, Menestrina G, Serra MD, Potrich C, Tomazzolli R, Meyer-Klaucke W, Morante S (2006) Metal binding in amyloid β-peptides shows intra- and inter-peptide coordination modes. Eur Biophys J 35, 340–351. [DOI] [PubMed] [Google Scholar]
- [110].Minicozzi V, Stellato F, Comai M, Serra MD, Potrich C, Meyer-Klaucke W, Morante S (2008) Identifying the minimal copper- and zinc-binding site sequence in amyloid-β peptides. J Biol Chem 283, 10784–10792. [DOI] [PubMed] [Google Scholar]
- [111].Smith DG, Cappai R, Barnham KJ (2007) The redox chemistry of the Alzheimer’s disease amyloid β peptide. Biochim Biophys Acta 1768, 1976–1990. [DOI] [PubMed] [Google Scholar]
- [112].El Khoury Y, Dorlet P, Faller P, Hellwig P (2011) New insights into the coordination of Cu(II) by the amyloid-B 16 peptide from fourier transform ir spectroscopy and isotopic labeling. J Phys Chem B 115, 14812–14821. [DOI] [PubMed] [Google Scholar]
- [113].Rickard GA, Gomez-Balderas R, Brunelle P, Raffa DF, Rauk A (2005) Binding affinities for models of biologically available potential Cu(II) ligands relevant to Alzheimer’s disease: An ab initio study. J Phys Chem A 109, 8361–8370. [DOI] [PubMed] [Google Scholar]
- [114].La Penna G, Hureau C, Andreussi O, Faller P (2013) Identifying, by first-principles simulations, Cu[amyloid-β] species making fenton-type reactions in Alzheimer’s disease. J Phys Chem B 117, 16455–16467. [DOI] [PubMed] [Google Scholar]
- [115].La Penna G, Li MS (2018) Towards high-throughput modelling of copper reactivity induced by structural disorder in amyloid peptides. Chemistry 24, 5259–5270. [DOI] [PubMed] [Google Scholar]
- [116].Pedersen JT, Østergaard J, Rozlosnik N, Gammelgaard B, Heegaard NHH (2011) Cu(II) mediates kinetically distinct, non-amyloidogenic aggregation of amyloid-β peptides. J Biol Chem 286, 26952–26963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Ramesh BN, Raichurkar KP, Shamasundar NM, Rao TSS, Rao KSJ (2011) Aβ(42) induced MRI changes in aged rabbit brain resembles AD brain. Neurochem Int 59, 637–642. [DOI] [PubMed] [Google Scholar]
- [118].Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Barnwell E, Padmaraju V, Baranello R, Pacheco-Quinto J, Crosson C, Ablonczy Z, Eckman E, Eckman CB, Ramakrishnan V, Greig NH, Pappolla MA, Sambamurti K (2014) Evidence of a novel mechanism for partial γ-secretase inhibition induced paradoxical increase in secreted amyloid β protein. PLoS One 9, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Wijsman EM, Daw EW, Yu X, Steinbart EJ, Nochlin D, Bird TD, Schellenberg GD (2005) APOE and other loci affect age-at-onset in Alzheimer’s disease families with PS2 mutation. Am J Med Genet B Neuropsychiatr Genet 132B, 14–20. [DOI] [PubMed] [Google Scholar]
- [121].Ashburn TT, Han H, McGuinness BF, Lansbury PT (1996) Amyloid probes based on Congo Red distinguish between fibrils comprising different peptides. Chem Biol 3, 351–358. [DOI] [PubMed] [Google Scholar]
- [122].Gupta VB, Rao KSJ (2007) Anti-amyloidogenic activity of S-allyl-l-cysteine and its activity to destabilize Alzheimer’s β-amyloid fibrils in vitro. Neurosci Lett 429, 75–80. [DOI] [PubMed] [Google Scholar]
- [123].Gadad BS, Britton GB, Rao KS (2011) Targeting oligomers in neurodegenerative disorders: Lessons from α-synuclein, tau, and amyloid-β peptide. J Alzheimers Dis 24, 223–232. [DOI] [PubMed] [Google Scholar]
- [124].Ono K, Hamaguchi T, Naiki H, Yamada M (2006) Anti-amyloidogenic effects of antioxidants: Implications for the prevention and therapeutics of Alzheimer’s disease. Biochim Biophys Acta 1762, 575–586. [DOI] [PubMed] [Google Scholar]
- [125].Hayden EY, Teplow DB (2013) Amyloid β-protein oligomers and Alzheimer’s disease. Alzheimers Res Ther 5, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Faller P, Hureau C, La Penna G (2014) Metal ions and intrinsically disordered proteins and peptides: From Cu/Zn amyloid-β to general principles. Acc Chem Res 47, 2252–2259. [DOI] [PubMed] [Google Scholar]
- [127].Hu WP, Chang GL, Chen SJ, Kuo YM (2006) Kinetic analysis of β-amyloid peptide aggregation induced by metal ions based on surface plasmon resonance biosensing. J Neurosci Methods 154, 190–197. [DOI] [PubMed] [Google Scholar]
- [128].Ren H, Zhang Y, Guo S, Lin N, Deng L, Yue T, Huang F (2017) Identifying the Cu(II)-amyloid β peptide binding intermediates in the early stage of aggregation by resonance Raman spectroscopy: A simulation study. Phys Chem Chem Phys 19, 31103–31112. [DOI] [PubMed] [Google Scholar]
- [129].Faller P (2009) Copper and zinc binding to amyloid-β: Coordination, dynamics, aggregation, reactivity and metal-ion transfer. Chembiochem 10, 2837–2845. [DOI] [PubMed] [Google Scholar]
- [130].Fitzpatrick AWP, Debelouchina GT, Bayro MJ, Clare DK, Caporini MA, Bajaj VS, Jaroniec CP, Wang L, Ladizhansky V, Müller SA, MacPhee CE, Waudby CA, Mott HR, De Simone A, Knowles TPJ, Saibil HR, Vendruscolo M, Orlova EV, Griffin RG, Dobson CM (2013) Atomic structure and hierarchical assembly of a cross-β amyloid fibril. Proc Natl Acad Sci U S A 110, 5468–5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Mitra S, Prasad P, Chakraborty S (2018) A unified view of assessing the pro-oxidant versus antioxidant nature of amyloid beta conformers. Chembiochem 19, 2360–2371. [DOI] [PubMed] [Google Scholar]
- [132].Gu M, Bode DC, Viles JH (2018) Copper redox cycling inhibits aβ fibre formation and promotes fibre fragmentation, while generating a dityrosine Aβ dimer. Sci Rep 8, 16190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Mezentsev YV, Medvedev AE, Kechko OI, Makarov AA, Ivanov AS, Mantsyzov AB, Kozin SA (2016) Zinc-induced heterodimer formation between metal-binding domains of intact and naturally modified amyloid-beta species: Implication to amyloid seeding in Alzheimer’s disease? J Biomol Struct Dyn 34, 2317–2326. [DOI] [PubMed] [Google Scholar]
- [134].Istrate AN, Kozin SA, Zhokhov SS, Mantsyzov AB, Kechko OI, Pastore A, Makarov AA, Polshakov VI (2016) Interplay of histidine residues of the Alzheimer’s disease Aβ peptide governs its Zn-induced oligomerization. Sci Rep 6, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Innocenti M, Salvietti E, Guidotti M, Casini A, Bellandi S, Foresti ML, Gabbiani C, Pozzi A, Zatta P, Messori L (2010) Trace copper(II) or zinc(II) ions drastically modify the aggregation behavior of Amyloid-β1–42: An AFM study. J Alzheimers Dis 19, 1323–1329. [DOI] [PubMed] [Google Scholar]
- [136].Lanza V, Bellia F, Rizzarelli E (2018) An inorganic overview of natural Aβ fragments: Copper(II) and zinc(II)-mediated pathways. Coord Chem Rev 369, 1–14. [Google Scholar]
- [137].Miller Y, Ma B, Nussinov R (2010) Polymorphism in Alzheimer Aβ amyloid organization reflects conformational selection in a rugged energy landscape. Chem Rev 110, 4820–4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Miller Y, Ma B, Nussinov R (2010) Zinc ions promote Alzheimer Abeta aggregation via population shift of polymorphic states. Proc Natl Acad Sci U S A 107, 9490–9495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Pham DQH, Li MS, La Penna G (2018) Copper binding induces polymorphism in amyloid-β peptide: Results of computational models. J Phys Chem B 122, 7243–7252. [DOI] [PubMed] [Google Scholar]
- [140].Sitkiewicz E, Kłoniecki M, Poznański J, Bal W, Dadlez M (2014) Factors influencing compact-extended structure equilibrium in oligomers of Aβ1–40 peptide—an ion mobility mass spectrometry study. J Mol Biol 426, 2871–2885. [DOI] [PubMed] [Google Scholar]
- [141].Kanti Das T, Wati MR, Fatima-Shad K (2015) Oxidative stress gated by Fenton and Haber Weiss reactions and its association with Alzheimer’s disease. Arch Neurosci 2, e60038. [Google Scholar]
- [142].Rowley DA, Halliwell B (1983) Superoxide-dependent and ascorbate-dependent formation of hydroxyl radicals in the presence of copper salts: A physiologically significant reaction? Arch Biochem Biophys 225, 279–284. [DOI] [PubMed] [Google Scholar]
- [143].Halliwell B, Gutteridge JMC (1984) Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem J 219, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Gutteridge JMC, Halliwell B (1995) Antioxidants in nutrition, health, and disease. Oxford University Press, Oxford, UK, pp 249–268. [Google Scholar]
- [145].Huang J, Agus DB, Winfree CJ, Kiss S, Mack WJ, McTaggart RA, Choudhri TF, Kim LJ, Mocco J, Pinsky DJ, Fox WD, Israel RJ, Boyd TA, Golde DW, Connolly ES (2001) Dehydroascorbic acid, a blood-brain barrier transportable form of vitamin C, mediates potent cerebroprotection in experimental stroke. Proc Natl Acad Sci U S A 98, 11720–11724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Guilloreau L, Combalbert S, Sournia-Saquet A, Mazarguil H, Faller P (2007) Redox chemistry of copper-amyloid-β: The generation of hydroxyl radical in the presence of ascorbate is linked to redox-potentials and aggregation state. Chembiochem 8, 1317–1325. [DOI] [PubMed] [Google Scholar]
- [147].Cheignon C, Tomas M, Bonnefont-Rousselot D, Faller P, Hureau C, Collin F (2018) Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol 14, 450–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Fang CL, Wu WH, Liu Q, Sun X, Ma Y, Zhao YF, Li YM (2010) Dual functions of β-amyloid oligomer and fibril in Cu(II)-induced H2O2 production. Regul Pept 163, 1–6. [DOI] [PubMed] [Google Scholar]
- [149].Reybier K, Ayala S, Alies B, Rodrigues JV, Bustos Rodriguez S, La Penna G, Collin F, Gomes CM, Hureau C, Faller P (2016) Free superoxide is an intermediate in the production of H2O2 by copper(I)-Aβ peptide and O2. Angew Chem Int Ed Engl 55, 1085–1089. [DOI] [PubMed] [Google Scholar]
- [150].Mirats A, Alí-Torres J, Rodríguez-Santiago L, Sodupe M, La Penna G (2015) Dioxygen activation in the Cu-amyloid β complex. Phys Chem Chem Phys 17, 27270–27274. [DOI] [PubMed] [Google Scholar]
- [151].Arrigoni F, Prosdocimi T, Mollica L, De Gioia L, Zampella G, Bertini L (2018) Copper reduction and dioxygen activation in Cu-amyloid beta peptide complexes: Insight from molecular modelling. Metallomics 10, 1618–1630. [DOI] [PubMed] [Google Scholar]
- [152].La Penna G, Li MS (2019) Computational models explain how copper binding to amyloid-β peptide oligomers enhances oxidative pathways. Phys Chem Chem Phys 21, 8774–8784. [DOI] [PubMed] [Google Scholar]
- [153].Bousejra-ElGarah F, Bijani C, Coppel Y, Faller P, Hureau C (2011) Iron(II) binding to amyloid-β, the Alzheimer’s peptide. Inorg Chem 50, 9024–9030. [DOI] [PubMed] [Google Scholar]
- [154].Valensin D, Migliorini C, Valensin G, Gaggelli E, La Penna G, Kozlowski H, Gabbiani C, Messori L (2011) Exploring the reactions of β-amyloid (Aβ) peptide 1–28 with Al(III) and Fe(III) ions. Inorg Chem 50, 6865–6867. [DOI] [PubMed] [Google Scholar]
- [155].Plascencia-Villa G, Ponce A, Collingwood JF, Arellano-Jiménez MJ, Zhu X, Rogers JT, Betancourt I, José-Yacamán M, Perry G (2016) High-resolution analytical imaging and electron holography of magnetite particles in amyloid cores of Alzheimer’s disease. Sci Rep 6, 24873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Bu X-L, Rao PPN, Wang Y-J (2016) Anti-amyloid aggregation activity of natural compounds: Implications for Alzheimer’s drug discovery. Mol Neurobiol 53, 3565–3575. [DOI] [PubMed] [Google Scholar]
- [157].Sharma A, Pachauri V, Flora SJS (2018) Advances in multi-functional ligands and the need for metal-related pharmacology for the management of Alzheimer disease. Front Pharmacol 9, 1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Kenche VB, Barnham KJ (2011) Alzheimer’s disease & metals: Therapeutic opportunities. Br J Pharmacol 163, 211–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, Barnham KJ, Volitakis I, Fraser FW, Kim YS, Huang X, Goldstein LE, Moir RD, Lim JT, Beyreuther K, Zheng H, Tanzi RE, Masters CL, Bush AI (2001) Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 30, 665–676. [DOI] [PubMed] [Google Scholar]
- [160].Wax PM (2013) Current use of chelation in american health care. J Med Toxicol 9, 303–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].World Health Organization (2018) World Health Organization model list of essential medicines. https://www.who.int/selection_medicines/list/en/ [Google Scholar]
- [162].Andersen O (2004) Chemical and biological considerations in the treatment of metal intoxications by chelating agents. Mini Rev Med Chem 4, 11–21. [DOI] [PubMed] [Google Scholar]
- [163].Aaseth J, Skaug MA, Cao Y, Andersen O (2015) Chelation in metal intoxication-Principles and paradigms. J Trace Elem Med Biol 31, 260–266. [DOI] [PubMed] [Google Scholar]
- [164].Ono K, Naiki H, Yamada M (2006) The development of preventives and therapeutics for Alzheimers disease that inhibit the formation of β-amyloid fibrils (fAβ), as well as destabilize preformed fAβ. Curr Pharm Des 12, 4357–4375. [DOI] [PubMed] [Google Scholar]
- [165].Bolognin S, Drago D, Messori L, Zatta P (2009) Chelation therapy for neurodegenerative diseases. Med Res Rev 29, 547–570. [DOI] [PubMed] [Google Scholar]
- [166].Budimir A (2011) Metal ions, Alzheimer’s disease and chelation therapy. Acta Pharm 61, 1–14. [DOI] [PubMed] [Google Scholar]
- [167].Yang G-J, Liu H, Ma DL, Leung CH (2019) Rebalancing metal dyshomeostasis for Alzheimer’s disease therapy. J Biol Inorg Chem 24, 1159–1170. [DOI] [PubMed] [Google Scholar]
- [168].Di Vaira M, Bazzicalupi C, Orioli P, Messori L, Bruni B, Zatta P (2004) Clioquinol, a drug for Alzheimer’s disease specifically interfering with brain metal metabolism: Structural characterization of its zinc(II) and copper(II) complexes. Inorg Chem 43, 3795–3797. [DOI] [PubMed] [Google Scholar]
- [169].Robert A, Liu Y, Nguyen M, Meunier B (2015) Regulation of copper and iron homeostasis by metal chelators: A possible chemotherapy for Alzheimer’s disease. Acc Chem Res 48, 1332–1339. [DOI] [PubMed] [Google Scholar]
- [170].Ritchie CW, Bush AI, Masters CL (2004) Metal-protein attenuating compounds and Alzheimer’s disease. Expert Opin Investig Drugs 13, 1585–1592. [DOI] [PubMed] [Google Scholar]
- [171].Nuñez M, Chana-Cuevas P (2018) New perspectives in iron chelation therapy for the treatment of neurodegenerative diseases. Pharmaceuticals 11, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Cukierman DS, Pinheiro AB, Castiñeiras-Filho SLP, da Silva ASP, Miotto MC, De Falco A, de P. Ribeiro T, Maisonette S, da Cunha ALMC, Hauser-Davis RA, Landeira-Fernandez J, Aucélio RQ, Outeiro TF, Pereira MD, Fernández CO, Rey NA (2017) A moderate metal-binding hydrazone meets the criteria for a bioinorganic approach towards Parkinson’s disease: Therapeutic potential, blood-brain barrier crossing evaluation and preliminary toxicological studies. J Inorg Biochem 170, 160–168. [DOI] [PubMed] [Google Scholar]
- [173].Masters CL, Beyreuther K (2006) Alzheimer’s centennial legacy: Prospects for rational therapeutic intervention targeting the Abeta amyloid pathway. Brain 129, 2823–2839. [DOI] [PubMed] [Google Scholar]
- [174].Hegde ML, Bharathi P, Suram A, Venugopal C, Jagannathan R, Poddar P, Srinivas P, Sambamurti K, Rao KJ, Scancar J, Messori L, Zecca L, Zatta P (2009) Challenges associated with metal chelation therapy in Alzheimer’s disease. J Alzheimers Dis 17, 457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Tahmasebinia F, Emadi S (2017) Effect of metal chelators on the aggregation of beta-amyloid peptides in the presence of copper and iron. Biometals 30, 285–293. [DOI] [PubMed] [Google Scholar]
- [176].Crouch PJ, Barnham KJ (2012) Therapeutic redistribution of metal ions to treat Alzheimer’s disease. Acc Chem Res 45, 1604–1611. [DOI] [PubMed] [Google Scholar]
- [177].Mancino AM, Hindo SS, Kochi A, Lim MH (2009) Effects of clioquinol on metal-triggered amyloid-β aggregation revisited. Inorg Chem 48, 9596–9598. [DOI] [PubMed] [Google Scholar]
- [178].Chang PT, Kung FL, Talekar RS, Chen CS, Lai SY, Lee HY, Chern JW (2009) An improved screening model to identify inhibitors targeting zinc-enhanced amyloid aggregation. Anal Chem 81, 6944–6951. [DOI] [PubMed] [Google Scholar]
- [179].Dong J, Atwood CS, Anderson VE, Siedlak SL, Smith MA, Perry G, Carey PR (2003) Metal binding and oxidation of amyloid-β within isolated senile plaque cores: Raman microscopic evidence. Biochemistry 42, 2768–2773. [DOI] [PubMed] [Google Scholar]
- [180].Barnham KJ, Bush AI (2014) Biological metals and metal-targeting compounds in major neurodegenerative diseases. Chem Soc Rev 43, 6727–6749. [DOI] [PubMed] [Google Scholar]
- [181].Telpoukhovskaia MA, Rodríguez-Rodríguez C, Cawthray JF, Scott LE, Page BDG, Alí-Torres J, Sodupe M, Bailey GA, Patrick BO, Orvig C (2014) 3-hydroxy-4-pyridinone derivatives as metal ion and amyloid binding agents. Metallomics 6, 249–262. [DOI] [PubMed] [Google Scholar]
- [182].Faux NG, Ritchie CW, Gunn A, Rembach A, Tsatsanis A, Bedo J, Harrison J, Lannfelt L, Blennow K, Zetterberg H, Ingelsson M, Masters CL, Tanzi RE, Cummings JL, Herd CM, Bush AI (2010) PBT2 rapidly improves cognition in Alzheimer’s disease: Additional phase II analyses. J Alzheimers Dis 20, 509–516. [DOI] [PubMed] [Google Scholar]
- [183].Angus D, Herd C, Stone C, Stout J, Wieler M, Reilmann R, Ritchie CW, Dorsey ER, Helles K, Kayson E, Oakes D, Rosas HD, Vaughan C, Panegyres PK, Ames D, Goh A, Agarwal P, Churchyard A, Murathodizic M, Chua P, Germaine D, Lim DL, Mack H, Loy C, Griffith J, Mitchell P, Corey-Bloom J, Gluhm S, Goldstein J, Levi L, Rosas HD, Margolis R, Yoritomo N, Janicki S, Marder K, Clouse R, Singer C, Moore H, Padron N, Kostyk S, Daley A, Segro V, Kumar R, Anderson K, Drazinic C, Hennig B, Nance M, Molho E, Criswell S, LeDoux MS, Guyot S, Iannaccone A, Jennings B, Leavitt BR, Feigin A, Evans S, Wray S, Casaceli C, Orme C, Gao S, Watts A, Baker K, Labuschagne I, El-Dairi M, Fekrat S, Hersch S, Moscovitch-Lopatin M, Angus D, Herd C, Stone C, Ritchie CW, Tanzi R, Targum S (2015) Safety, tolerability, and efficacy of PBT2 in Huntington’s disease: A phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol 14, 39–47. [DOI] [PubMed] [Google Scholar]
- [184].Pérez-Jiménez J, Neveu V, Vos F, Scalbert A (2010) Systematic analysis of the content of 502 polyphenols in 452 foods and beverages: An application of the phenol-explorer database. J Agric Food Chem 58, 4959–4969. [DOI] [PubMed] [Google Scholar]
- [185].Tsao R (2010) Chemistry and biochemistry of dietary polyphenols. Nutrients 2, 1231–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Kim Y, Keogh JB, Clifton PM (2016) Polyphenols and glycemic control. Nutrients 8, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Cory H, Passarelli S, Szeto J, Tamez M, Mattei J (2018) The role of polyphenols in human health and food systems: A mini-review. Front Nutr 5, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Mennen LI, Walker R, Bennetau-Pelissero C, Scalbert A (2005) Risks and safety of polyphenol consumption. Am J Clin Nutr 81, 326S–329S. [DOI] [PubMed] [Google Scholar]
- [189].Piccolella S, Crescente G, Candela L, Pacifico S (2019) Nutraceutical polyphenols: New analytical challenges and opportunities. J Pharm Biomed Anal 175, 112774. [DOI] [PubMed] [Google Scholar]
- [190].Gollucke AP, Peres RC, Odair A Jr, Ribeiro DA (2013) Polyphenols: A nutraceutical approach against diseases. Recent Pat Food Nutr Agric 5, 214–219. [DOI] [PubMed] [Google Scholar]
- [191].Quero J, Mármol I, Cerrada E, Rodríguez-Yoldi MJ (2020) Insight into the potential application of polyphenol-rich dietary intervention in degenerative disease management. Food Funct 11, 2805–2825. [DOI] [PubMed] [Google Scholar]
- [192].Lee WH, Loo CY, Bebawy M, Luk F, Mason R, Rohanizadeh R (2013) Curcumin and its derivatives: Their application in neuropharmacology and neuroscience in the 21st century. Curr Neuropharmacol 11, 338–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Iranshahi M, Chini MG, Masullo M, Sahebkar A, Javidnia A, Chitsazian Yazdi M, Pergola C, Koeberle A, Werz O, Pizza C, Terracciano S, Piacente S, Bifulco G (2015) Can small chemical modifications of natural pan-inhibitors modulate the biological selectivity? The case of curcumin prenylated derivatives acting as HDAC or mPGES-1 inhibitors. J Nat Prod 78, 2867–2879. [DOI] [PubMed] [Google Scholar]
- [194].Takahashi M, Ishiko T, Kamohara H, Hidaka H, Ikeda O, Ogawa M, Baba H (2007) Curcumin (1,7-bis(4-hydroxy-3-methoxyphenyl)-1, 6-heptadiene-3,5-dione) blocks the chemotaxis of neutrophils by inhibiting signal transduction through IL-8 receptors. Mediators Inflamm 2007, 10767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Jurenka JS (2009) Anti-inflammatory properties of curcumin, a major constituent of Curcuma longa: A review of preclinical and clinical research. Altern Med Rev 14, 141–53. [PubMed] [Google Scholar]
- [196].Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB (2007) Bioavailability of curcumin: Problems and promises. Mol Pharm 4, 807–818. [DOI] [PubMed] [Google Scholar]
- [197].Lakey-Beitia J, González Y, Doens D, Stephens DE, Santamaría R, Murillo E, Gutiérrez M, Fernández PL, Rao KS, Larionov OV, Durant-Archibold AA (2017) Assessment of novel curcumin derivatives as potent inhibitors of inflammation and amyloid-β aggregation in Alzheimer’s disease. J Alzheimers Dis 60(Suppl 1), S59–S68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Mishra S, Palanivelu K (2008) The effect of curcumin (turmeric) on Alzheimer’s disease: An overview. Ann Indian Acad Neurol 11, 13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Elmegeed GA, Ahmed HH, Hashash MA, Abd-Elhalim MM, El-Kady DS (2015) Synthesis of novel steroidal curcumin derivatives as anti-Alzheimer’s disease candidates: Evidences-based on in vivo study. Steroids 101, 78–89. [DOI] [PubMed] [Google Scholar]
- [200].Ferrari E, Benassi R, Sacchi S, Pignedoli F, Asti M, Saladini M (2014) Curcumin derivatives as metal-chelating agents with potential multifunctional activity for pharmaceutical applications. J Inorg Biochem 139, 38–48. [DOI] [PubMed] [Google Scholar]
- [201].Mary CPV, Vijayakumar S, Shankar R (2018) Metal chelating ability and antioxidant properties of curcumin-metal complexes - A DFT approach. J Mol Graph Model 79, 1–14. [DOI] [PubMed] [Google Scholar]
- [202].Perron NR, Brumaghim JL (2009) A review of the antioxidant mechanisms of polyphenol compounds related to iron binding. Cell Biochem Biophys 53, 75–100. [DOI] [PubMed] [Google Scholar]
- [203].Singh NA, Mandal AKA, Khan ZA (2015) Potential neuroprotective properties of epigallocatechin-3-gallate (EGCG). Nutr J 15, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Lee Y-J, Choi D-Y, Yun Y-P, Han SB, Oh K-W, Hong JT (2013) Epigallocatechin-3-gallate prevents systemic inflammation-induced memory deficiency and amyloido-genesis via its anti-neuroinflammatory properties. J Nutr Biochem 24, 298–310. [DOI] [PubMed] [Google Scholar]
- [205].Weinreb O, Amit T, Youdim MBH (2007) A novel approach of proteomics and transcriptomics to study the mechanism of action of the antioxidant-iron chelator green tea polyphenol (−)-epigallocatechin-3-gallate. Free Radic Biol Med 43, 546–556. [DOI] [PubMed] [Google Scholar]
- [206].Weinreb O, Amit T, Mandel S, Youdim MBH (2009) Neuroprotective molecular mechanisms of (−)-epigallocatechin-3-gallate: A reflective outcome of its antioxidant, iron chelating and neuritogenic properties. Genes Nutr 4, 283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Hyung SJ, DeToma AS, Brender JR, Lee S, Vivekanandan S, Kochi A, Choi JS, Ramamoorthy A, Ruotolo BT, Lim MH (2013) Insights into antiamyloidogenic properties of the green tea extract (−)-epigallocatechin-3-gallate toward metal-associated amyloid- species. Proc Natl Acad Sci U S A 110, 3743–3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Zhang B, Cheng XR, da Silva IS, Hung VWS, Veloso AJ, Angnes L, Kerman K (2013) Electroanalysis of the interaction between (−)-epigallocatechin-3-gallate (EGCG) and amyloid-β in the presence of copper. Metallomics 5, 259. [DOI] [PubMed] [Google Scholar]
- [209].Savelieff MG, DeToma AS, Derrick JS, Lim MH (2014) The ongoing search for small molecules to study metal-associated amyloid-β species in Alzheimers disease. Acc Chem Res 47, 2475–2482. [DOI] [PubMed] [Google Scholar]
- [210].Jomova K, Lawson M, Drostinova L, Lauro P, Poprac P, Brezova V, Michalik M, Lukes V, Valko M (2017) Protective role of quercetin against copper(II)-induced oxidative stress: A spectroscopic, theoretical and DNA damage study. Food Chem Toxicol 110, 340–350. [DOI] [PubMed] [Google Scholar]
- [211].Tay WM, da Silva GFZ, Ming L-J (2013) Metal binding of flavonoids and their distinct inhibition mechanisms toward the oxidation activity of Cu2+-β-amyloid: Not just serving as suicide antioxidants! Inorg Chem 52, 679–690. [DOI] [PubMed] [Google Scholar]
- [212].Zhao B, Guengerich FP, Bellamine A, Lamb DC, Izumikawa M, Lei L, Podust LM, Sundaramoorthy M, Kalaitzis JA, Reddy LM, Kelly SL, Moore BS, Stec D, Voehler M, Falck JR, Shimada T, Waterman MR (2005) Binding of two flaviolin substrate molecules, oxidative coupling, and crystal structure of streptomyces coelicolor A3(2) cytochrome P450 158A2. J Biol Chem 280, 11599–11607. [DOI] [PubMed] [Google Scholar]
- [213].Hider RC, Liu ZD, Khodr HH (2001) Metal chelation of polyphenols. Methods Enzymol 335, 190–203. [DOI] [PubMed] [Google Scholar]
- [214].Li Y, Li Z, Hou H, Zhuang Y, Sun L (2018) Metal chelating, inhibitory DNA damage, and anti-inflammatory activities of phenolics from rambutan (nephelium lappaceum) peel and the quantifications of geraniin and corilagin. Molecules 23, 2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Kontoghiorghe C, Kolnagou A, Kontoghiorghes G (2015) Phytochelators intended for clinical use in iron overload, other diseases of iron imbalance and free radical pathology. Molecules 20, 20841–20872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Belščak-Cvitanović A, Durgo K, Huđjek A, Bačun-Družina V, Komes D (2018) Overview of polyphenols and their properties. In Polyphenols: Properties, Recovery, and Applications. Woodhead Publishing, Duxford, UK, pp. 3–44. [Google Scholar]
- [217].Wei Y, Guo M (2014) Zinc-binding sites on selected flavonoids. Biol Trace Elem Res 161, 223–230. [DOI] [PubMed] [Google Scholar]
- [218].Fraga CG, Galleano M, Verstraeten SV Oteiza PI (2010) Basic biochemical mechanisms behind the health benefits of polyphenols. Mol Aspects Med 31, 435–445. [DOI] [PubMed] [Google Scholar]
- [219].Korshavn KJ, Jang M, Kwak YJ, Kochi A, Vertuani S, Bhunia A, Manfredini S, Ramamoorthy A, Lim MH (2015) Reactivity of metal-free and metal-associated amyloid-β with glycosylated polyphenols and their esterified derivatives. Sci Rep 5, 17842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Pearson RG (1963) Hard and soft acids and bases. J Am Chem Soc 85, 3533–3539. [Google Scholar]
- [221].Telpoukhovskaia MA, Orvig C (2013) Werner coordination chemistry and neurodegeneration. Chem Soc Rev 42, 1836–1846. [DOI] [PubMed] [Google Scholar]
- [222].Santos MA, Chand K, Chaves S (2016) Recent progress in multifunctional metal chelators as potential drugs for Alzheimer’s disease. Coord Chem Rev 327–328, 287–303. [Google Scholar]
- [223].Haas KL, Franz KJ (2009) Application of metal coordination chemistry to explore and manipulate cell biology. Chem Rev 109, 4921–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Chan S, Kantham S, Rao VM, Palanivelu MK, Pham HL, Shaw PN, McGeary RP, Ross BP (2016) Metal chelation, radical scavenging and inhibition of Aβ42 fibrillation by food constituents in relation to Alzheimer’s disease. Food Chem 199, 185–194. [DOI] [PubMed] [Google Scholar]
- [225].Priyadarsini KI (2014) The chemistry of curcumin: From extraction to therapeutic agent. Molecules 19, 20091–20112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Tsukahara T, Nagaoka K, Morikawa K, Mawatari K, Kitamori T (2015) Keto-enol tautomeric equilibrium of acetylacetone solution confined in extended nanospaces. J Phys Chem B 119, 14750–14755. [DOI] [PubMed] [Google Scholar]
- [227].Nam G, Ji Y, Lee HJ, Kang J, Yi Y, Kim M, Lin Y, Lee YH, Lim MH (2019) Orobol: An isoflavone exhibiting regulatory multifunctionality against four pathological features of Alzheimer’s disease. ACS Chem Neurosci 10, 3386–3390. [DOI] [PubMed] [Google Scholar]
- [228].Fernandez MT, Mira ML, Florêncio MH, Jennings KR (2002) Iron and copper chelation by flavonoids: An electrospray mass spectrometry study. J Inorg Biochem 92, 105–111. [DOI] [PubMed] [Google Scholar]
- [229].Ríha M, Karlícková J, Filipský T, Jahodár L, Hrdina R, Mladenka P (2014) In vitro copper-chelating properties of flavonoids. Free Radic Biol Med 75, S46. [DOI] [PubMed] [Google Scholar]
- [230].Mladěnka P, Macáková K, Filipský T, Zatloukalová L, Jahodář L, Bovicelli P, Silvestri IP, Hrdina R, Saso L (2011) In vitro analysis of iron chelating activity of flavonoids. J Inorg Biochem 105, 693–701. [DOI] [PubMed] [Google Scholar]
- [231].Bors W, Heller W, Michel C, Saran M (1990) Flavonoids as antioxidants: Determination of radical-scavenging efficiencies. Methods Enzymol 186, 343–355. [DOI] [PubMed] [Google Scholar]
- [232].Picciano AL, Vaden TD (2013) Complexation between Cu(II) and curcumin in the presence of two different segments of amyloid β. Biophys Chem 184, 62–67. [DOI] [PubMed] [Google Scholar]
- [233].Huy PDQ, Vuong Q Van, La Penna, Faller P, Li MS(2016) Impact of Cu(II) binding on structures and dynamics of aβ42 monomer and dimer: Molecular dynamics study. ACS Chem Neurosci 7, 1348–1363. [DOI] [PubMed] [Google Scholar]
- [234].Kozmon S, Tvaroška I (2015) Molecular dynamic studies of amyloid-beta interactions with curcumin and Cu2+ ions. Chem Pap 69, 1262–1276. [Google Scholar]
- [235].Renaud J, Martinoli M-G (2019) Considerations for the use of polyphenols as therapies in neurodegenerative diseases. Int J Mol Sci 20, 1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Utsuki T, Yu Q, Davidson D, Chen D, Holloway HW, Brossi A, Sambamurti K, Lahiri DK, Greig NH, Giordano T (2006) Identification of novel small molecule inhibitors of amyloid precursor protein synthesis as a route to lower Alzheimer’s disease amyloid-beta peptide. J Pharmacol Exp Ther 318, 855–862. [DOI] [PubMed] [Google Scholar]
- [237].Yan J, Hu J, Liu A, He L, Li X, Wei H (2017) Design, synthesis, and evaluation of multitarget-directed ligands against Alzheimer’s disease based on the fusion of donepezil and curcumin. Bioorganic Med Chem 25, 2946–2955. [DOI] [PubMed] [Google Scholar]