

Abstract
Objectives
Francisella tularensis, the causative agent of tularaemia, is an exceptionally infectious bacterium, potentially fatal for humans if left untreated and with the potential to be developed as a bioweapon. Both natural infection and live‐attenuated vaccine strain (LVS) confer good protection against tularaemia. LVS vaccination is traditionally administered by scarification, and the formation of a cutaneous reaction or take at the vaccination site is recognised as a clinical correlate of protection. Although previous studies have suggested that high antibody titres following vaccination might serve as a useful surrogate marker, the immunological correlates of protection remain unknown.
Methods
We investigated the host T‐cell‐mediated immune (T‐CMI) responses elicited following immunisation with LVS vaccine formulated by the DynPort Vaccine Company (DVC‐LVS) or the United States Army Medical Research Institute of Infectious Diseases (USAMRIID‐LVS). We compared T‐CMI responses prompted by these vaccines and correlated them with take size.
Results
We found that both LVS vaccines elicited similar T‐CMI responses. Interestingly, take size associated with the T cells’ ability to proliferate, secrete IFN‐γ and mobilise degranulation, suggesting that these responses play an essential role in tularaemia protection.
Conclusions
These results renew the appreciation for vaccination through the scarification as a prime route of inoculation to target pathogens driving specific T‐CMI responses and provide further evidence that T‐CMI plays a role in protection from tularaemia.
Keywords: human, T cells, take, tularaemia, vaccination
The actual immunological correlate(s) of protection from tularaemia in humans remain(s) unknown. As with smallpox, take is widely accepted as a clinical correlate of protection against tularaemia. We report, for the first time, a correlation between take‐lesion size and cellular‐mediated immune responses in humans after tularaemia immunization.

Introduction
Francisella tularensis (F. tularensis) is an aerobic, gram‐negative, intracellular bacterium and the causative agent of tularaemia, a zoonotic disease. 1 , 2 Although F. tularensis does not spread from person to person through direct contact, transmission may occur when infected insects bite humans, or when humans inadvertently inhale the bacteria, or handle, eat or drink contaminated products. 2 Francisella tularensis can survive for weeks in water, soil, moist hay and decaying animal carcasses. 3 The worldwide incidence of human tularaemia has declined markedly over the past decades. 4 In the United States, these numbers have changed from several thousand in the 1930s to currently 90–154 cases annually. 4 , 5 , 6 Nevertheless, F. tularensis is very infectious and potentially fatal if left untreated 2 , 7 , 8 , and there is a long history of developing it as a bioweapon. 6 , 9 As few as 10–50 inhaled organisms might result in clinical tularaemia. 10 , 11
Seminal tularaemia vaccine studies have shown that live‐attenuated vaccine strain (LVS) is immunologically superior to the killed virulent strain for vaccination in mice, guinea pigs, monkeys and humans. 12 , 13 , 14 While killed tularaemia vaccine (Foshay) does protect against systemic infection and decreases the severity of the disease, it does not prevent local infection. 10 In contrast, natural infection with F. tularensis or vaccination with LVS protects against tularaemia. 10 , 15 , 16 , 17 Our group and others have found that LVS vaccination elicits a broad spectrum of humoral and cellular‐mediated immune responses 18 , 19 , 20 , 21 , 22 , including specific memory T cells which produce IFN‐γ and exhibit diverse homing characteristics. 23 The LVS vaccine is commonly administered by scarification using a bifurcated needle. A common reaction is the formation of vaccine‐related major cutaneous reaction or ‘take’ at the vaccination site. Similar to smallpox 24 , 25 , 26 , take is recognised as a clinical correlate of protection against tularaemia. 11 , 18
To replenish its limited and ageing supply of LVS vaccine, the US government has funded both the production of new lots of LVS vaccine and a phase 2 clinical trial to compare the new lots produced by DynPort Vaccine Company (DVC) to older lots prepared by the United States Army Medical Research Institute of Infectious Diseases (USAMRIID). In this phase 2 clinical trial, seroconversion, as measured by a tularaemia‐specific microagglutination assay, was observed in over 90% of subjects and was similar for both vaccines. 18 However, serological responses overestimated vaccine efficacy if the take was used as the gold standard for protection. Furthermore, no correlation was found between take and seroconversion for either vaccine. 18
We hypothesise that cellular T‐cell‐mediated immune responses (T‐CMI) rather than humoral responses correlate with take after LVS vaccination. This hypothesis is based on previous work showing that B‐cell depletion did not affect the size of skin lesions induced by the smallpox vaccine. 27 Also, scarification is critical for the generation of protective T‐CMI following traditional vaccination for smallpox. 28 Here, we utilised specimens from the phase 2 clinical trial to investigate the host T‐CMI responses following tularaemia vaccination and correlated them with take.
Results
No evidence of differences between the magnitude of proliferation rates by treatment group
First, we investigated whether there was a difference in the number of responders among the two vaccines, DVC‐LVS and USAMRIID‐LVS. To this end, we measured F. tularensis‐specific cells that proliferated in the presence of Schu‐S4, as determined by the incorporation of the radioactive compound [3H]‐thymidine. The more cells are proliferating, the more radioactivity is incorporated into these expanding cells. Thus, the number of proliferating cells is directly proportional to the amount of radiation in the sample. We tested a total of 228 subjects at six time points, days 0, 8, 14, 28, 56 and 180. We found that for the per‐protocol (PP) population, the proportion of positive responders was 90.2% (92/102) for DVC‐LVS and 88.0% (95/108) for USAMRIID‐LVS P = 0.663, two‐sided Fisher’s exact test) (Table 1). Thus, there is no evidence of a significant difference in the proliferative responses.
Table 1.
Positive responses based on cell proliferation after Schu‐S4 exposure measured by incorporation of [3H]‐thymidine
|
Time Post‐Vaccination |
Group a | |||
|---|---|---|---|---|
| DVC‐LVS | USAMRIID‐LVS | |||
| n b /N (%) | 95% CI | n/N (%) | 95% CI | |
| Day 8 | 21/100 (21.0) | 13.5, 30.3 | 16/107 (15.0) | 8.8, 23.1 |
| Day 14 | 32/99 (32.3) | 23.3, 42.5 | 26/104 (25.0) | 17.0, 34.4 |
| Day 28 | 59/99 (59.6) | 49.3, 69.3 | 64/103 (62.1) | 52.0, 71.5 |
| Day 56 | 70/99 (70.7) | 60.7, 79.4 | 71/102 (69.6) | 59.7, 78.3 |
| Day 180 | 58/89 (65.2) | 54.3, 75.0 | 67/94 (71.3) | 61.0, 80.1 |
| Any (8–180) | 92/102 (90.2) | 82.7, 95.2 | 95/108 (88.0) | 80.3, 93.4 |
Per‐protocol population.
Positive criteria: (1) Visit (SCHU‐S4 [cpm]/Media [cpm]) ≥ 3, (2) Visit (SCHU‐S4 [cpm] − Media [cpm]) > 500 cpm and (3) Visit (SCHU‐S4 [cpm]/Media [cpm]) ≥ 2 × baseline (day 0) (SCHU‐S4 [cpm]/Media [cpm]).
Similarly, there were no significant differences between DVC‐LVS and USAMRIID‐LVS in the proportion of positive responders when compared separately for each visit (Day 0, 8, 14, 28, 56, and 180). For both vaccines, when analysing the cell proliferation kinetics over 180 days of follow‐up, we detected proliferative responses as early as 8 days with a plateau about days 56 and 180 after immunisation (Figure 1). Finally, for both vaccines, the magnitude of the peak responses, defined to be the maximum response regardless of the time point, was higher than the values on days 56 and 180 (Figure 1).
Figure 1.
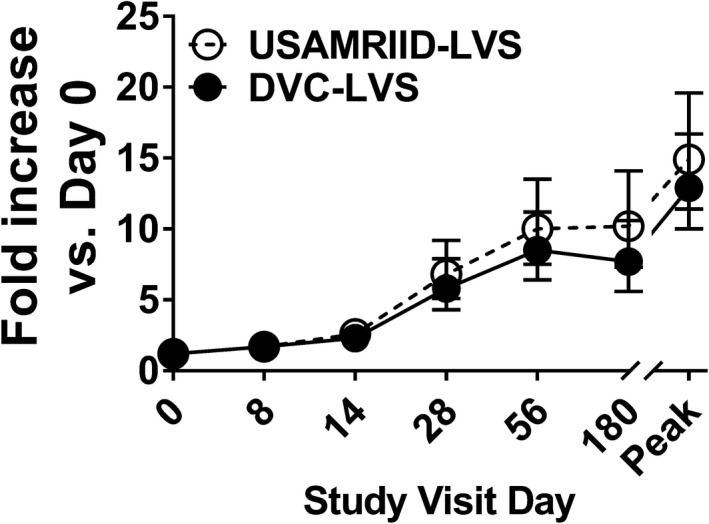
No evidence of differences between proliferation magnitude rates by treatment group. Cells from 228 subjects at six time points (days 0, 8, 14, 28, 56 and 180) were stimulated with Schu‐S4. After 6 days of stimulation, cells were harvested, and cell proliferation determined by [3H]‐thymidine incorporation. Symbols and error bars represent the geometric mean of the fold increase with 95% confidence intervals. Peak, peak fold increase as defined by the maximum fold increase among all the available measures on time points post‐vaccination. Per‐Protocol Population. Data are representative of 66 independent experiments with 4 replicates.
Next, a linear mixed‐effects model was fit to describe the geometric mean fold rise (GMFR) in proliferation as a function of treatment, visit, gender and age. The highest‐order interaction (visit*treatment*age) was not significant, indicating that the two groups did not behave differently across visits for different age categories. There were no significant interactions between treatment and gender or age or visit. Also, the interaction between the treatment and visit (visit*treatment) was not significant. Thus, the main treatment effects were averaged over levels of the visit factor. The two vaccine types were not significantly different (P‐value = 0.350, F‐Test). The estimate was 3.7 (3.1, 4.5) for DVC‐LVS and 4.2 (3.5, 5.1) for USAMRIID‐LVS. GMFR was significantly different for age across visits (visit*age) (P‐value = 0.042, F‐Test). Thus, age effects were analysed separately for each visit. The hypothesis of equality of proliferation GMFR across age groups was rejected with alpha = 0.05 for: day 8 (P‐value = 0.035, GMFR ‘25–29 years’ > ‘30–45 years’ > ‘18–24 years’), and day 56 (P‐value = 0.014, GMFR ‘30–45 years’ > ‘18–24 years’ > ‘25–29 years’).
Kinetics of IFN‐γ‐secreting cells after LVS immunisation
Since recent data suggested that IFN‐γ response is required for anti‐tularaemia immune protection, 22 , 23 , 29 , 30 , 31 , 32 we measured IFN‐γ production in the cell culture supernatants 4 days after Schu‐S4 stimulation using the Meso Scale Discovery (MSD) assay. We tested a total of 228 subjects at six time points, days 0, 8, 14, 28, 56 and 180, and found that for the PP population, the proportion of positive responders was 90.2% (92/102) for DVC‐LVS and 90.7% (98/108) for USAMRIID‐LVS. There was no evidence in the data of a significant difference (P > 0.99, two‐sided Fisher’s exact test) (Table 2).
Table 2.
Positive responses based on the secretion of IFN‐γ after Schu‐S4 stimulation measured by MSD assay
|
Time Post‐Vaccination |
Group a | |||
|---|---|---|---|---|
| DVC‐LVS | USAMRIID‐LVS | |||
| n b /N (%) | 95% CI | n/N (%) | 95% CI | |
| Day 8 | 27/100 (27.0) | 18.6, 36.8 | 30/107 (28.0) | 19.8, 37.5 |
| Day 14 | 44/99 (44.4) | 34.5, 54.8 | 39/104 (37.5) | 28.2, 47.5 |
| Day 28 | 61/99 (61.6) | 51.3, 71.2 | 65/103 (63.1) | 53.0, 72.4 |
| Day 56 | 75/99 (75.8) | 66.1, 83.8 | 69/102 (67.6) | 57.7, 76.6 |
| Day 180 | 65/89 (73.0) | 62.6, 81.9 | 65/94 (69.1) | 58.8, 78.3 |
| Any (8–180) | 92/102 (90.2) | 82.7, 95.2 | 98/108 (90.7) | 83.6, 95.5 |
Per‐protocol population.
Positive criteria: (1) Visit (SCHU‐S4 [pg mL−1]/Media [pg mL−1]) ≥ 2 and (2) Visit (SCHU‐S4 [pg mL−1]/Media [pg mL−1]) ≥ 2 × baseline (day 0) (SCHU‐S4 [pg mL−1]/Media [pg mL−1]).
Similarly, there was no difference in the proportions of positive responders using geometric mean titres compared separately for each visit (days 0, 8, 14, 28, 56 and 180) using a two‐sided Fisher’s exact test. For the PP population, the null hypothesis of equality of the geometric means could not be rejected with P > 0.35 for any of the six visits.
A linear mixed‐effects model was fit to describe the GMFR in IFN‐γ as a function of treatment, visit, gender and age. The highest‐order interaction (visit*treatment*age) was not significant, indicating that the two groups did not behave differently across visits for different age categories. There were no significant interactions between treatment and gender or age. Also, the interaction between the treatment and visit (visit*treatment) was not significant. Thus, treatment main effects (treatment) were averaged over levels of the visit factor. The two vaccine types were not significantly different (PP: P‐value = 0.255, F‐Test). The GMFR (95% CI) estimate (averaged over all visit levels) was 5.9 (4.6, 7.7) for DVC‐LVS and 4.8 (3.7, 6.2) for USAMRIID‐LVS. The model indicated that IFN‐γ GMFR did not differ by gender. Like proliferation, GMFR was significantly different for age, with the oldest age group obtaining the highest GMFR (PP: P‐value = 0.014, F‐Test, GMFR ‘30–45 years’ > ‘18–24 years’ > ‘25–29 years’). For both vaccines, DVC‐LVS and USAMRIID‐LVS, the peak response occurred at day 56 (Figure 2a). A positive correlation was found between proliferation magnitude and IFN‐γ levels for both vaccines (Figure 2b, c).
Figure 2.
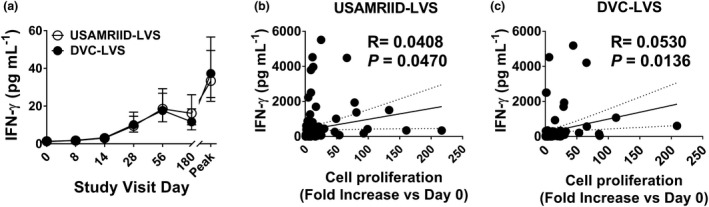
Levels of IFN‐γ secretion directly correlated with cell proliferation magnitude. Cells from 228 subjects at six time points (days 0, 8, 14, 28, 56 and 180) were stimulated with Schu‐S4. (a) After 4 days of stimulation, supernatants were harvested and used to measure IFN‐γ production using MSD technology. Symbols and error bars represent the geometric mean of the IFN‐γ levels (pg mL−1) with 95% confidence interval. Peak, peak increase as defined by the maximum level of IFN‐γ increase among all the available measures on time points post‐vaccination. Data are representative of 32 independent experiments with one replicate. Correlation between IFN‐γ levels and cell proliferation magnitudes, for both vaccines, USAMRIID‐LVS (b) and DVC‐LVS (c). Trendlines (solid lines), the correlation coefficient R and P‐values are shown. P‐values of < 0.05 were considered statistically significant. Dashed lines represent 95% confidence intervals. Per‐Protocol Population.
Cumulative CD4+ and CD8+ T‐cell responses over 180 days after immunisation
Our group and others have shown that IFN‐γ‐secreting CD4+ and CD8+ T‐cell subsets are elicited in humans following ex vivo and in vivo exposure to F. tularensis and are likely to be critical for protection. 23 , 29 , 33 To investigate the relationship between IFN‐γ production and the frequency and function of the CD4+, and CD8+ T‐cell subsets, ex vivo PBMC from LVS vaccinees were exposed to Schu‐S4 for ~20 h. After stimulation, their production of IL‐2, IFN‐γ and TNF‐α cytokines and/or expression of CD107a and b molecules was assessed by flow cytometry. The 43 volunteers recruited at the University of Maryland site were evaluated, and analyses were performed at six time points, days 0, 8, 14, 28, 56 and 180. For the PP population, we found that although the proportion of positive responders based on CD4+ T‐cell expression of IFN‐γ was 38.1% (8/21) for DVC‐LVS and 27.8% (5/18) for USAMRIID‐LVS, it did not achieve statistical significance (P = 0.734, two‐sided Fisher’s exact test) (Table 3). We also found that the proportion of positive responders based on CD8+ T‐cell expression of IFN‐γ was 57.1% (12/21) for DVC‐LVS and 38.9% (7/18) for USAMRIID‐LVS (Table 3). However, this was not statistically significant (P = 0.341, two‐sided Fisher’s exact test). Supplementary figure 1 shows CD4+ and CD8+ T‐cell responses from a representative volunteer.
Table 3.
Positive responses based on the IFN‐γ production after Schu‐S4 stimulation measured by flow cytometry
|
Time Post‐Vaccination |
Group a | |||||||
|---|---|---|---|---|---|---|---|---|
| CD4+ T cells | CD8+ T cells | |||||||
| DVC‐LVS | USAMRIID‐LVS | DVC‐LVS | USAMRIID‐LVS | |||||
| n b /N (%) | 95% CI | n/N (%) | 95% CI | n/N (%) | 95% CI | n/N (%) | 95% CI | |
| Day 8 | 2/21 (9.5) | 1.2, 30.4 | 2/18 (11.1) | 1.4, 34.7 | 3/21 (14.3) | 3.0, 36.3 | 4/18 (22.2) | 6.4, 47.6 |
| Day 14 | 2/20 (10.0) | 1.2, 31.7 | 1/18 (5.6) | 0.1, 27.3 | 3/20 (15.0) | 3.2, 37.9 | 1/18 (5.6) | 0.1, 27.3 |
| Day 28 | 3/21 (14.3) | 3.0, 36.3 | 2/17 (11.8) | 1.5, 36.4 | 2/21 (9.5) | 1.2, 30.4 | 2/17 (11.8) | 1.5, 36.4 |
| Day 56 | 3/21 (14.3) | 3.0, 36.3 | 0/17 (0.0) | 0.0, 19.5 | 6/21 (28.6) | 11.3, 52.2 | 0/17 (0.0) | 0.0, 19.5 |
| Day 180 | 6/21 (28.6) | 11.3, 52.2 | 2/16 (12.5) | 1.6, 38.3 | 6/21 (28.6) | 11.3, 52.2 | 1/16 (6.3) | 0.2, 30.2 |
| Any (8–180) | 8/21 (38.1) | 18.1, 61.6 | 5/18 (27.8) | 9.7, 53.5 | 12/21 (57.1) | 34.0, 78.2 | 7/18 (38.9) | 17.3, 64.3 |
Per‐protocol population.
Positive criteria: (1) Visit (SCHU‐S4 [% cells] − Media [cells %]) ≥ 0.1%, (2) Visit (SCHU‐S4 [% cells] − Media [% cells]) − Baseline (SCHU‐S4 [% cells] − Media [% cells]) ≥ 0.1% and (3) P‐value < 0.01 and Visit (SCHU‐S4 [cells]) > 10.
Since both vaccines appeared to induce similar T‐CMI responses with no evidence of significant differences, we combined all individuals into a composite and then stratified them into two groups based on the take results: (1) take positive (Take, n = 37) or (2) take negative (non‐take, n = 6). For the positive take group, when comparing CD4+ and CD8+ T‐cell responses, we observed different patterns in the IFN‐γ, CD107a/b and TNF‐α expression in the 14 days following immunisation, with levels consistently being above baseline (Figure 3 and Supplementary figures 2 and 3). Interestingly, the curves representing IL‐2 expression showed that CD8+ T‐cell levels had a sharp decline when compared to those of CD4+ T cells at the early time points following immunisation (Supplementary figure 4). For volunteers without a take, regardless of the T‐cell subset, T‐cell levels steadily declined 14 days after vaccination, a timeframe used to capture the early events occurring after LVS immunisation. Successful vaccination is characterised by the induction of robust and persistent immune responses. 34 , 35 , 36 We used the area under the curve (AUC) analysis as a tool to cumulatively measure the rise and persistence of T‐cell immune responses above baseline (day 0) after immunisation. Except for CD107a/b expression, the AUC values were consistently higher for CD4+ T cells than for CD8+ T cells (Supplementary table 1).
Figure 3.
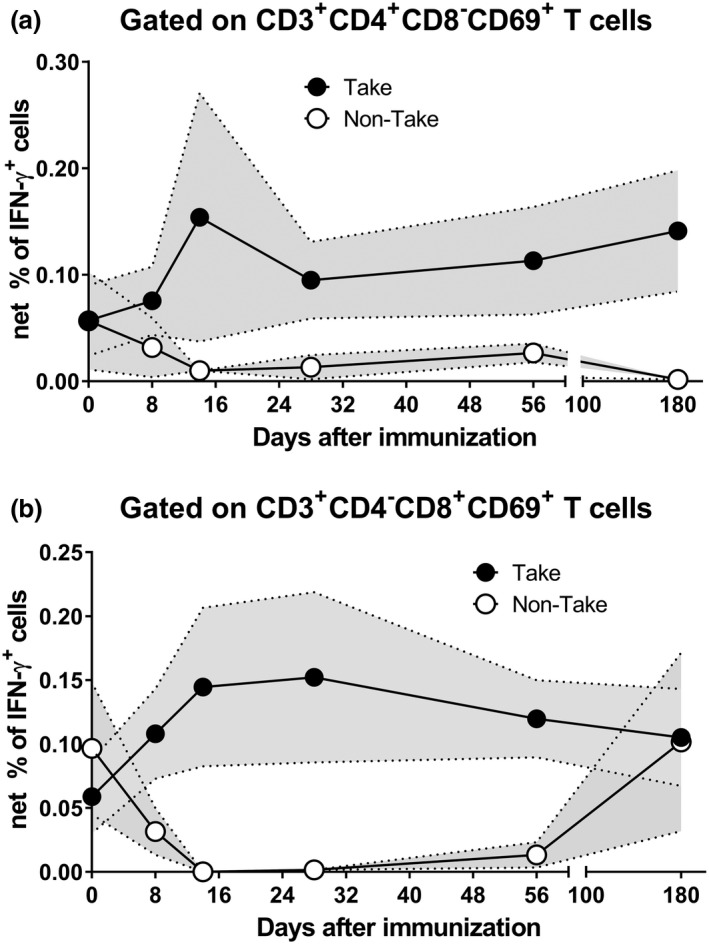
Kinetics of F. tularensis‐specific T cells over 180 days after immunisation. Cells from 43 subjects at six time points, days 0, 8, 14, 28, 56 and 180, were stimulated with Schu‐S4 in the presence of CD107 ‘a’ and ‘b’ monoclonal antibodies. After an overnight incubation, PBMC were stained with ViViD, followed by surface staining with mAbs to CD3, CD4, CD8, CD14 and CD19. After fixation and permeabilisation, cells were stained intracellularly for CD69, as well as to IL‐2, IFN‐γ and TNF‐α cytokines and analysed by flow cytometry. IFN‐γ‐expressing CD4 (a) and CD8 (b) T cells are shown. Symbols represent the means, and the filled area denotes the standard error bands. Subjects were divided into two groups based on their take results: (1) take positive (Take) and (2) take negative (non‐Take). Per‐Protocol Population. Data are representative of 21 independent experiments with one replicate.
Major cutaneous reaction (Take) correlates with cellular‐mediated immune responses after tularaemia immunisation
LVS vaccination by scarification induces a skin papule that progresses to an erythematous papule, vesicle, and/or eschar with or without underlying induration. 18 This lesion is known as take. In this study, LVS vaccination‐induced skin lesions reached maximum size within 8 days post‐scarification (geometric mean of 5.85 mm in diameter with 5.41–6.33 95% CI) and were slow to resolve (Supplementary figure 5).
Since previous studies have shown that take is a clinical correlate of protection against tularaemia, 11 , 18 and smallpox, 24 , 25 , 26 and no correlation was found between take and seroconversion for both DVC‐LVS and USAMRIID‐LVS vaccines, 18 we next performed a correlation between investigator take assessment and cell‐mediated responses (CMI). First, the relationship between CMI responses and the take (lesion size) was investigated as continuous variables at all time points in which take and CMI were measured (i.e. days 0, 8, 14, 28, 56 and 180). We found that except for day 180, cell proliferative responses to Schu‐S4, as measured by the [3H]‐thymidine incorporation assay, correlated with lesion size at all time points (Table 4). Levels of IFN‐γ secretion also correlated with lesion size at days 14 and 56 (Table 4). Surprisingly, expression of CD107 a/b by CD4+ T cells, but not CD8+ T cells, correlated with lesion size at day 14 (Table 4). Subsequently, we investigated whether the maximum lesion size could serve as a predictive factor of protective CMI responses. To this end, we correlated the maximum lesion size and the peak of CMI responses. We found significant correlations between maximum lesion size and the peak of cell proliferative responses, IFN‐γ secretion, and expression of CD107a/b by either CD4+ and CD8+ T cells. Interestingly, in contrast to CD8+ T cells, we found that the peak level of IFN‐γ‐expressing CD4+ T cells was significantly associated with the maximum lesion size. Of note, we did not observe positive correlations between take and either IL‐2 or TNF‐α production.
Table 4.
Statistically significant (Spearman’s) correlations of variables and lesion size by per‐protocol population – both vaccines
| Assay | Take | Number of observations |
Spearman's Corr. (95%CI) |
P‐value | ||
|---|---|---|---|---|---|---|
| Type | Timing | Variable | ||||
| [3H]‐thymidine Assay | Day 8 | Cell proliferation | Day 8 | 202 | 0.16 (0.02, 0.29) | 0.023 |
| Day 14 | Day 14 | 193 | 0.14 (0.00, 0.28) | 0.047 | ||
| Day 28 | Day 28 | 180 | 0.20 (0.05, 0.33) | 0.008 | ||
| Day 56 | Day 56 | 157 | 0.23 (0.07, 0.37) | 0.004 | ||
| Peak Response | Max. Lesion Size | 207 | 0.14 (0.01, 0.28) | 0.037 | ||
| MSD a | Day 14 | IFN‐γ | Day 14 | 193 | 0.16 (0.02, 0.30) | 0.023 |
| Day 56 | Day 56 | 157 | 0.26 (0.11, 0.40) | 0.001 | ||
| Peak Response | Max. Lesion Size | 207 | 0.16 (0.03, 0.29) | 0.018 | ||
| Flow Cytometry | ||||||
| CD4+ T cells | Day 14 | CD107a/b | Day 14 | 36 | −0.36 (−0.62, −0.04) | 0.027 |
| Peak Response | Max. Lesion Size | 39 | −0.46 (−0.68, −0.17) | 0.002 | ||
| Peak Response | IFN‐γ | Max. Lesion Size | 39 | −0.32 (−0.58, −0.00) | 0.045 | |
| CD8+ T cells | Peak Response | CD107a/b | Max. Lesion Size | 39 | −0.32 (−0.57, −0.00) | 0.047 |
Meso scale discovery assay.
Multifunctional patterns of T‐cell responses elicited by F. tularensis
Our previous work 23 and the results presented above argue that multifunctional cells able both to secrete cytokines and proliferate may play an important role in protection from tularaemia. Thus, we next simultaneously measured the four T‐cell functions (CD107 mobilisation and IFN‐γ, TNF‐α and IL‐2 production) using the automated FCOM deconvolution tool to identify populations exhibiting four, three, two or one functions. This functional signature was then evaluated in the two T‐cell subsets, CD4+ and CD8+ T cells, using unsupervised principal component analysis (PCA). To increase statistical power, FCOM data from all replicates were merged for combined analysis, generating a matrix of 15 possible FCOM phenotypes for T cells from the 43 University of Maryland volunteers at 6 different time points (3870 points). The PCA analysis revealed that the first principal component (PC1) accounted for most of the total variance (63.4% for CD4+ T cells and 54% for CD8+ T cells) (Supplementary figure 6). Analysis of PC1 vs. PC2 loadings showed that CD4+ T‐cell subsets displayed tighter clustering than CD8+ T‐cell subsets. While most of the CD4+ T‐cell subsets cluster together, monofunctional/bi‐functional CD8+ T cells were scattered in the PCA plot (Supplementary figure 6). To confirm the variances, we next performed cell hierarchical clustering analyses (Figure 4). Based on the clustering tightness, the PCA arrangement of CD4+ T‐cell phenotypes suggested a connection between cells that are monofunctional for IFN‐γ+ with bi‐functional (e.g. TNF‐α and IFN‐γ) or tri‐functional (e.g. IFN‐γ+ TNF‐α+ CD107a/b+) (Figure 4a). Interestingly, CD8+ T‐cell clustering suggested a connection between monofunctional CD107a/b‐expressing cells and those cells double positive for TNF‐α and CD107a/b or triple positive for IFN‐γ, TNF‐α and CD107a/b (Figure 4b).
Figure 4.
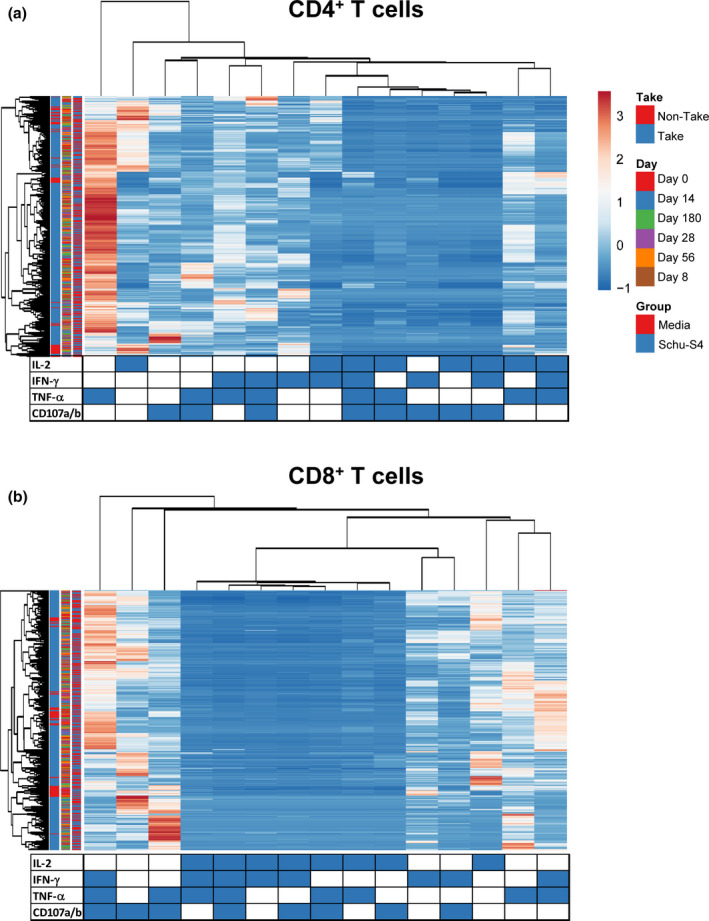
Hierarchical clustering of T‐cell functions using principal component analysis (PCA). The four T‐cell functions (IL‐2, IFN‐γ, TNF‐α and CD107a/b expression) were analysed using the FCOM deconvolution tool. FCOM data of the 15 possible combinations were used to perform an unsupervised PCA analysis. PCA compared CD4 (a) and CD8 (b) T‐cell changes before and after exposure to Schu‐S4 at different time points (0, 8, 14, 28 56 and 180) in cells from individuals with (1) take positive (Take, n = 37) and (2) take negative (non‐Take, n = 6). Cells cultured with media only were used as controls (media). PCA was performed using the ClustVis web tool. Rows were centred, and unit variance scaling applied to rows. Rows were clustered using Euclidean distance and average linkage. Columns were clustered using correlation distance and average linkage. Trees ordering for both rows and columns display the tightest cluster first.
DISCUSSION
Although previous studies have suggested that high antibody titres following tularaemia vaccination might serve as a useful surrogate marker of vaccine efficacy, 32 , 37 , 38 efforts to treat humans with tularaemia immune sera have provided inconclusive results. While some studies showed that immune serum could successfully treat individuals suffering from tularaemia, others do not demonstrate this effect in humans or animals. 37 , 39 , 40 Thus, the actual immunological correlate(s) of protection from tularaemia in humans remain(s) unknown. Here, we report a correlation between take lesion size and T‐CMI responses after tularaemia immunisation.
As with smallpox, 24 , 25 , 26 take is widely accepted as a clinical correlate of protection against tularaemia. 11 , 18 We found that lesion size induced by LVS tularaemia correlates with the frequency of IFN‐γ secretion as early as day 14 and as late as day 56 after vaccination. In contrast, volunteers who did not develop a take exhibited low numbers of IFN‐γ‐secreting T cells in the first 8 days after immunisation when compared to those who developed a positive take. These results are in agreement with previous work showing that the tularaemia vaccine elicited gene expression with signatures similar to other replicating vaccines, such as for smallpox, and induced early upregulation of interferon‐inducible genes. 41 The results described in this report also confirm previous observations by Bosio and colleagues showing in a mouse model that after subcutaneous LVS vaccination followed by SchuS4 challenge, survival correlated with IFN‐γ‐producing T cells. 29 , 42 In contrast, one human study in which subcutaneous and scarification data were combined found no correlation between the maximum erythema measurements and IFN‐γ levels produced in response to LVS vaccination. 22 These results suggest that protection involving IFN‐γ might be related to the route of immunisation. Indeed, when comparing the effects of smallpox vaccination using the subcutaneous route of administration with those involving scarification, the latter was superior as judged by the induction of protective memory T cells against challenge with lethal doses of the pathogenic vaccinia virus strain WR. 28 , 43 , 44
It is important to note that our studies suggest a strong correlation between the ability of the cells to proliferate and secrete IFN‐γ after stimulation with Schu‐S4. These are in agreement with our previous observations that proliferative responses were relatively lower among tularaemia vaccinees who did not exhibit IFN‐γ secretion. 23 The current results also argue in favor of the possibility that multifunctional cells able to produce concomitantly high levels of various cytokines, as well as proliferate, may play an essential role in the host’s response to tularaemia infection. In this regard, hierarchical clustering of T‐cell functions using PCA demonstrated that contrary to CD4+ T‐cell subsets, CD8+ T‐cell subsets with different multifunctionalities might represent distinct sub‐populations.
Another important finding was the association between take and CD107a/b expression. CD107a/b is a surrogate marker of degranulation, a mechanism crucial for the killing of infected cells by cytotoxic T cells. 45 , 46 Cummings and colleagues have demonstrated that the lack of viraemia after smallpox vaccination was due to T‐cell responses restraining infected cells at the vaccination site. 47 Surprisingly, the expression of CD107 a/b by CD4+ T cells, but not CD8+ T cells, correlated with lesion size on day 14 after immunisation. We hypothesised that the lack of correlation by CD8+ T cells was due to their inability to secrete IL‐2 and, subsequently, proliferate during the early stages after vaccination. However, a rescue IL‐2 response at later time points triggered the observed correlation between ‘maximum’ lesion size and the peak levels of CD107a/b‐expressing CD8+ T cells.
This study provides a strong rationale for further pursuing mechanistic studies to conclusively link take with the development of protective T‐cell responses. Although take size predicts T‐cell response levels in our study, it did not provide a mechanistic basis for this phenomenon. Thus, our results cannot determine which factor is influencing the other. Moreover, it is not possible to eliminate the possibility that other drivers such as innate and skin‐resident T cells shape both the take size and the circulating T‐cell responses. The Mackay and Masopust groups, for instance, found that protective resident T cells are expandable and persisted long term within the skin. 48 , 49 Consequently, how scarification can elicit protective T‐cell responses is a fundamental vaccinology question. The relatively small amount of vaccine required and the easiness by which volunteers can be vaccinated through scarification in the world's most remote places make this a particularly valuable route of immunisation.
Finally, although the Schu‐S4 antigen used in our studies is closely related to the vaccines used to immunise the volunteers, it is not possible to exclude that some of the observations in these studies were due to the nature of the stimulant used. Previous findings from our group have shown that CD4+ and CD8+ T‐cell responses against bacterial antigens are likely to depend on the nature of the stimulant. 45 , 50 , 51 , 52 , 53 For example, we found that CD4+, but not CD8+ T‐cell responses, to live infected target cells were significantly associated with their counterpart responses to purified proteins. 53
In summary, we provide the first direct evidence of an association between take formation and T‐CMI responses such as the production of IFN‐γ and expression of CD107a/b. These results reinforce the importance of the choice of the route of immunisation to achieve successful vaccination. These results also renewed the appreciation for vaccination through scarification as a prime route of inoculation.
METHODS
Study design
This study is an ancillary laboratory study conducted within a parent phase 2, multi‐centre, double‐blind, randomised, clinical trial evaluating the safety and immunogenicity between two live, attenuated tularaemia vaccines: (1) Francisella tularensis Live Vaccine Strain (LVS) produced by DynPort Vaccine Company (DVC‐LVS), and (2) LVS made by the United States Army Medical Research Institute of Infectious Diseases (USAMRIID‐LVS). 18 This parent study was conducted at five sites: The Hope Clinic of the Emory Vaccine Center, University of Maryland School of Medicine, Saint Louis University Medical School, University of Iowa, and Baylor College of Medicine. A total of 228 males and non‐pregnant females, aged 18–45 years, were randomly assigned to receive a single dose of either DVC‐LVS (n = 113) or USAMRIID‐LVS (n = 115) at ~1:1 ratio. After vaccination, subjects were followed for safety, immunological responses, and take. Over 70% of volunteers had positive takes, as defined by the development of an erythematous papule, vesicle and/or eschar with or without underlying induration, by study visit 5 (7–9 days post‐vaccination). 18 This parent study is registered with Clinical trials.gov #NCT01150695. Before initiating study procedures, all volunteers were explained the purpose of this study and signed informed consent. The protocol and consent form were reviewed by the US Food and Drug Administration. The human experimentation guidelines of the US Department of Health and Human Services and those of the sites’ Institutional Review Boards were followed in the conduct of the clinical research. Blood was collected before and at days 8, 14, 28, 56 and 180 after immunisation. PBMC were isolated from the blood by standard density gradient centrifugation and cryopreserved in liquid N2 until use. 23
Vaccination
Lyophilised vials were reconstituted with sterile water for injection (WFI), U.S. Pharmacopeia (USP). When reconstituted, each vial contained approximately 1 × 109 colony‐forming units (CFU) mL−1 of live, attenuated F. tularensis in 10% sucrose, 1.9 gelatin, and 10 mm potassium phosphate. Both vaccines were delivered via scarification, which consisted of the use of a sterile bifurcated needle to make 15 superficial punctures through a droplet of 100 µL (~108 colony‐forming units of organisms) on a skin area of approximately 0.5 cm to permit percutaneous penetration of the vaccine (a process known as multiple puncture technique). 54 The cutaneous reaction at the site of vaccination was measured in millimetres by study staff. Any erythema or satellite lesions surrounding the cutaneous lesions were measured and recorded. Take was defined to be the development of an erythematous papule, vesicle and/or eschar with or without underlying induration following vaccination.
Antigens and cell culture conditions
Similar to our previous work, 23 cells were stimulated with heated and formalin‐killed F. tularensis strain Schu‐S4 (Schu‐S4) (107 CFU mL−1 concentration, ATCC, Manassas, VA), obtained through the Division of Microbiology and Infectious Diseases (DMID), NIAID, NIH, and cultured at 37°C, 5% CO2. Of note, attenuated Schu‐S4 strain was used to prepare both LVS vaccines. Cultures with media only or with Staphylococcus enterotoxin B (SEB) (Sigma, St. Louis, MO, 10 μg mL−1) were used as negative and positive controls, respectively. The culture medium consisted of RPMI (Gibco, Grand Island, New York) supplemented with 100 U mL−1 penicillin, 100 µg mL−1 streptomycin, 50 µg mL−1 gentamicin, 2 mm L‐glutamine, 2.5 mm sodium pyruvate, 10 mm HEPES buffer, 1% non‐essential amino acids, and 2% heat‐inactivated AB human serum.
Proliferation assays by [3H]‐thymidine incorporation
The ability of PBMC to proliferate after antigenic stimulation was measured by [3H]‐thymidine incorporation. 55 To this end, cells were stimulated with Schu‐S4 for 4 days. After stimulation, half of the culture media was removed and fresh media added to the culture. Removed culture media was kept at –20°C for future IFN‐γ measurements. Cultures were allowed to progress for 2 extra days. Six days after initiation of the cultures, 1 μCi per well of [3H]‐thymidine was added to each well, and the cultures continued for an additional 18 h. Cultures were then harvested and [3H]‐thymidine incorporation determined by a Beta Counter (1450 MicroBeta TriLuxMicroplate Scintillation and Luminescence Counter, PerkinElmer Life and Analytical Sciences, Shelton CT). A positive responder was defined to be follows: (1) the stimulation index is 3‐fold or higher than media cultures at a particular time point, (2) experimental (Schu‐S4) minus media control exceeds 500 counts per minutes (CPM), and (3) the fold values (Schu‐S4 divided by media) at a particular time point after immunisation are 2‐fold or higher than those at Day 0 (baseline) in at least one post‐vaccination visit.
Quantification of IFN‐γ by Meso Scale Discovery (MSD) assay
Cell culture supernatants were used to measure IFN‐γ production using MSD technology (Meso Scale Discovery, Gaithersburg MD). MSD assays were carried out following the manufacturer’s instructions. Briefly, PBMC were stimulated with LVS, and the supernatants harvested after 96 h and kept at −70°C until assayed. Based on linearity, the levels of sensitivity for IFN‐γ ranged from 0.47 to 2.91 pg mL−1. Responses were scored positive if (1) cytokine production was 2‐fold or higher than media cultures at a particular time point, and (2) the fold values (Schu‐S4 divided by media) at a specific point of time after immunisation were 2‐fold or higher than those at Day 0 (baseline) in at least one post‐vaccination visit.
Monoclonal antibodies for surface and intracellular staining
The following mAb to surface molecules were used to stain PBMC: CD3 (clone UCHT1), CD69 (clone TPI‐55‐3) (Beckman‐Coulter, Miami, FL), CD4 (clone SK3), CD8 (clone HIT8a), CD14 (clone M5E2), CD107a (eBioscience, clone eBioH4A3), CD107b (eBioscience clone eBioH4B4), IL‐2 (clone 5344.111), IFN‐γ (clone B27), TNF‐α (clone MAb11) (BD Pharmingen, San Diego, CA, USA) and CD19 (clone SJ25‐C1) (Invitrogen, Carlsbad, CA). Monoclonal antibodies conjugated to the following fluorochromes were used in these studies: Fluorescein isothiocyanate (FITC), Phycoerythrin (PE), ECD (Energy Coupled Dye), PE‐Cy5.5, PE‐Cy7, Alexa 647, allophycocyanin (APC)‐Alexa 700, APC‐Fluor 780, Pacific blue, Quantum Dot (QD) 655, and biotin followed by streptavidin‐Pacific Orange (Molecular Probes, Eugene, OR).
Cell staining, data acquisition, and analyses
Flow cytometric assays were performed as previously described with small modifications. 45 Briefly, cells were stimulated with SchuS4 in the presence of mAbs to CD107a and CD107b (2 μg/106 cells each). These mAbs were used to detect degranulation, a mechanism crucial for the killing of target cells by the cytotoxic T cells. 45 , 46 PBMC cultured with media only or SEB were used as negative and positive controls, respectively. After 2 h, protein transport blockers, Monensin (1 μg mL−1; Sigma) and brefeldin‐A (BFA) (2 μg mL−1; Sigma), were added to the culture. After an additional 16–18 h (overnight) incubation, cells were harvested, stained with a dead‐cell discriminator, violet fluorescent viability dye (ViVid; Invitrogen), followed by two successive 30‐minute incubations with human Fc receptor blocking IgG and an antibody cocktail for surface markers to identify CD3+, CD4+, CD8+ T‐cell subsets, as well as CD19 and CD14 to exclude B cells and monocytes from analyses. Cells were then fixed and permeabilised with Fix & Perm cell buffers (Invitrogen, Carlsbad, CA), and intracellularly stained with mAbs specific to IL‐2, IFN‐γ, TNF‐α and CD69. Finally, cells were fixed with 1% paraformaldehyde and analysed by flow cytometry on an LSR‐II instrument (BD Biosciences). Data were analysed with WinList 9.0 (Verity Software House, Topsham, ME) (http://www.vsh.com/products/winlist/index.asp).
During sample acquisition, 100 000–500 000 events were collected in the forward and side scatter (FS/SS) lymphocyte gate. Single lymphocytes were gated based on forward scatter height vs. forward scatter area. A ‘dump’ channel was used to eliminate dead cells (ViVid+) as well as macrophages/monocytes (CD14+), and B lymphocytes (CD19+) from the analysis. Additional gating on CD3, CD4 and CD8 and CD69‐positive cells was performed to identify cytokine‐producing (IL‐2, IFN‐γ, and TNF‐α) and degranulation, CD107 expressing cells. Multifunctionality analyses were performed using the FCOM function of WinList software to determine the proportion of all possible 16 combinations for the 4 markers, 3 for cytokines (i.e. IL‐2, IFN‐γ and TNF‐α) and one for degranulation (i.e. CD107a/b). Flow cytometry experiments were performed at the Flow Cytometry and Mass Cytometry Core Facility of the University of Maryland School of Medicine Center for Innovative Biomedical Resources (CIBR), Baltimore, Maryland. Positive responders were based on the IFN‐γ expression and defined to be as follows: (1) minimum number of IFN‐γ‐secreting cells collected in experimental cultures (Schu‐S4) > 10 events, (2) differential in the number of IFN‐γ‐secreting cells in experimental cultures is significantly higher than the number of events in the negative control cultures (media) as determined by using a one‐tailed z test (P‐value < 0.01), and (3) net experimental % value (Schu‐S4 minus media) at a particular time point ≥ 0.1% than that measured at Day 0 (baseline) in at least one post‐vaccination visit.
Statistical analysis
Fisher’s exact test or logistic regression for a binary outcome, and the two‐sample t‐test or Wilcoxon rank‐sum test for continuous outcome, were used to compare the distributions between the two groups. The outcomes at different time points from the same subject were expected to be correlated. To get efficient estimates of the regression parameters, linear mixed‐effect models were fitted on the log‐transformed immunogenicity outcomes to define an adequate error covariate structure. The covariates for the fixed effects in the models could include group, baseline and time post‐vaccination. Other demographic variables, such as sex and age, were considered. All attempts were made to collect all data per protocol. No imputations were performed for missing values. The area under the curve (AUC) values were computed using the trapezoid rule, which connected a straight line to every set of adjacent points defining the curve and sums up the areas beneath these areas starting from day 0 and ending at day 180.
To visualise the variance of 15 possible FCOM T‐cell phenotypes, we performed principal component analysis (PCA) as described previously with small modifications. 56 Briefly, the calculation of principal components was performed by ClustVis web tools. 57 Columns were grouped using correlation distance (Pearson correlation subtracted from 1) and average linkages calculated. Rows were centred, and unit variance scaling was applied to the rows. Rows were clustered using Euclidean distance (square root of the sum of squared distances) and average linkage (average distance of all possible pairs) calculated. Trees ordering for both rows and columns display the tightest cluster first.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
Rosangela Salerno‐Goncalves: Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Project administration; Supervision; Validation; Visualization; Writing‐original draft. Wilbur Chen: Funding acquisition; Investigation; Project administration; Supervision; Writing‐review & editing. Mark J. Mulligan: Funding acquisition; Investigation; Project administration; Supervision; Writing‐review & editing. Sharon E. Frey: Funding acquisition; Investigation; Project administration; Supervision; Writing‐review & editing. Jack T. Stapleton: Funding acquisition; Investigation; Project administration; Supervision; Writing‐review & editing. Wendy A. Keitel: Funding acquisition; Investigation; Project administration; Supervision; Writing‐review & editing. Jason Bailey: Formal analysis; Methodology; Validation; Writing‐review & editing. Eli Sendra: Formal analysis; Methodology; Validation; Writing‐review & editing. Heather Hill: Formal analysis; Methodology; Validation; Writing‐review & editing. Robert A. Johnson: Conceptualization; Investigation; Project administration; Resources; Supervision; Writing‐review & editing. Marcelo B. Sztein: Conceptualization; Funding acquisition; Investigation; Project administration; Resources; Supervision; Validation; Writing‐review & editing.
Supporting information
Acknowledgments
We are indebted to the volunteers who allowed us to perform this study. We thank Dr Haiyan Chen, Mr Guillermo Sahaniuk, Ms Regina Harley and Catherine Storrer for excellent technical assistance. This work was supported by the National Institute of Allergy And Infectious Diseases of the National Institutes of Health under Award Numbers N01‐AI‐30028 (Immunology Research Unit ‐IRU‐ of the Food and Waterborne Diseases Integrated Research Network ‐FWD‐IRN); HHSN272200800001C, HHSN272201300022I, HHSN272200800005C, HHSN272201300018I, HHSN272200800008C, HHSN272201300020I, HHSN272200800003C, HHSN272201300021I, HHSN272200800002C, HHSN272201300015I (Vaccine and Treatment Evaluation Unit ‐VTEU); Emmes Contract HHSN272201500002C; and NCRR grant K12‐RR023250. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases, the National Institutes of Health, the Biomedical Advanced Research and Development Authority, the National Health Service, the National Institute for Health Research (NIHR) or UMB.
References
- 1. Zellner B, Huntley JF. Ticks and tularemia: do we know what we don't know? Front Cell Infect Microbiol 2019; 9: 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Roberts LM, Powell DA, Frelinger JA. Adaptive immunity to Francisella tularensis and considerations for vaccine development. Front Cell Infect Microbiol 2018; 8: 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Balestra A, Bytyci H, Guillod C, Braghetti A, Elzi L. A case of ulceroglandular tularemia presenting with lymphadenopathy and an ulcer on a linear morphoea lesion surrounded by erysipelas. Int Med Case Rep J 2018; 11: 313–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Farlow J, Wagner DM, Dukerich M et al Francisella tularensis in the United States. Emerg Infect Dis 2005; 11: 1835–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Centers for Disease Control and Prevention (CDC) . Tularemia – United States, 2001‐2010. MMWR Morb Mortal Wkly Rep 2013; 62: 963–966. [PMC free article] [PubMed] [Google Scholar]
- 6. Ellis J, Oyston PC, Green M, Titball RW. Tularemia. Clin Microbiol Rev 2002; 15: 631–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gill V, Cunha BA. Tularemia pneumonia. Semin Respir Infect 1997; 12: 61–67. [PubMed] [Google Scholar]
- 8. Dienst FT Jr. Tularemia: a perusal of three hundred thirty‐nine cases. J Louisiana State Med Soc 1963; 115: 114–127. [PubMed] [Google Scholar]
- 9. Centers for Disease Control and Prevention (CDC), Department of Health and Human Services (HHS) . Possession, use, and transfer of select agents and toxins; biennial review of the list of select agents and toxins and enhanced biosafety requirements. Final rule. Fed Regist 2017; 82: 6278–6294. [PubMed] [Google Scholar]
- 10. Saslaw S, Eigelsbach HT, Wilson HE, Prior JA, Carhart S. Tularemia vaccine study. I. Intracutaneous challenge. Arch Intern Med 1961; 107: 689–701. [DOI] [PubMed] [Google Scholar]
- 11. Saslaw S, Eigelsbach HT, Prior JA, Wilson HE, Carhart S. Tularemia vaccine study: II. Respiratory Challenge. Arch Intern Med 1961; 107: 702–714. [DOI] [PubMed] [Google Scholar]
- 12. Eigelsbach HT, Downs CM. Prophylactic effectiveness of live and killed tularemia vaccines. I. Production of vaccine and evaluation in the white mouse and guinea pig. J Immunol 1961; 87: 415–425. [PubMed] [Google Scholar]
- 13. Downs CM, Woodward JM. Studies on pathogenesis and immunity in tularemia; immunogenic properties for the white mouse of various strains of Bacterium tularense. J Immunol 1949; 63: 147–163. [PubMed] [Google Scholar]
- 14. Foshay L, Hesselbrock WH, Wittenberg HJ, Rodenberg AH. Vaccine prophylaxis against tularemia in man. Am J Public Health Nation's Health 1942; 32: 1131–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Burke DS. Immunization against tularemia: analysis of the effectiveness of live Francisella tularensis vaccine in prevention of laboratory‐acquired tularemia. J Infect Dis 1977; 135: 55–60. [DOI] [PubMed] [Google Scholar]
- 16. Hornick RB, Eigelsbach HT. Aerogenic immunization of man with live Tularemia vaccine. Bacteriol Rev 1966; 30: 532–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tigertt WD. Soviet viable Pasteurella tularensis vaccines. A review of selected articles. Bacteriol Rev 1962; 26: 354–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mulligan MJ, Stapleton JT, Keitel WA et al Tularemia vaccine: Safety, reactogenicity, "Take" skin reactions, and antibody responses following vaccination with a new lot of the Francisella tularensis live vaccine strain – a phase 2 randomized clinical trial. Vaccine 2017; 35: 4730–4737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Waag DM, Galloway A, Sandstrom G et al Cell‐mediated and humoral immune responses induced by scarification vaccination of human volunteers with a new lot of the live vaccine strain of Francisella tularensis . J Clin Microbiol 1992; 30: 2256–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Waag DM, Sandström G, England MJ, Williams JC. Immunogenicity of a new lot of Francisella tularensis live vaccine strain in human volunteers. FEMS Immunol Med Microbiol 1996; 13: 205–209. [DOI] [PubMed] [Google Scholar]
- 21. McMurry JA, Moise L, Gregory SH, De Groot AS. Tularemia vaccines – an overview. Med Health R I 2007; 90: 311–314. [PubMed] [Google Scholar]
- 22. El Sahly HM, Atmar RL, Patel SM et al Safety, reactogenicity and immunogenicity of Francisella tularensis live vaccine strain in humans. Vaccine 2009; 27: 4905–4911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Salerno‐Goncalves R, Hepburn MJ, Bavari S, Sztein MB. Generation of heterogeneous memory T cells by live attenuated tularemia vaccine in humans. Vaccine 2009; 28: 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Henderson DA. Smallpox: clinical and epidemiologic features. Emerg Infect Dis 1999; 5: 537–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Breman JG, Henderson DA. Diagnosis and management of smallpox. N Engl J Med 2002; 346: 1300–1308. [DOI] [PubMed] [Google Scholar]
- 26. Walsh SR, Dolin R. Vaccinia viruses: vaccines against smallpox and vectors against infectious diseases and tumors. Expert Rev Vaccines 2011; 10: 1221–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gordon SN, Cecchinato V, Andresen V et al Smallpox vaccine safety is dependent on T cells and not B cells. J Infect Dis 2011; 203: 1043–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu L, Zhong Q, Tian T, Dubin K, Athale SK, Kupper TS. Epidermal injury and infection during poxvirus immunization is crucial for the generation of highly protective T cell‐mediated immunity. Nat Med 2010; 16: 224–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Anderson RV, Crane DD, Bosio CM. Long lived protection against pneumonic tularemia is correlated with cellular immunity in peripheral, not pulmonary, organs. Vaccine 2010; 28: 6562–6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cowley SC, Elkins KL. Immunity to Francisella. Front Microbiol 2011; 2: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Paranavitana C, DaSilva L, Vladimirova A, Pittman PR, Velauthapillai M, Nikolich M. Transcriptional profiling of recall responses to Francisella live vaccine strain. Pathog Dis 2014; 70: 141–152. [DOI] [PubMed] [Google Scholar]
- 32. Rhinehart‐Jones TR, Fortier AH, Elkins KL. Transfer of immunity against lethal murine Francisella infection by specific antibody depends on host gamma interferon and T cells. Infect Immun 1994; 62: 3129–3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yee D, Rhinehart‐Jones TR, Elkins KL. Loss of either CD4+ or CD8+ T cells does not affect the magnitude of protective immunity to an intracellular pathogen, Francisella tularensis strain LVS. J Immunol 1996; 157: 5042–5048. [PubMed] [Google Scholar]
- 34. Levine MM, Sztein MB. Vaccine development strategies for improving immunization: the role of modern immunology. Nat Immunol 2004; 5: 460–464. [DOI] [PubMed] [Google Scholar]
- 35. Robinson HL, Amara RR. T cell vaccines for microbial infections. Nat Med 2005; 11: S25–32. [DOI] [PubMed] [Google Scholar]
- 36. Salerno‐Goncalves R, Sztein MB. Cell‐mediated immunity and the challenges for vaccine development. Trends Microbiol 2006; 14: 536–542. [DOI] [PubMed] [Google Scholar]
- 37. Fulop M, Mastroeni P, Green M, Titball RW. Role of antibody to lipopolysaccharide in protection against low‐ and high‐virulence strains of Francisella tularensis . Vaccine 2001; 19: 4465–4472. [DOI] [PubMed] [Google Scholar]
- 38. Drabick JJ, Narayanan RB, Williams JC, Leduc JW, Nacy CA. Passive protection of mice against lethal Francisella tularensis (live tularemia vaccine strain) infection by the sera of human recipients of the live tularemia vaccine. Am J Med Sci 1994; 308: 83–87. [DOI] [PubMed] [Google Scholar]
- 39. Tärnvik A. Nature of protective immunity to Francisella tularensis . Rev Infect Dis 1989; 11: 440–451. [PubMed] [Google Scholar]
- 40. Foshay L. A comparative study of the treatment of tularemia with immune serum, hyperimmune serum and streptomycin. Am J Med 1946; 1: 180–188. [DOI] [PubMed] [Google Scholar]
- 41. Natrajan MS, Rouphael N, Lai L et al Systems vaccinology for a live attenuated tularemia vaccine reveals unique transcriptional signatures that predict humoral and cellular immune responses. Vaccines 2019; 8: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Elkins KL, Cowley SC, Bosio CM. Innate and adaptive immune responses to an intracellular bacterium, Francisella tularensis live vaccine strain. Microbes Infect 2003; 5: 135–142. [DOI] [PubMed] [Google Scholar]
- 43. Koplan JP, Marton KI. Smallpox vaccination revisited. Some observations on the biology of vaccinia. Am J Trop Med Hyg 1975; 24: 656–663. [PubMed] [Google Scholar]
- 44. Kupper TS. Old and new: recent innovations in vaccine biology and skin T cells. J Invest Dermatol 2012; 132: 829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Salerno‐Goncalves R, Tettelin H, Luo D et al Differential functional patterns of memory CD4+ and CD8+ T‐cells from volunteers immunized with Ty21a typhoid vaccine observed using a recombinant Escherichia coli system expressing S. Typhi proteins. Vaccine 2020; 38: 258–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Betts MR, Brenchley JM, Price DA et al Sensitive and viable identification of antigen‐specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods 2003; 281: 65–78. [DOI] [PubMed] [Google Scholar]
- 47. Cummings JF, Polhemus ME, Hawkes C, Klote M, Ludwig GV, Wortmann G. Lack of vaccinia viremia after smallpox vaccination. Clin Infect Dis 2004; 38: 456–458. [DOI] [PubMed] [Google Scholar]
- 48. Beura LK, Mitchell JS, Thompson EA et al Intravital mucosal imaging of CD8+ resident memory T cells shows tissue‐autonomous recall responses that amplify secondary memory. Nat Immunol 2018; 19: 173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Park SL, Zaid A, Hor JL et al Local proliferation maintains a stable pool of tissue‐resident memory T cells after antiviral recall responses. Nat Immunol 2018; 19: 183–191. [DOI] [PubMed] [Google Scholar]
- 50. Salerno‐Goncalves R, Wyant TL, Pasetti MF et al Concomitant induction of CD4+ and CD8+ T cell responses in volunteers immunized with Salmonella enterica serovar typhi strain CVD 908‐htrA. J Immunol 2003; 170: 2734–2741. [DOI] [PubMed] [Google Scholar]
- 51. Wahid R, Salerno‐Goncalves R, Tacket CO, Levine MM, Sztein MB. Cell‐mediated immune responses in humans after immunization with one or two doses of oral live attenuated typhoid vaccine CVD 909. Vaccine 2007; 25: 1416–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sztein MB, Salerno‐Goncalves R, McArthur MA. Complex adaptive immunity to enteric fevers in humans: lessons learned and the path forward. Front Immunol 2014; 5: 516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Salerno‐Goncalves R, Tettelin H, Lou D et al Use of a novel antigen expressing system to study the Salmonella enterica serovar Typhi protein recognition by T cells. PLoS Negl Trop Dis 2017; 11: e0005912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Centers for Disease Control and Prevention NCfEaZIDN, Division of High‐Consequence Pathogens and Pathology (DHCPP) . Vaccine Administration. [updated 2018]. Available from: https://www.cdc.gov/smallpox/clinicians/vaccination‐administration2.html.
- 55. Froebel KS, Pakker NG, Aiuti F et al Standardisation and quality assurance of lymphocyte proliferation assays for use in the assessment of immune function. European Concerted Action on Immunological and Virological Markers of HIV Disease Progression. J Immunol Methods 1999; 227: 85–97. [DOI] [PubMed] [Google Scholar]
- 56. Sztein MB, Bafford AC, Salerno‐Goncalves R. Salmonella enterica serovar Typhi exposure elicits ex vivo cell‐type‐specific epigenetic changes in human gut cells. Sci Rep 2020; 10: 13581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Metsalu T, Vilo J. ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res 2015; 43: W566–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials