

Abstract
Although hypertrophic scars and keloids both generate excessive scar tissue, keloids are characterized by their extensive growth beyond the borders of the original wound, which is not observed in hypertrophic scars. Whether or not hypertrophic scars and keloids are two sides of the same coin or in fact distinct entities remains a topic of much debate. However, proper comparison between the two ideally occurs within the same study, but this is the exception rather than the rule. For this reason, the goal of this review was to summarize and evaluate all publications in which both hypertrophic scars and keloids were studied and compared to one another within the same study. The presence of horizontal growth is the mainstay of the keloid diagnosis and remains the strongest argument in support of keloids and hypertrophic scars being distinct entities, and the histopathological distinction is less straightforward. Keloidal collagen remains the strongest keloid parameter, but dermal nodules and α‐SMA immunoreactivity are not limited to hypertrophic scars alone. Ultimately, the current hypertrophic scars‐keloid differences are mostly quantitative in nature rather than qualitative, and many similar abnormalities exist in both lesions. Nonetheless, the presence of similarities does not equate the absence of fundamental differences, some of which may not yet have been uncovered given how much we still have to learn about the processes involved in normal wound healing. It therefore seems pertinent to continue treating hypertrophic scars and keloids as separate entities, until such a time as new findings more decisively convinces us otherwise.
Keywords: diagnosis, histopathology, hypertrophic, keloid, scar
1. INTRODUCTION
Wound healing comprises a series of carefully orchestrated processes that ideally culminate in the development of a relatively inconspicuous, flat and thin‐lined normotrophic scar (Figure 1). In the event of excessive wound healing however, abnormal scars may develop instead. Two types of abnormal scars are commonly recognized: hypertrophic scars and keloids (Figure 1). Both these abnormal scar types involve excessive collagen deposition leading to the formation of raised scar tissue, but in keloids, scar formation extends beyond the boundaries of the original wound and shows no regression.[ 1 , 2 , 3 ]
FIGURE 1.
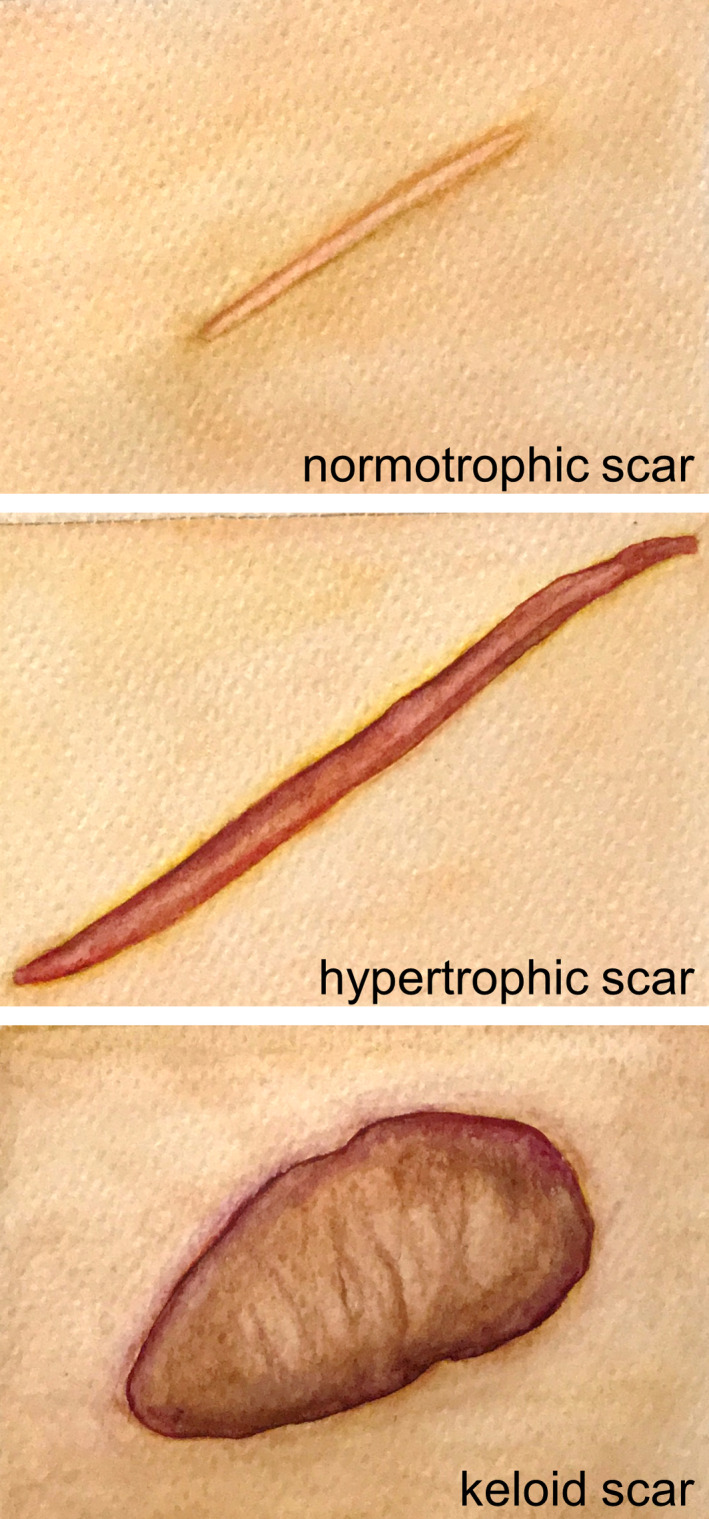
Scar spectrum. Watercolour illustration of a normotrophic, a (linear) hypertrophic and a (major) keloid scar
Hypertrophic scars and keloids have long been a topic of heated debate among researchers, with experts divided on whether or not these abnormal scars are two sides of the same coin, or actually distinct entities. Essential clinical differences have been observed between hypertrophic and keloid scars with respect to i. growth pattern, ii. natural progression over time and iii. association with scar contracture (Figure 2). Invasive horizontal growth remains the most defining characteristic on which the clinical diagnosis of a keloid is based, in contrast to the non‐invasive growth of hypertrophic scars contained within or just around the original wound edges. Hypertrophic and keloid scars also differ with respect to their natural progression over time. Hypertrophic scars usually arise within 4‐8 weeks after wound closure, develop over the next 6‐8 months, after which progression usually halts and they become quiescent. Similar to normotrophic scars, hypertrophic scars also go through the cycle of matrix proliferation, stabilization and maturation; even if not all hypertrophic scars will mature to the same extent as their normotrophic scar counterparts. In contrast, keloids may develop anywhere from 3 months to several years after injury, rarely mature and do not follow the same pattern of evolution, stabilization and involution as the normal and hypertrophic scars. Thirdly, only hypertrophic scars are associated with scar contractures, which cause reduced joint mobility by way of tissue shortening.[ 1 , 2 , 4 , 5 , 6 , 7 ]
FIGURE 2.
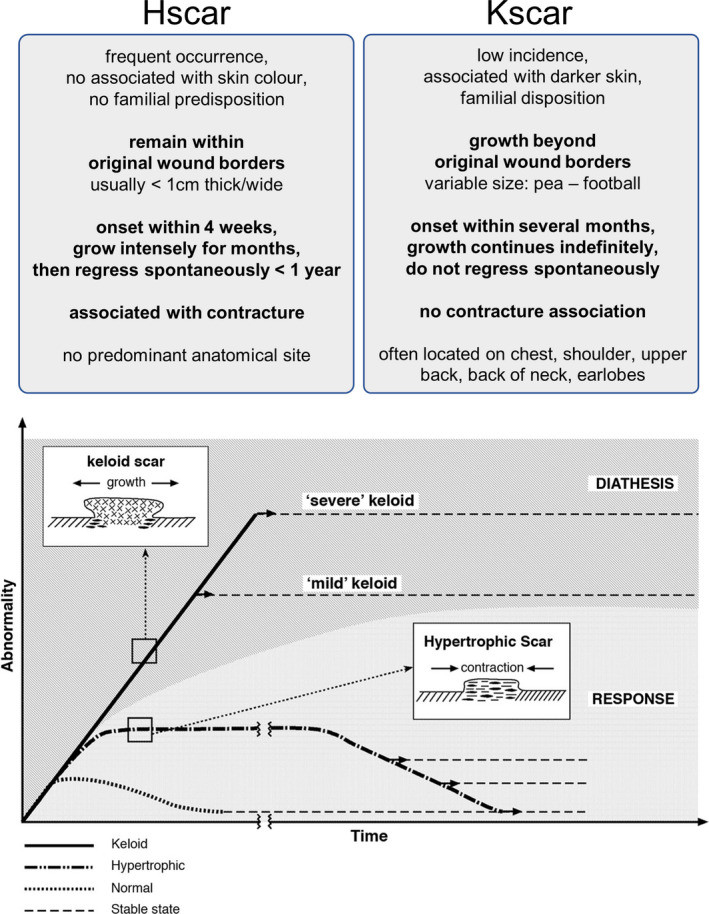
Natural progression of hypertrophic and keloid scars over time. Graph in figure taken from Ref.[2] reproduced with permission from publisher. Hypertrophic and keloid scars show distinct clinical behaviour. The bottom graph illustrates the differential cellular and matrix arrangement of hypertrophic and keloid scars, together with their contrary biological behaviour. Unlike normotrophic and hypertrophic scars, keloids rarely mature, but mild and severe subtypes exist with gross morphologic differences. Hscar: hypertrophic scar. Kscar: keloid scar
Other important differentiating features relate to overall incidence, race association and nature of the antecedent trauma. Hypertrophic scars occur more commonly than keloids, but only keloid incidence is associated with increased racially determined skin pigmentation.[ 1 , 2 , 4 , 5 , 6 , 7 ] Keloids often develop at certain anatomical predilection sites, most of which are associated with the upper torso (see Figure 2). These predilection areas have also been linked to increased skin tension and constant stretching during normal movement.[ 8 , 9 , 10 , 11 , 12 ] Although hypertrophic scars are indeed known to occur when scars cross joints or skin creases at a right angle,[ 1 ] the absence of an overall anatomical association has been put forward by both Burd & Huang[ 2 ] as well as Seifert et al.[ 13 ] The occurrence of hypertrophic scars at joints may very well reflect their known association with scar contracture,[ 2 ] which is not observed in keloids. Aside from these mechanical factors, young age, the nature of the inciting injury (thermal burns especially) and infection are all considered contributing factors to hypertrophic scar development.[ 1 , 2 , 4 , 5 , 6 ] Notably, the risk of hypertrophic scar formation is associated with the depth of the inciting injury.[ 14 , 15 ] Lastly, hypertrophic scars are usually not therapy‐resistant and will not recur as frequently after surgery as keloids.[ 2 , 16 , 17 ]
To offer our perspective on the question if keloids are merely hypertrophic scar exacerbations or actually distinct entities, we focused our attention on the scientific publications (accessible via PubMed) in which both hypertrophic scars and keloids were both included for analysis. This was the key requirement for inclusion in this review, Tables 1 and 2 summarize the main findings of the included studies.
TABLE 1.
Similarities between hypertrophic and keloid scars
| Parameter | Hypertrophic and keloid scar | References |
|---|---|---|
| In vivo | ↑ High‐frequency conductance (skin surface hydration) | [18, 19] |
| Persistent ↑ TEWL (SC barrier function) | ||
| Faster SC turnover | ||
| Epidermis | ↑ But inconsistently, ↑ μm (in all scars), ↑ cell layers | [28, 29],[42],[38, 79] |
| Rete ridges ‐ (all scars) | [22, 26, 28, 38, 79] | |
| ↓ Length cuboidal desmosomes | [42] | |
| Epidermal appendages ‐ (in all scars), displaced | [28, 29],[26, 29] | |
| ↑ K2e | [20] | |
| ↑ K1/K10, normal K10 | [20],[79] | |
| Normal loricrin, filaggrin, SKALP, SPRR2 | [79] | |
| Ki67 ↑, normal Ki67 | [20],[38, 79] | |
| Normal Bcl‐2 +, c‐jun + expression, p53 − | [109] | |
| Diffuse ↑ TGF‐β1 (in 90%), ↑ S100A12 | [110],[111] | |
| Keratinocytes (in vitro) | Normal proliferation rates | [79] |
| Dermal cells | p53 −; normal Bcl‐2 +, c‐jun +, c‐fos +, Ki67 +; ↑ apoptosis | [109];[112] |
| CD34 −, FXIIIa −, S‐100 − (in all scars); TGF‐β1 + (all scars) | [46] c ;[110] | |
| CD34−/α‐SMA+/p16 + population | [38] b | |
| ECM | ↑ Connective tissue; non‐fibrotic PD ‐ (in all scars) | [26, 28, 29];[45] |
| Parallel collagen orientation | [30] | |
| Nodules +, nodules (48%‐50%) | [29, 32],[38, 41] | |
| Normal collagen synthesis | [21] | |
| Normal collagen content in μg/mg wet tissue (in all scars) | [113] | |
| ↑ Collagen synthesis (sensitive to tranilast inhibition) | [21, 22, 23, 24] | |
| ↑ Collagen I (in all scars) | [43] | |
| ↑ Fibronectin; ↓ fibrillin‐1 (in all scars), ↓ elastin in superficial dermis | [25, 26, 27];[44] | |
| ↑ Periostin; + hyaluronic acid in RD | [114];[115] | |
| Normal levels lysyl oxidase (cross‐linking enzyme) | [116] | |
| Normal active collagenase, no collagen degradation resistance | [117] | |
| Normal MMP‐9; normal MMP‐2 secretion, ↑ MMP‐2 | [118];[113, 118, 119] | |
| ↑ TIMP‐1, ↑ TIMP‐2 | [119] | |
| Cellular density | ↑ | [26, 28, 29] |
| ↑ ATP and protein levels, ↑ fibroblasts, ↓ fibrocytes | [120] | |
| Vasculature | ↓ Vessels, similar pattern of vascularization | [40] |
| ↑ Blood vessel density | [26, 28, 29] | |
| ↑ Occluded microvessels | [31, 32] | |
| ↑ Endothelin‐1; ↑ HIF‐1α (margin Kscar) | [121];[122] | |
| Immune cells | Variable inflammatory infiltrate + | [41] |
| Mast cells + | [123] | |
| CD68 − | [124] | |
| ↑ FXIIIa + DCs in RD (in all scars) | [125] | |
| ↑ HLA‐DR+/CD1a + DCs | [126] | |
| Fibroblasts | ↑ Vimentin | [38] |
| ↑ Apoptosis; ↑ caspase‐3, ↑ caspase‐9 | [127];[128] | |
| PAR‐1 −, PAR‐2 − | [29] | |
| ↑ DNMT in HsF (90%) and KF (100%) | [129] | |
| Fibroblasts (in vitro) | Normal proliferation (MTT), normal apoptosis rates | [65] |
| ↑ Type I fibroblast (migratory, small spindle shaped) | [62] a | |
| ↑ Collagen I and III, collagen processing genes | [37] | |
| ↑ Cancer, cellular movement, cellular growth and proliferation, tissue development, connective tissue function, cell death genes | ||
| ↓ Cancer, reproductive system disease, tissue development, cell growth and proliferation, cell‐to‐cell signalling genes | ||
| ↑ Fibronectin, ↑ fibronectin (in all scars) | [36] a , c ,[25, 27] a | |
| ↑ Transcriptional activity of α1(I) procollagen gene | [73] a | |
| ↑ Collagen I, ↑ PAI‐2, ↓ MMP‐3 | [75] a , c | |
| Fas +, Bcl‐2 +, Bax +; ceramide‐induced apoptosis + | [67, 68] a ;[68] a | |
| ↑ DNMT1, ↑ TGF‐β1, ↓ Smad7; reversible by DNA methylation inhibition | [129] | |
| ↓ Glucose consumption; COX‐1 +, COX‐2 − | [130];[131] a | |
| ↑ Migration, CTGF, PAI‐1, TGF‐β1/2, collagen I, fibronectin, hydroxyproline | [33] | |
| + TGF‐β1: ↓ MMP1, ↑ collagen I (similar to NF) | [43] | |
| + Sucrose: ↓ collagen I, ↓ collagen I:III ratio | [132] a | |
| + Tacrolimus: ↓ NME/NM23 NDK1, heterogenous NRP H3‐2H9 | [133] | |
| + miR‐188‐5p mimic transfection: ↑ proliferation | [134] | |
| + BM‐MSC supernatant: ↓ proliferation, migration, CTGF, PAI‐1, TGF‐β1/2, collagen I, fibronectin, hydroxyproline; ↑ TGF‐β3, decorin | [33] | |
| Fibroblasts (3D) | ↑↑ Contraction in collagen gel with TGF‐β2, without TGF‐β2 | [34],[35] |
| Continued ↑ collagen I and III (normal expression in monolayers) | [135] c | |
| Skin equivalents (in vitro) | ↑ Contraction, (↑) dermal thickness, ↑ α‐SMA, ↑ p16, ↓ HAS1, ↓ MMP3 | [39] |
| Myofibroblasts | Predominant cell type | [26, 136] |
| ↑ Cross‐linking in 3D collagen structure | [136] | |
| PAR‐1 +, PAR‐2 +; ↑ LH2b | [29];[136] | |
| Nerve cells | Normal nerve fibre density in epidermis (α1‐AR/α‐SMA, α1‐AR/PGP9.5) | [137] |
| Explants (in vitro) | ↓ MMP‐3, collagen fibres composed of thick bundles | [138] |
| + PDT: ↓ collagen I and III | [138] | |
| Other | ↑ TGFβRI/II, Smad2/3/4, p‐Smad 2 | [139] |
| ↑ Collagen I and III, fibronectin, α‐SMA | ||
| ↑ COX‐1 in dermal cells | [140] | |
| ↓ COX‐2, normal | [140],[141] | |
| ↑ FGF‐2, LTBP‐2 | [142] | |
| ↑ SIP1 | [43] | |
| CD34−/ proline‐4‐hydroxylase +, FVIII −, FXIIIa − | [124, 143] | |
| ↑ mTOR | [144] | |
| ↑ Bcl‐2 in basal keratinocytes & in dermis, p53 − | [109] | |
| Normal p63 | [145] | |
| Mathematical modelling of NO in wound healing: ↑ vascularity, hypoxia, ↑ blood vessel occlusion | [146] |
Similar abnormalities in hypertrophic and keloid scars, parameter expression listed as compared to control groups (normal skin and/or normal scar). Table contains all publications in which both mature hypertrophic and keloid scars are studied, and in this table shared abnormalities of hypertrophic and keloid scars are listed. Legend; + (located after parameter): present, normal expression or values; ↑: increased; −: absent; ↓: decreased; ≈: similar to normal skin and/or normal scar.
Abbreviations: ATP, adenosine triphosphate; Bcl‐2, B‐cell lymphoma 2; BM‐MSC, bone marrow‐derived mesenchymal stem cells; COX, cyclo‐oxygenase; DCs, dendritic cells; DNMT1, DNA methyltransferase 1 (catalyses DNA methylation); FGF‐2, (basic) fibroblast growth factor 2; FVIII, factor VIII; FXIIIa, factor XIIIa; HAS1, hyaluronan synthase 1 gene expression; heterogenous NRP H3‐2H9, heterogenous nuclear ribonucleoprotein H3‐2H9 (RNA binding protein, involved in epithelial‐mesenchymal interactions and post‐translational control of collagen I and III expression); HIF‐1α, hypoxia‐inducible factor 1 alpha; HLA, human leucocyte antigen; HsF, hypertrophic scar fibroblasts; IHC, immunohistochemistry; K, keratin; KF, keloid fibroblasts; LH2b, lysyl hydroxylase (collagen cross‐linking); LTBP‐2, latent‐transforming growth factor beta‐binding protein 2; MMP, matrix metalloproteinase; MMP3, matrix metalloproteinase 3 gene expression; mTOR, mammalian target of rapamycin; NF, normal skin fibroblasts; NME/NM23 NDK1, NME/NM23 nucleoside diphosphate kinase 1 (metastasis suppressor gene, involved in cell movement and adhesion); NO, nitric oxide; P4H, proline‐4‐hydroxylase (marker for active collagen synthesis); PAR, protease‐activated receptor; PDT, photodynamic therapy; PGP9.5, protein gene product 9.5 (neuronal marker); p‐Smad, phosphorylated Smad; RD, reticular dermis; SC, stratum corneum; SIP1, Smad interacting protein 1; TEWL, transepidermal water loss; TGF‐β, transforming growth factor beta; TIMP, tissue inhibitor of metalloproteinase. NB any additional information on parameters listed in abbreviations are all derived from cited literature in table; α1‐AR, alpha 1 adrenergic receptor; α‐SMA, alpha smooth muscle actin.
Explant fibroblast cell isolation; unless stated otherwise, fibroblasts were isolated via enzymatic digestion;
Gradient differences between hypertrophic scars and keloids, see also Table 2;
Results based on n = 1.
TABLE 2.
Differences between hypertrophic and keloid scars
| Category | Parameter | Hscar | Kscar | References |
|---|---|---|---|---|
| Incidence | Occurrence | ↑ | ↓ | [1, 2] |
| Association with darker skin | − | + | [1, 2] | |
| Familial predisposition | − | + | [1, 2] | |
| Growth pattern | Relation to original wound borders | within | beyond | [2, 4] |
| Natural progression | Onset in | weeks | months | [2, 4] |
| Growth period | months | indefinitely | [2, 4] | |
| Spontaneous regression | + | − | [2, 4] | |
| Scar contracture | Association | + | − | [2, 4] |
| Location | Predilection sites | − | + b | [2, 4] |
| In vivo | Allergy symptoms | ↑ | ↑↑ | [147] |
| Optical coherence tomography: hyper‐reflectivity of epidermis, dermis | bands, bands | vague, disarray | [80] | |
| Optical coherence tomography: vascularity | ↑ | ↓ | [80] | |
| Vascular structures (dermoscopy) | − (73%) | ↑↑ (90%) | [61] | |
| Epidermis | Epidermal thickness | ↑, ↑↑, + | +, ↑, ↑ | [22, 40],[76],[60] |
| Epidermal thickness in μm; in cell layers | ↑; ↑/↑↑ | ↑↑; ↑↑ | [42];[38, 79] | |
| Rete ridges | −, + (60%) | +, + (8%) | [40],[41] | |
| Lengthening of rete ridges | − | + | [60] | |
| Epidermal appendages | − | + | [40] | |
| K5/K14, K6/K16, involucrin | ↑, ↑↑, +/↑ | ↑↑, ↑, ↑ | [148],[20],[79] | |
| Hemidesmosome density | ↓ | ↓↓ | [42] | |
| Hyaluronic acid | +/− | ↑ | [115] | |
| PAR‐1, PAR‐2; NICD | +, +; ↓ | ↑↑, ↑↑; ↑ | [29];[149] | |
| Keratinocytes (in vitro) | Involucrin | + | ↑↑ | [79] |
| Dermis | Cellularity, nodules, α‐SMA & immune cells dependent on scar maturity | yes | no | [26] |
| IGF‐1R; COX‐1, COX‐2 | ↑; ↑, + | ↑↑↑; +, ↑ | [150] | |
| Dermal cells | Cell density, diffuse cellularity | ↑, − | +, ↑ | [21, 40],[26] |
| CD34−/α‐SMA+/p16 + population | −/↑↑/↑ | −/↑/↑↑ | [38] | |
| ATP with scar maturity | ↓ | persistent ↑ | [120] | |
| CXCL1, CXCR2 | −, − | ↑, ↑ | [151] | |
| Fibroblasts | Mean cell size | small spindles | ↑ | [116] |
| Cell type activity | active | quiescent | [32] | |
| Caspase‐2, caspase‐3 | ↑ | ↑↑ | [127] | |
| MCP‐1, CCR2 | +, + | ↑, ↑ | [152] | |
| RUNX2; NICD | +; ↑ | ↑; ↑↑ | [153];[149] | |
| Fibroblasts (in vitro) | Proliferation; PCNA | ↑, +, ↓; + | ↓, ↑↑↑, ↓↓; ↑ | [62] a ,[66],[75] a ;[65] |
| Proliferation & metabolic activity | +, + | ↑, ↑ | [22] | |
| Apoptosis resistance | − | + | [67,68] a [64] | |
| p63ΔN, p53, Fas, Bcl‐2 | +, ↑, <, + | ↑, ↓, ↑, ↑ | [154],[64,154],[68] a ,[65] | |
| Dead cells, viability | ↑, ↓ | +, ↑ | [64] | |
| Fibroblast type II c , epithelial outgrowth | −, ↑ | ↑, − | [62] a | |
| Fibroblast length, width, size, nucleus size | ↑ | ↑↑ | [63] | |
| Vacuoles, dense bodies | ↑ | ↑↑ | [63] | |
| Collagen I and III, collagen I:III ratio | +, + | ↑, ↑ | [73] a | |
| Mucin synthesis | − | ↑ | [62] a | |
| MMP1, MMP19 | +, + | ↓, ↑ | [75] a , c [76] | |
| TGF‐β1; TGF‐β2; TGF‐β3 | +, ↑; ↑; +, ↑ | ↑, +; +; ↓, + | [69,76],[70];[69];[69],[76] | |
| TGFβRI, TGFβRII | ↓, ↓ | +, ↓ | [69] | |
| CTGF; GDF‐9; IGF‐1R | trend ↑; ↓; + | ↑; ↑; ↑ | [71];[72];[150] | |
| HAS2, IL‐32, IGFBP4, STAT1 | >,>, <, < | ↓, ↓, ↑, ↑ | [37] f | |
| CGRP, HSP27, PAI‐2, α2β1‐integrin | +, +, +, + | ↑, ↑, ↑, ↑ | [76] | |
| Mannose & glucose levels | < | ↓ | [130] a | |
| COX‐1; COX‐1 response to sugar | +; ↑ | ↑; − | [141];[131] a | |
| Gap junctions, connexin 43 | ↓, ↓↓ | ↓↓, ↓ | [155] a | |
| +BMP4: adipogenic conversion | − a | + | [77] | |
| + TGF‐β1/2/3: CTGF | ↑↑ | trend ↑ | [71] | |
| +TGF‐β1: SIP1 | ↓ | − | [43] | |
| +sialic acid: collagen I and III | − | ↑ | [74] a | |
| +mTOR inhibitor: ERK/Akt/NFκB/inflam. targets | ↑ | − | [37] f | |
| + mTOR inhibitor: VEGF/PDGF/catenin/rac1/oestrogen | − | ↑ | [37] f | |
| + SP: apoptosis, proliferation; SP‐blocking | +, +; complete | ↑, ↑, partial | [65] | |
| Fibroblasts (3D) | Contraction | +, ↑ | ↑, + | [34,156] a ,[70] a |
| + TGF‐β: contraction; +anti‐TGF‐β: contraction | ↑; + | ↑↑; − | [70] a ;[70] a | |
| Collagen synthesis, TGF‐β sensitive | ↑, − | ↑↑, + | [157] a | |
| Skin equivalents (in vitro) | CCL5, HGF secretion | ↓, (↓) | +, ↓↓ | [39] |
| LAMA1, COL4A2, ITGA5, MMP1 | ↑, +, +, ↓ | +, ↓, ↓, ↑ | ||
| Myofibroblasts | α‐SMA | + (100%), − | −, + (33%) | [28, 60],[46] e |
| + (33%‐100%) | + (50%‐81%) | [26, 32, 40, 41, 45] | ||
| ECM | Water, collagen, PGs | ↑ | ↑↑ | [22] |
| Nodules | + | −, +/− − | [28,40,45],[40] | |
| + (100%) | + (58%) | [26] | ||
| +/−, large | +/−, small | [38,41],[38] | ||
| Keloidal collagen | − | +, + (55%) | [26,28,29,45],[41] d | |
| +/− − | + | [38, 53, 59] | ||
| Amorphous pericellular material (EM finding) | − | + | [28] | |
| Non‐fibrotic PD | + (80%) | − (40%) | [41] | |
| Tongue‐like advancing edge | − (100%) | + (100%) | [41, 45] | |
| Horizontal fibrous band in upper RD | − (100%) | + (>93%) | [41, 45] | |
| Whorled bundles in RD | + | − | [41, 60] | |
| Prominent fascia‐like band | − (100%) | + (100%) | [41] | |
| Collagen bundles | crisp | glazing | [32] | |
| Collagen bundles size | < | larger, irregular | [30, 32] | |
| Interfibrillar distance | < | ↓ | [32] | |
| Collagen fibre and fibre bundle organization | ↓ | ↓↓ | [116] | |
| Cross‐linked collagen, abnormal fibril assembly | +, − | ↓, + | [22] | |
| Collagen orientation | parallel | haphazard | [26, 45] | |
| Collagen:non‐collagen protein synthesis ratio | + | ↑ | [21] | |
| Collagen I | + PD, + RD | + PD, ↓ RD | [42] | |
| Collagen III | ↑ PD, + RD | ↓ PD, + RD | [42] | |
| Elastin, elastic fibres | +, −, + | ↑ in C, +/−, − | [44],[46] e ,[60] | |
| Hyaluronic acid in PD | ↓ | ↓↓ | [115] | |
| α1β1 integrin collagen receptor; TIMP‐1 | ↑; ↑ | ↑↑; ↑↑ | [60];[119] | |
| Vasculature | Vascular density | +; + | ↑, ↓; ↑ P, ↓ C | [158,159],[159];[122] |
| Capillary density | < | ↓ | [160] | |
| Vascular lumen | < | flatter, narrow | [159, 160] | |
| Microvessel patency | ↓ ≈ granulation | ↓ ≈ Nscar | [31, 32] | |
| Prominent vertically oriented vessels | + | − | [41, 45] | |
| Small aggregating vessels subepidermal | − | + | [41, 45] | |
| Localized differences in vasculature | − | + | [122] | |
| Lactate contents; HIF‐1α, VEGF; NICD | ↑; ↑, ↑; + | ↑↑; ↑↑, ↑↑; ↑↑ | [159];[122];[149] | |
| Blood | Antinuclear Ab in lymphoid cell eluates | − | ↑ | [161] |
| HLA‐B14, HLA‐Bw16 | −, − | ↑, ↑ | [162] | |
| Immune cells | Mast cells | + (0%‐30%) | ↑ (70%‐73%) | [42,25,163] [46] a |
| FXIIIa (DC) | + | ↑ | [125] | |
| CD1a/CD36/HLA‐DR/ICAM‐1/CD54 (DC) | + | ↑ | [26] | |
| CD3/CD45RO/CD4/HLA‐DR/LF‐1 (T‐cells) | + | ↑ | [26] | |
| NICD | ↑ | ↑↑ | [149] | |
| Nerve fibres | α1‐AR/PGP9.5 nerve fibres (dermis) | + | ↑ | [137] |
| Explants (in vitro) | Elastin and elastic fibres | ↓ | ↓↓↓ | [164] |
| Epidermal apoptosis, proliferation | ↑, + | +, ↑ | [138] | |
| Elastin, collagen I, collagen III | +, ↑, ↑ | ↑, ↑↑, ↑↑ | [138] | |
| + PDT: epidermal apoptosis, elastin, MMP‐3 | ↑, ↑, ↑ | −, ↓, − | [138] | |
| Other | Caveolin‐1 activation sNskin:scar ratio | = | ↑ | [165] |
| General protein synthesis | + | ↓ | [21] | |
| Improvement after Nd:YAG laser treatment | ↑ | + | [166] | |
| AgNORs (cellular activity & proliferation) | ↑ | ↑↑ | [167] | |
| PAR‐1 and PAR‐2; p53, p73; leptin | +; ↑, ↑; ↑ | ↑↑; ↑↑, +; ↑↑ | [29];[145];[168] | |
| Active enzyme:proenzyme MMP‐2 ratio, MMP‐9 | ↑, ↑ | ↑↑, + | [113] | |
| Mathematical modelling of NO in wound healing: hypercellular/ regressive; acellular/ ↑ collagen | +; − | −; + | [146] | |
| # miRNAs downregulated, miR‐188‐5p | 9, + | 28, ↓ | [134] |
Differences between hypertrophic and keloid scars, parameter expression listed as compared to control groups (normal skin and/or normal scar). Table contains all publications in which both mature hypertrophic (Hscar) and keloid scars (Kscar) are studied and were found to exhibit differences. +, present, normal expression or values; ↑, increased; −, absent; +/− −, little to no expression; ↓, decreased; ≈, similar to normal skin and/or normal scar; =, no difference; #, number;
Abbreviations: Ab, antibody; AgNORs, silver‐stained nucleolar organizer regions; Akt, protein kinase B pathway; ATP, adenosine triphosphate; Bcl‐2, B‐cell lymphoma 2; BMP4, bone morphogenetic protein 4 (both BMP4 and a special adipogenic cocktail was added to the in vitro keloid fibroblast cultures); C, central keloid region; catenin, cytoskeletal protein; CCL5, C‐C motif chemokine ligand 5; CCR2, C‐C chemokine receptor type 2 (receptor for MCP‐1); CGRP, calcitonin gene‐related peptide (neuropeptide); COL4A2, collagen type IV alpha 2 chain gene expression; COX, cyclo‐oxygenase; CTGF, connective tissue growth factor; DC, dendritic cell; ECM, extracellular matrix; EM, electron microscopy; ERK, extracellular signal–regulated kinase pathway; FXIIIa, factor XIIIa; GDF‐9, growth differentiation factor 9; HAS2, hyaluronan synthase 2; HGF, hepatocyte growth factor; HIF‐1α, hypoxia‐inducible factor 1 alpha; HLA, human leucocyte antigen; Hscar, hypertrophic scar; HSP27, heat shock protein 27; IGF‐1, insulin‐like growth factor 1; IGF‐1R, insulin‐like growth factor 1 receptor; IGFBP, insulin‐like growth factor binding protein; IL, interleukin; inflam. targets, inflammatory targets (in context of this paper, (eg PDGF, IL‐1, IL‐8, TGFA); ITGA5, integrin alpha 5 gene expression; K, keratin; Kscar, keloid scar; LAMA1, laminin subunit alpha 1 gene expression; MCP‐1, monocyte chemoattractant protein 1 (also known as CCL2, C‐C motif chemokine ligand 2); miRNAs, microRNAs; MMP, matrix metalloproteinase; MMP1, matrix metalloproteinase 1 gene expression; NICD, Notch intracellular domain (involved in cell fate determination, modulates, for example proliferation, apoptosis, migration); NO, nitric oxide; Nscar, normotrophic scar; P, peripheral keloid region; PAI‐2, plasminogen activator inhibitor 2; PAR, protease‐activated receptor; PCNA, proliferating cell nuclear antigen; PD, papillary dermis; PDGF, platelet‐derived growth factor; PDT, photodynamic therapy; PGP9.5, protein gene product 9.5 (neuronal marker); PGs, proteoglycans; rac1, cytoskeletal protein; RD, reticular dermis; RUNX2, Runt‐related transcription factor 2 (involved in osteogenesis, chondrogenesis); SIP1, Smad interacting protein 1 (suppresses TGF‐β1); SP, substance P (neuropeptide); STAT1, signal transducer and activator of transcription 1; TGF‐β, transforming growth factor beta; TGF‐βR, transforming growth factor beta receptor; TIMP, tissue inhibitor of metalloproteinase; VEGF, vascular endothelial growth factor. NB any additional information on parameters listed in abbreviations are all derived from cited literature in table; α1‐AR, alpha 1 adrenergic receptor; α2β1‐integrin, collagen receptor; α‐SMA, alpha smooth muscle actin.
Explant fibroblast cell isolation; unless stated otherwise, fibroblasts were isolated via enzymatic digestion;
Chest, shoulder, back, neck and earlobes are known keloid predilection sites;
Type II fibroblasts are more adhesive, large with dendrites;
Hypertrophic scars were said to have no keloidal collagen at all, but both 0 and small fragments of keloidal collagen were scored as negative; < or>: less than Hscar or Kscar, used for parameters when Hscar and Kscar were compared to each other and neither were compared to normal skin or normotrophic scar;
Results based on n = 1 normotrophic and/or hypertrophic scar;
Hypertrophic and keloid scars showed differential transcriptional profiling of at least 50 genes including pathways involved in c21‐steroid hormone metabolism, immune cell cytokines, eicosanoid signalling, arachidonic acid metabolism (the table only lists the most commonly known ECM and wound healing mediators); normal font: gradient differences; italic font: absolute, qualitative differences.
2. “KELOIDS ARE HYPERTROPHIC SCAR EXACERBATIONS”
We will start with a discussion of the literature in favour of considering keloids as an exacerbation of hypertrophic scars (see Table 1). Although there appear to be more reported differences between hypertrophic and keloid scars (Table 2) than similarities (see Table 1), the latter table does suggest that that hypertrophic and keloid scars share several pathogenetic mechanisms. For example, both abnormal scar types show stratum corneum barrier dysfunction,[ 18 , 19 ] as well as upregulation of epidermal differentiation and proliferation markers.[ 20 ] In the dermis, overexpression of connective tissue components,[ 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 ] parallel orientation of collagen bundles[ 30 ] and microvessel occlusion[ 31 , 32 ] represent a few of the shared abnormal scar abnormalities. With respect to the dermal cell population, both hypertrophic and keloid scar‐derived fibroblasts showed increased migration,[ 33 ] contraction,[ 34 , 35 ] and expression of wound healing mediators[ 33 ] and ECM components.[ 33 , 36 , 37 ] In our own comparative analysis of hypertrophic scars and keloids, we also identified shared abnormalities in both these abnormal scar types.[ 38 ] For example, the increased epidermal thickness combined with abnormal involucrin overexpression and the CD34−/α‐SMA+/p16+ dermal cell population in keloids were also observed in hypertrophic scars, albeit in slightly different degrees. When reconstructed in vitro,[ 39 ] both hypertrophic scars and keloids showed increased α‐SMA and p16 immunoreactivity with reduced dermal hyaluronan synthase 1 and matrix metalloproteinase 1 gene expression, as well as a non‐significant trend of increased dermal thickness.
At times, the reported similarities have directly contradicted each other. This was the case in both collagen synthesis and blood vessel density, which were both reported as normal[ 21 , 40 ] and increased.[ 21 , 22 , 23 , 24 , 26 , 28 , 29 ] Dermal nodules were also reported to be present in both abnormal scars[ 29 , 32 ] or variably present in 48%‐50% of either scar type.[ 41 ] Furthermore, certain features were observed in all scar types (normal and abnormal), such as increased thickness[ 42 ] and flattening of the epidermis,[ 22 , 26 , 28 ] loss of epidermal appendages[ 28 , 29 ]; but also increased collagen I,[ 43 ] fibronectin[ 25 , 27 ] and reduced fibrillin‐1[ 44 ] levels, absence of a non‐fibrotic papillary dermis[ 45 ] and a CD34−/FXIIIa−/S‐100− immunohistochemical profile.[ 46 ] The reason for these apparently conflicting findings is not known, but could be explained—at least in part, by differences in tissue sample collection with respect to: scar maturity, location within scar (scars may be heterogenous) or even lack of proper discernment between hypertrophic scars and keloids.
Several theories have been put forward in which hypertrophic and keloid are considered the same entity with different degrees of keloid triad components,[ 47 ] endothelial cell dysfunction,[ 48 ] inflammation[ 49 , 50 ] or extent of microvessel injury.[ 9 , 32 ] In the keloid triad hypothesis,[ 47 ] the difference between hypertrophic scars and keloids is dependent on the number of major and minor aetiological factors (eg genetics, infective agent, surgery) present. Keloid scars were thought to develop when at least one major factor (African ethnicity, age 10‐30 years, familial susceptibility or keloid‐prone upper part of body site) and two minor factors (orientation of incisions/sutures with respect to relaxed skin tension lines, wound or sutures under tension, healing by secondary intention, type of infection) were present, while hypertrophic scars were more likely to develop if only minor factors of all three categories or only two major factors were present.
Based on the shared histology of increased fibroblast density and collagen deposition, and the co‐existence of hypertrophic dermal nodules together with keloidal collagen within keloid scars, Huang and Ogawa[ 49 , 50 ] proposed that hypertrophic and keloid scars may represent successive stages of the same fibroproliferative skin disorder with different degrees of inflammation. The concentration of inflammation features (eg presence of microvessels, lymphocytes and fibroblasts) at the leading invasive edge of keloids is evidence in line with this theory. In an extension of this theory, Ogawa et al[ 48 ] proposed that local factors such as stretching tension together with genetic factors can both act to induce endothelial cell dysfunction during the inflammatory phase of wound healing. Vascular hyperpermeability prolongs the influx of inflammatory cells and factors, thereby also prolonging the inflammatory phase. The consequent dysfunction of the fibroblast cell population then leads to the formation of either hypertrophic or keloid scars. Evidence in favour of this hypothesis includes that most systemic factors associated with abnormal scar formation are also associated with vascular hyperpermeability and the fact that all effective treatment modalities (such as radiotherapy, compression, steroids, laser) act at least in part by acting on the vasculature to suppress endothelial dysfunction.
Kischer et al[ 32 ] suggested another mechanism by which microvessel abnormalities could generate either hypertrophic scars or keloids depending on the extent of microvessel injury. During regeneration of the microvessels after injury, the pericytes of the newly regenerating microvessels may act as the source of the fibroblasts that generate the excessive collagen in these abnormal scars. As keloid‐forming patients are thought to have a greater volume of microvessels in their skin, hypertrophic and keloid scars then only differ in the volume of injured microvessels. In this way, Kischer suggested that keloids are most probably quantitative exacerbations of hypertrophic scars. In line with this hypothesis, the increased predisposition of darker skinned individuals to keloid scarring could be explained by the presence of a greater volume of microvessels and therefore increased microvessel destruction compared with Caucasians with similar injuries and ultimately more regeneration, more pericytes which produce more fibroblasts and therefore more collagen.[ 9 ]
Stretch and tension probably play a greater role in hypertrophic scarring, but it also appears to have a role in keloid scarring.[ 51 ] Ogawa et al[ 52 ] speculated that both hypertrophic and keloid scarring may be directly caused by either hyperresponsive or deranged responsiveness of mechanosensors or mechanosensitive nociceptor of sensory endings in response to mechanical force stimuli, particularly stretching tension. In summary, findings of shared abnormalities across the entire spectrum of the cellular and connective tissue constituents of the epidermis and dermis support the notion that a keloid is simply a quantitative exacerbation of a hypertrophic scar. Additionally, elaborate theories have been proposed that suggest keloids and hypertrophic scars only differ in quantitative degrees of either endothelial cell dysfunction[ 48 ] or injury,[ 9 , 32 ] inflammation,[ 49 , 50 ] aetiological factors present[ 47 ] or abnormal mechanosensory responsiveness.[ 52 ]
3. “HYPERTROPHIC AND KELOID SCARS ARE DISTINCT ENTITIES”
From a clinical perspective, horizontal growth beyond the margins of the original wound is the defining characteristic which separates keloids from hypertrophic scars. One of the essential characteristics separating hypertrophic scars from keloids is the hypertrophic scar growth pattern, which involves reaching an eventual plateau after months of intense growth, usually followed by some degree of regression (see Figure 2).[ 2 ] This is demonstrated by Santucci et al,[ 26 ] who showed that the immunohistochemical profile depended on the age of the lesion with progressive normalization in hypertrophic scars, but not keloids. The abnormalities demonstrated in keloids were maintained irrespective of lesion age, and this absence of age‐related changes does suggest that maturation does not occur in keloids.
The histopathological distinction is far less straightforward and has hinged mostly on the presence of keloidal collagen in keloid scars.[ 53 , 54 ] The abnormal extracellular matrix and its genesis by the aberrant keloid fibroblasts has received much of the attention in current keloid research, particularly in an attempt to identify pathognomonic differences between hypertrophic and keloid scars (Table 2). Currently, the presence of keloidal collagen, whorls of thickened collagen bundles[ 26 , 53 , 55 ] versus the presence of α‐SMA‐positive myofibroblasts[ 28 ] and dermal nodules[ 28 , 50 ] are considered distinguishing features of keloids or hypertrophic scars, respectively. In fact, for keloids the presence of keloidal collagen is often used by pathologist to exclude a hypertrophic scar diagnosis.[ 53 ] Yet α‐SMA‐positive nodules[ 28 ] have long since lost their value as features differentiating hypertrophic scars from keloids as α‐SMA[ 26 , 41 , 46 ] and dermal nodules[ 26 , 41 , 50 , 53 , 56 ] have also been observed in both keloids and hypertrophic scars, with α‐SMA absence even reported as characteristic for hypertrophic scars.[ 45 ] In our experience, keloids generally showed less α‐SMA immunoreactivity compared with hypertrophic scars, but all keloid samples stained positive for α‐SMA. Conversely, while keloidal collagen was seldomly observed in hypertrophic scars, it was also not always found in all keloid scars.[ 57 , 58 ] We found that keloidal collagen was present constitutively and abundantly in all our keloid samples, but could also be observed in one of the hypertrophic scars we studied.[ 38 ] This has been reported for clinically diagnosed hypertrophic scars before,[ 53 , 59 ] but also further complicates its usefulness as a keloid marker. For keloids without keloidal collagen, Lee et al[ 41 ] also identified additional keloid‐specific features which could help identify keloid scars and have been corroborated by others[ 45 , 60 ]: a tongue‐like advancing edge underneath a normal‐appearing epidermis, a prominent horizontal fascia‐like band and a horizontal cellular fibrous band in the upper reticular dermis. Other studies focused specifically on identifying differences between hypertrophic and keloid scars have reported distinct collagen bundle thickness,[ 30 ] collagen deposition patterns[ 22 ] and vascular structure visibility.[ 61 ] Keloids showed lower levels of collagen fibril cross‐linking and abnormal collagen fibril assembly,[ 22 ] as well as significantly thicker collagen bundles.[ 30 ] Additionally, vascular structures as identified by in vivo dermoscopy were 24 times more likely to be observed in keloid scars compared with hypertrophic scars.[ 61 ]
In vitro studies have also been performed with the goal of identifying differences between the two abnormal scar types. As early as 1960, an attempt was made to differentiate hypertrophic scars and keloids by tissue culture.[ 62 ] Keloids possess larger, more adhesive fibroblasts[ 62 , 63 ] with increased proliferative capacity,[ 22 , 64 , 65 , 66 ] apoptosis resistance,[ 64 , 67 , 68 ] and increased secretion of wound healing factors TGF‐β1,[ 69 , 70 ] CTGF[ 71 ] and GDF‐9.[ 72 ] Increased production of extracellular matrix (ECM) or ECM‐associated factors has also been observed in keloid fibroblasts for mucin,[ 62 ] collagen I and III,[ 73 , 74 ] MMP1 and MMP19,[ 75 , 76 ] hyaluronan synthase.[ 37 ] Keloid fibroblasts not only display a more aggressive phenotype, they also respond differently to various stimuli compared to hypertrophic scar fibroblasts, such as TGF‐β1,[ 43 ] sialic acid,[ 74 ] mTOR inhibitor[ 37 ] and rapamycin.[ 37 ] Fibroblasts from keloids also show greater capacity for transdifferentiation. Stimulation with BMP4 or indirect hair follicle cell co‐culture was able to induce adipogenic conversion in keloid fibroblasts, but not their hypertrophic scar counterparts.[ 77 ]
Our in vitro reconstructed hypertrophic scars and keloids also showed differential expression of several scar parameters.[ 39 ] Only the keloid models significantly reduced epidermal laminin subunit α1 gene expression compared with normotrophic scars and significantly reduced dermal collagen IV α2 chain gene expression compared with normal skin. In direct comparison to one another, reconstructed hypertrophic scars showed higher dermal integrin α5 gene expression than the keloids, while dermal matrix metalloproteinase 1 gene expression was increased in keloids. Furthermore, hypertrophic scar models secreted reduced levels of several inflammatory mediators such as CCL5, with similar non‐significant trends for CXCL1, CXCL8 and IL‐6. Lastly, HGF secretion was only significantly reduced in the keloid model, although the hypertrophic scar model showed a similar non‐significant pattern. This was also the case with respect to the reduced collagen type IV α2 chain gene expression observed in the keloid scar model. In short, our in vitro reconstruction of keloid scars identified several new keloid‐specific markers with differential expression from hypertrophic scars.
In summary, studies specifically focused on identifying differences between hypertrophic and keloid scars reported differential keloidal collagen expression,[ 26 , 28 , 29 , 41 , 45 , 53 , 59 ] differences in ECM structure,[ 41 ] as well as distinct ultrastructural patterns of collagen deposition and assembly,[ 22 ] collagen bundle thickness,[ 30 ] vascular structures as visualized by dermoscopy[ 61 ]; differential ECM gene expression and CCL5 secretion when reconstructed in vitro[ 39 ]; in addition to differential fibroblast phenotypes,[ 62 ] transcriptional response to rapamycin treatment[ 37 ] and adipogenic conversion capabilities[ 77 ] in keloids compared with hypertrophic scars. These features, together with others listed in Table 2, strongly support the notion of hypertrophic scars and keloids being separate entities. However, the strongest argument for their distinction remains their emphatically different clinical behaviour in growth pattern and progression over time,[ 2 , 59 , 78 ] as is clearly illustrated in the graph of Figure 2 which was taken from a Burd and Huang's review[ 2 ] on the differences between the two abnormal scars.
4. DISTINGUISHING KELOIDS FROM HYPERTROPHIC SCARS
We combined our ex vivo findings[ 38 , 79 ] with what has already been reported in current literature in the decision tree in Figure 3A. This decision tree may aid the clinician in determining whether a raised scar is a hypertrophic scar or a keloid. A raised scar growing within the borders can immediately be classified as a hypertrophic scar and similarly, invasive growth defines keloids. When it is unclear whether growth is beyond the original borders, the histopathological diagnosis is best be made based on a combination of features more likely to occur in one or the other abnormal scar type. As observed by Kamath et al,[ 46 ] the absence of CD34 in suspected abnormal scars already helps to rule out other CD34‐positive disorders from the keloid differential diagnosis, such as dermatofibrosarcoma protuberans. Further negative staining with Factor XIIIa and S‐100 protein can help exclude dermatofibroma (Factor XIIIa+), desmoplastic malignant melanoma (S‐100+, note that this diagnosis is not automatically excluded by S‐100 absence) and other neural neoplasms (S‐100+).[ 46 ] Needless to say, the definitive diagnosis of either abnormal scar and therefore exclusion of other potential differential diagnoses should always be based on the clinical picture in conjunction with the histopathological findings. After the diagnosis of abnormal scar has been confirmed by the absence of CD34 combined with α‐SMA and p16 presence, we can then attempt to differentiate between the two scar types. For example, the presence of significant, constitutive keloidal collagen (see Figure 3B), smaller dermal nodules if present at all, higher levels of p16 but limited α‐SMA immunoreactivity which is strongest around the margins of the scar dermis, together with a thickened and strongly abnormal involucrin‐stained epidermis, favour a keloid scar diagnosis. Conversely, a strong case for a hypertrophic scar diagnosis is made by the combination of normal/increased epidermal involucrin expression, strong and diffuse α‐SMA immunoreactivity which is concentrated in larger dermal nodules if present, and the absence or negligible quantities of keloidal collagen.
FIGURE 3.
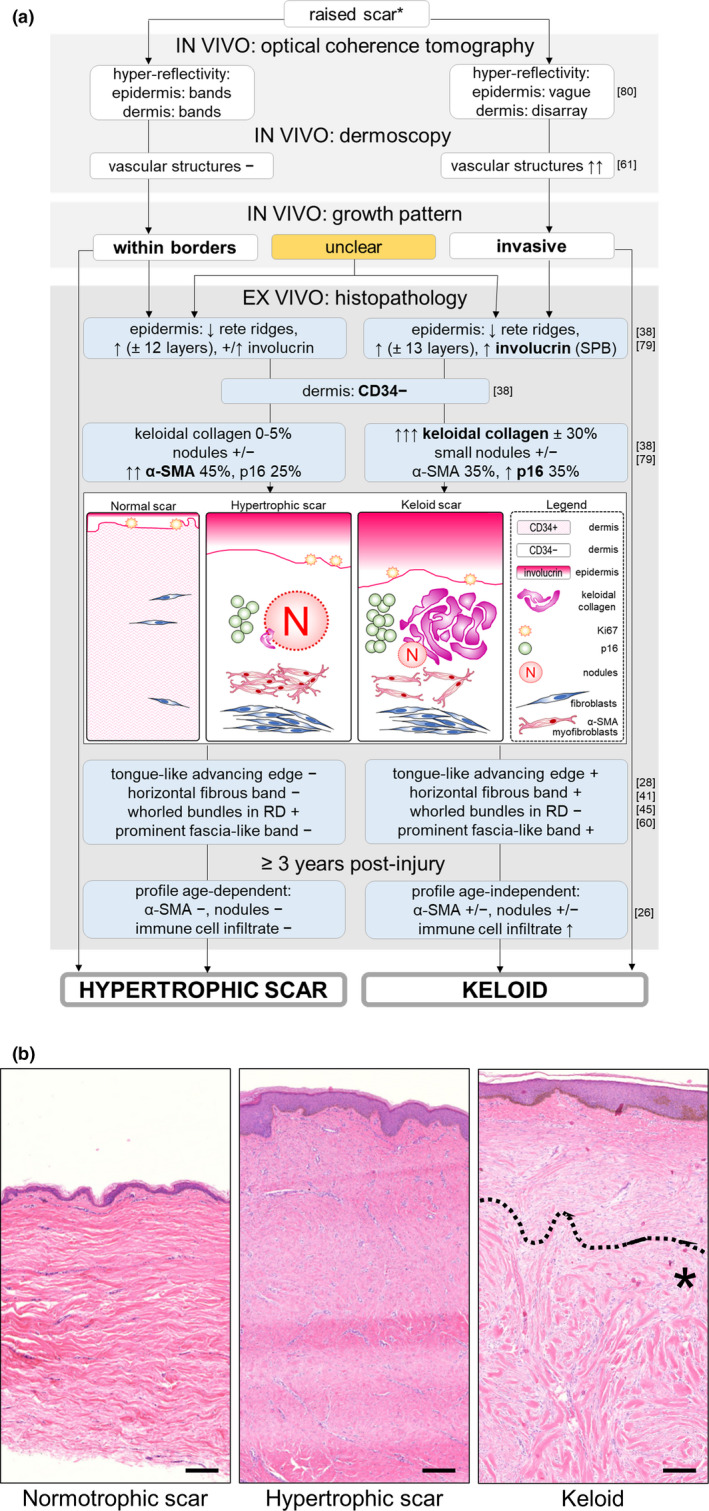
A, Histopathology decision tree: hypertrophic or keloid scar? This decision tree can be used to distinguish between hypertrophic scars and keloids, * after other differential diagnoses (eg dermatofibroma, dermatofibrosarcoma protuberans) have already been excluded or deemed highly unlikely based on clinical findings. Legend; white text boxes: in vivo findings; blue text boxes: ex vivo findings; associated references are listed to the right of each text box; bold font: strongest discriminating features; +: present, normal expression or values; +/↑: variable expression, both normal and increased expression observed; +/−: variable expression, both presence and absence of expression observed; ↑: increased; −: absent; ↓: decreased; ±: %: percentages of. Abbreviations listed in alphabetical order; α‐SMA: alpha smooth muscle actin; RD: reticular dermis; SPB: suprabasal expression. B, Haematoxylin and eosin staining of a normotrophic scar, a hypertrophic scar and a keloid. Area with keloidal collagen is marked with an asterisk (*) under the dotted line in the keloid panel. Scale bar = 200 μm
If this proves insufficient for adequate differentiation or in the event that keloidal collagen is absent, additional information may be obtained by evaluating the dermis for the presence of the following previously established markers: a tongue‐like advancing edge, a horizontal fibrous band, a prominent fascia‐like band and the presence of amorphous pericellular material are only found in keloid scars, while whorled bundles of collagen in the reticular dermis are specific to hypertrophic scars.[ 28 , 41 , 45 , 60 ] If this still does not provide a decisive answer, it may serve well to observe a waiting period as Santucci et al[ 26 ] observed that lesions ≥ 3 years old generated greater distinctive histopathological profiles. Keloids were unaffected by scar age and continued to show an increased immune cell infiltrate, while hypertrophic scars gradually lost this infiltrate. Additionally, optical coherence tomography is a non‐invasive imaging technique that can be performed on in vivo raised scars to differentiate between hypertrophic and keloid scars based on hyper‐reflectivity of the epidermis and dermis, although this technique may not be easily available in most clinical settings.[ 80 ] Alternatively, dermoscopy is a non‐invasive imaging tool at the dispense of every dermatologist's office and may identify vascular structures, which are significantly associated with keloids rather than hypertrophic scars.[ 61 ]
The diagnosis of a keloid scar still relies heavily on its clinical features, but these may not always offer a definitive answer. Following the decision tree in Figure 3A may aid clinicians in diagnosing hypertrophic or keloid scars when they are presented with raised scars with an ambiguous growth pattern.
5. HYPERTROPHIC SCARS AND KELOIDS: INTERCHANGEABLE TERMS OR DISTINCT ENTITIES?
As early as 1951,[ 81 ] it was recognized that differences between hypertrophic scars and keloids are often more clinical and quantitative in nature rather than qualitative. One could argue sufficient time has passed since then without the discovery of any major qualitative differences, to maintain that hypertrophic scars and keloids are separate and distinct entities. However, we have only barely scratched the surface of the processes involved in normal wound healing, let alone abnormal wound healing.[ 82 ] Furthermore, despite the numerous reports on similarities between hypertrophic and keloid scars (Table 1), the presence of similarities does not equate the absence of fundamental differences between the two abnormal scar types. There is bound to be a common initial pathway or likely multiple common initial pathways leading to the development of an abnormally raised scar, but an important divergence occurs at an unknown time point that leads to the development of decidedly different clinical behaviour and natural progression.[ 57 ] It is this clinical presentation that remains both the mainstay of the keloid diagnosis and in our opinion, the strongest argument against considering hypertrophic scars and keloids simply different stages of the same process. Over the past decades, a multitude of literature reviews on hypertrophic and keloid scars has been published. There are several reviews that do not particularly address the differences between the two,[ 8 , 83 , 84 , 85 , 86 , 87 ] or even deem the controversy insoluble and therefore sidestep the discussion entirely by grouping the two abnormal scar types into a single “hypertrophic keloid scar” entity.[ 88 ] However, the overwhelming majority of reviews discuss hypertrophic scars as separate from keloid scars[ 1 , 2 , 5 , 16 , 17 , 57 , 59 , 78 , 82 , 89 , 90 , 91 , 92 , 93 , 94 ]; and of these, four reviews by Burd and Huang,[ 2 ] Atiyeh et al,[ 78 ] Köse et al[ 57 ] and Ghazawi and colleagues[ 5 ] specifically addressed the controversy and concluded that in their opinion, the evidence was in favour of hypertrophic scars and keloids being separate, distinct entities instead of different stages of the same process. More importantly, the distinction is important because of the clinical implications. Keloids require a more aggressive therapeutic approach as they are known to be particularly therapy‐resistant compared with their hypertrophic counterparts,[ 1 , 2 , 16 , 17 , 78 ] and this is also reflected in the international scar management recommendations.[ 95 ]
The overview presented in this review is limited by the nature of the available studies that adhere to our single inclusion criterium: the inclusion of both hypertrophic scars and keloids within the same study. The majority of the studies in which similarities were observed between the two lesions (Table 1) were largely histopathological in nature. In contrast, studies that identified differences between the two groups (Table 2) involved a wider range of translational approaches, including the use of skin tissue engineering to develop in vitro scar models. Furthermore, several notable findings are currently excluded from our overview due to their “keloid only” or “hypertrophic scar only” experimental set‐up. These include novel findings in the area of keloid genetics, with laser capture microdissection of site‐specific regions within keloid biopsies demonstrating the presence of intralesional keloid heterogeneity[ 96 ] and the study of DNA methylation and histone acetylation in keloid‐derived fibroblasts supporting a role for epigenetic and transcriptome changes in the altered wound healing processes in keloids.[ 97 , 98 ] While most attempts at inducing keloid formation in animal models have inadvertently resulted in the development of a hypertrophic scar instead,[ 99 , 100 ] studies on animal scar models have also been excluded from this review due to their focus on either hypertrophic scars or keloids alone. Implanting reconstituted keloid‐derived cells into animal models has been more successful in humanizing the animal model and showing keloid tissue development.[ 101 , 102 , 103 , 104 ] Humanized animal hypertrophic scar models have been constructed in a similar fashion by transplanting healthy human split‐thickness skin grafts onto the backs of nude mice.[ 105 , 106 ] Interestingly, mechanomodulatory manipulation of both animal[ 107 ] and in vitro[ 108 ] hypertrophic scar models contributed to hypertrophic scarring mainly via apoptosis downregulation, and in Lee et al’s[ 102 ] humanized animal keloid model, high initial skin tension was mimicked by the use of a porous polyethylene ring to support implantation of the in vitro keloid construct. Ultimately, our limitations further illustrate the value and necessity of including both hypertrophic scars and keloids in experimental studies focused on identifying their underlying pathogenetic mechanisms and highlight essential areas of research that have thus far only been studied in these abnormal scars in isolation from each other. Furthermore, it is pertinent (and practical) to continue treating hypertrophic scars and keloids as separate entities, until such a time as new findings more decisively convince us otherwise.
CONFLICT OF INTEREST
The authors have no conflicts of interests to declare.
AUTHOR CONTRIBUTIONS
GCL conceived of the study, performed the literature study and wrote the manuscript. SG, RJS and FBN contributed to the manuscript outline and provided critical revisions to the manuscript content. All authors have read and approved the manuscript.
Limandjaja GC, Niessen FB, Scheper R, Gibbs S. Hypertrophic scars and keloids: Overview of the evidence and practical guide for differentiating between these abnormal scars. Exp Dermatol. 2021;30:146–161. 10.1111/exd.14121
REFERENCES
- 1. Wolfram D., Tzankov A., Pülzl P., Piza‐Katzer H., Dermatol. Surg. 2009, 35, 171. [DOI] [PubMed] [Google Scholar]
- 2. Burd A., Huang L., Plast. Reconstr. Surg. 2005, 116, 150e. [DOI] [PubMed] [Google Scholar]
- 3. Kumar V., Abbas A. K., Fausto N., In Robbins and Cotran, Pathologic Basis of Disease, 7th Edition (Eds: Kumar V., Abbas A. K., Fausto N.), Elsevier Saunders, Philadelphia, PA: 2004, Ch. 87‐118. [Google Scholar]
- 4. Sorg H., Tilkorn D. J., Hager S., Hauser J., Mirastschijski U., Eur. Surg. Res. 2017, 58, 81. [DOI] [PubMed] [Google Scholar]
- 5. Ghazawi F. M., Zargham R., Gilardino M. S., Sasseville D., Jafarian F., Adv. Skin. Wound Care 2018, 31, 582. [DOI] [PubMed] [Google Scholar]
- 6. Middelkoop E., Monstrey S., Téot L., Vranckx J. J., Editors. Scar Management: Practical Guidelines. Maca‐Cloetens, Elsene, Belgium: 2011, 11‐109. [Google Scholar]
- 7. Gauglitz G. G., Korting H. C., Pavicic T., Ruzicka T., Jeschke M. G., Mol. Med. 2011, 17, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rockwell W. B., Cohen I. K., Ehrlich H. P., Plast. Reconstr. Surg. 1998, 84, 827. [DOI] [PubMed] [Google Scholar]
- 9. Peacock E. E., Madden J. W., Trier W. C., South Med. J. 1970, 63, 755. [DOI] [PubMed] [Google Scholar]
- 10. Butler P. D., Longaker M. T., Yang G. P., J. Am. Coll. Surg. 2008, 206, 731. [DOI] [PubMed] [Google Scholar]
- 11. Bux S., Madaree A., Med. Hypotheses 2012, 78, 356. [DOI] [PubMed] [Google Scholar]
- 12. Ogawa R., Okai K., Tokumura F., Mori K., Ohmori Y., Huang C., Hyakusoku H., Akaishi S., Wound Repair Regen. 2012, 20, 149. [DOI] [PubMed] [Google Scholar]
- 13. Seifert O., Mrowietz U., Arch. Dermatol. Res. 2009, 301, 259. [DOI] [PubMed] [Google Scholar]
- 14. Honardoust D., Ding J., Varkey M., Shankowsky H. A., Tredget E. E., J. Burn Care Res. 2012, 33, 668. [DOI] [PubMed] [Google Scholar]
- 15. van den Broek L. J., Niessen F. B., Scheper R. J., Gibbs S., Altex 2012, 29, 389. [DOI] [PubMed] [Google Scholar]
- 16. Tuan T. L., Nichter L. S., Mol. Med. Today 1998, 4, 19. [DOI] [PubMed] [Google Scholar]
- 17. Niessen F. B., Spauwen P. H., Schalkwijk J., Kon M., Plast. Reconstr. Surg. 1999, 104, 1435. [DOI] [PubMed] [Google Scholar]
- 18. Sogabe Y., Akimoto S., Abe M., Ishikawa O., Takagi Y., Imokawa G., J. Dermatol. Sci. 2002, 29, 49. [DOI] [PubMed] [Google Scholar]
- 19. Suetake T., Sasai S., Zhen Y. X., Ohi T., Tagami H., Arch. Dermatol. 1996, 132, 1453. [PubMed] [Google Scholar]
- 20. Bloor B. K., Tidman N., Leigh I. M., Odell E., Dogan B., Wollina U., Ghali L., Waseem A., Am. J. Pathol. 2003, 162, 963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Craig P. R. D., Schofield J. D., Jackson S. S., Br. J. Surg. 1975, 62, 741. [DOI] [PubMed] [Google Scholar]
- 22. Meenakshi J., Jayaraman V., Ramakrishnan K. M., Babu M., Ann. Burn Fire Disasters 2005, 18, 83. [PMC free article] [PubMed] [Google Scholar]
- 23. Suzawa H., Kikuchi S., Arai N., Koda A., Jpn. J. Pharmacol. 1992, 60, 91. [DOI] [PubMed] [Google Scholar]
- 24. Venugopal J., Ramakrishnan M., Habibullah C. M., Babu M., Burns 1999, 25, 223. [DOI] [PubMed] [Google Scholar]
- 25. Kischer C. W., Hendrix M. J. C., Cell Tissue Res. 1983, 231, 29. [DOI] [PubMed] [Google Scholar]
- 26. Santucci M., Borgognoni L., Reali U. M., Gabbiani G., Virchows Arch. 2001, 438, 457. [DOI] [PubMed] [Google Scholar]
- 27. Sible J. C., Eriksson E., Smith S. P., Oliver N., Wound Repair Regen. 1994, 2, 3. [DOI] [PubMed] [Google Scholar]
- 28. Ehrlich H. P., Desmoulière A., Diegelmann R. F., Cohen I. K., Compton C. C., Garner W. L., Kapanci Y., Gabbiani G., Am. J. Pathol. 1994, 145, 105. [PMC free article] [PubMed] [Google Scholar]
- 29. Materazzi S., Pellerito S., Di Serio C., Paglierani M., Naldini A., Ardinghi C., Carraro F., Geppetti P., Cirino G., Santucci M., Tarantini F., Massi D., J. Pathol. 2007, 212, 440. [DOI] [PubMed] [Google Scholar]
- 30. Verhaegen P. D. H. M., van Zuijlen P. P. M., Pennings N. M., van Marle J., Niessen F. B., van der Horst C. M. A. M., Middelkoop E., Wound Repair Regen. 2009, 17, 649. [DOI] [PubMed] [Google Scholar]
- 31. Kischer C. W., Thies A. C., Chvapil M., Hum. Pathol. 1982, 13, 819. [DOI] [PubMed] [Google Scholar]
- 32. Kischer C., Scan. Electron. Microsc. 1984, (Pt 1), 423. [PubMed] [Google Scholar]
- 33. Fang F., Huang R.‐L., Zheng Y., Liu M., Huo R., J. Dermatol. Sci. 2016, 83, 95. [DOI] [PubMed] [Google Scholar]
- 34. Smith P., Mosiello G., Deluca L., Ko F., Maggi S., Robson M. C., J. Surg. Res. 1999, 82, 319. [DOI] [PubMed] [Google Scholar]
- 35. Sumi Y., Muramatsu H., Hata K.‐I., Ueda M., Muramatsu T., Exp. Cell Res. 2000, 256, 203. [DOI] [PubMed] [Google Scholar]
- 36. Kischer C. W., Pindur J., Cytotechnology 1990, 3, 231. [DOI] [PubMed] [Google Scholar]
- 37. Wong V. W., You F., Januszyk M., Gurtner G. C., Kuang A. A., Ann. Plast. Surg. 2014, 72, 711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Limandjaja G. C., Belien J. M., Scheper R. J., Niessen F. B., Gibbs S., Br. J. Dermatol. 2020, 182, 974. [DOI] [PubMed] [Google Scholar]
- 39. Limandjaja G. C., van den Broek L. J., Breetveld M., Waaijman T., Monstrey S., de Boer E. M., Scheper R. J., Niessen F. B., Gibbs S., Tissue Eng. Part C Methods 2018, 24, 242. [DOI] [PubMed] [Google Scholar]
- 40. Amadeu T., Braune A., Mandarim‐de‐Lacerda C., Porto L. C., Desmoulière A., Costa A., Pathol. Res. Pract. 2003, 199, 469. [DOI] [PubMed] [Google Scholar]
- 41. Lee J. Y. Y., Yang C. C., Chao S. C., Wong T. W., Am. J. Dermatopathol. 2004, 26, 379. [DOI] [PubMed] [Google Scholar]
- 42. Hellström M., Hellström S., Engström‐Laurent A., Bertheim U., J. Plast. Reconstr. Aesthetic Surg. 2014, 67, 1564. [DOI] [PubMed] [Google Scholar]
- 43. Zhang Z.‐F., Zhang Y.‐G., Hu D.‐H., Shi J.‐H., Liu J.‐Q., Zhao Z.‐T., Wang H.‐T., Bai X.‐Z., Cai W.‐X., Zhu H.‐Y., Tang C.‐W., Burns 2011, 37, 665. [DOI] [PubMed] [Google Scholar]
- 44. Amadeu T. P., Braune A. S., Porto L. C., Desmoulière A., Costa A. M. A., Wound Repair Regen. 2004, 12, 169. [DOI] [PubMed] [Google Scholar]
- 45. Moshref S., Mufti S. T., J. King Abdulaziz Univ. Med. Sci. 2009, 17, 3. [Google Scholar]
- 46. Kamath N. V., Ormsby A., Bergfeld W. F., House N. S., J. Cutan. Pathol. 2002, 29, 27. [DOI] [PubMed] [Google Scholar]
- 47. Agbenorku P. T., Kus H., Myczkowski T., Eur. J. Plast. Surg. 1995, 18, 301. [Google Scholar]
- 48. Ogawa R., Akaishi S., Med. Hypotheses 2016, 96, 51. [DOI] [PubMed] [Google Scholar]
- 49. Ogawa R., Int. J. Mol. Sci. 2017, 18, 1. [Google Scholar]
- 50. Huang C., Akaishi S., Hyakusoku H., Ogawa R., Int. Wound. J. 2014, 11, 517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Al‐Attar A., Mess S., Thomassen J. M., Kauffman C. L., Davison S. P., Plast. Reconstr. Surg. 2006, 117, 286. [DOI] [PubMed] [Google Scholar]
- 52. Ogawa R., Med Hypotheses 2008, 71, 493–500. [DOI] [PubMed] [Google Scholar]
- 53. Ogawa R., Akaishi S., Izumi M., Ann. Plast. Surg. 2009, 62, 104. [DOI] [PubMed] [Google Scholar]
- 54. Gulamhuseinwala N., Mackey S., Meagher P., Powell B., Ann. Plast. Surg. 2008, 60, 186. [DOI] [PubMed] [Google Scholar]
- 55. Cosman B., Crikelair G. F., Ju D. M. C., Gaulin J. C., Lattes R., Plast. Reconstr. Surg. 1961, 27, 335. [Google Scholar]
- 56. Bux S., Madaree A., Cells Tissues Organs 2010, 191, 213–34. [DOI] [PubMed] [Google Scholar]
- 57. Köse O., Waseem A., Dermatol. Surg. 2008, 34, 336. [DOI] [PubMed] [Google Scholar]
- 58. Robles D., Berg D., Clin. Dermatol. 2007, 25, 26. [DOI] [PubMed] [Google Scholar]
- 59. Muir I. F. K., Br. J. Plast. Surg. 1990, 43, 61. [DOI] [PubMed] [Google Scholar]
- 60. Szulgit G., Rudolph R., Wandel A., Tenenhaus M., Panos R., Gardner H., J. Invest. Dermatol. 2002, 118, 409. [DOI] [PubMed] [Google Scholar]
- 61. Yoo M. G., Kim I. H., Ann. Dermatol. 2014, 26, 603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Conway H., Gillette R., Smith J. W., Findley A., Plast. Reconstr. Surg. 1960, 25, 117. [DOI] [PubMed] [Google Scholar]
- 63. Ali S. S., Hajrah N. H., Ayuob N. N., Moshref S. S., Abuzinadah O. A., Saudi Med. J. 2010, 31, 874. [PubMed] [Google Scholar]
- 64. De Felice B., Garbi C., Santoriello M., Santillo A., Wilson R. R., Mol. Cell. Biochem. 2009, 327, 191. [DOI] [PubMed] [Google Scholar]
- 65. Jing C., Jia‐han W., Hong‐Xing Z., Wound Repair Regen. 2010, 18, 319. [DOI] [PubMed] [Google Scholar]
- 66. Concannon M. J., Barrett B. B., Adelstein E. H., Thornton W. H., Puckett C. L., J. Burn Care Rehabilit. 1993, 14, 141. [DOI] [PubMed] [Google Scholar]
- 67. Chodon T., Sugihara T., Igawa H. H., Funayama E., Furukawa H., Am. J. Pathol. 2000, 157, 1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lu F., Gao J., Ogawa R., Hyakusoku H., Ou C., Plast. Reconstr. Surg. 2007, 119, 1714. [DOI] [PubMed] [Google Scholar]
- 69. Bock O., Yu H., Zitron S., Bayat A., Ferguson M., Mrowietz U., Acta Derm. Venereol. 2005, 85, 216. [DOI] [PubMed] [Google Scholar]
- 70. Younai S., Venters G., Vu S., Nichter L., Nimni M. E., Tuan T.‐L., Ann. Plast. Surg. 1996, 36, 495. [DOI] [PubMed] [Google Scholar]
- 71. Colwell A. S., Phan T.‐T., Kong W., Longaker M. T., Lorenz P. H., Plast. Reconstr. Surg. 2005, 116, 1387. [DOI] [PubMed] [Google Scholar]
- 72. Gao Z., Wu X., Song N., Zhang L. U., Liu W., Burns 2010, 36, 1289. [DOI] [PubMed] [Google Scholar]
- 73. Friedman D. W., Boyd C. D., Mackenzie J. W., Norton P., Olson R. M., Deak S. B., J. Surg. Res. 1993, 55, 214. [DOI] [PubMed] [Google Scholar]
- 74. Kössi J., Aalto J., Haataja S., Niinikoski J., Peltonen J., Laato M., Ann. Chir. Gynaecol. 2001, 90, 25. [PubMed] [Google Scholar]
- 75. McFarland K. L., Glaser K., Hahn J. M., Boyce S. T., Supp D. M., J. Burn Care Res. 2011, 32, 498. [DOI] [PubMed] [Google Scholar]
- 76. Suarez E., Syed F., Alonso‐Rasgado T., Bayat A., Arch. Dermatol. Res. 2015, 307, 115. [DOI] [PubMed] [Google Scholar]
- 77. Plikus M. V., Guerrero‐Juarez C. F., Ito M., Li Y. R., Dedhia P. H., Zheng Y., Shao M., Gay D. L., Ramos R., Hsi T.‐C., Ji Won O., Wang X., Ramirez A., Konopelski S. E., Elzein A., Wang A., Supapannachart R. J., Lee H.‐L., Lim C. H., Nace A., Guo A., Treffeisen E., Andl T., Ramirez R. N., Murad R., Offermanns S., Metzger D., Chambon P., Widgerow A. D., Tuan T.‐L., Mortazavi A., Gupta R. K., Hamilton B. A., Millar S. E., Seale P., Pear W. S., Lazar M. A., Cotsarelis G., Science 2017, 355, 748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Atiyeh B. S., Costagliola M., Hayek S. N., Ann. Plast. Surg. 2005, 54, 676. [DOI] [PubMed] [Google Scholar]
- 79. Limandjaja G. C., Broek L. J., Waaijman T., Veen H. A., Everts V., Monstrey S., Scheper R. J., Niessen F. B., Gibbs S., Br. J. Dermatol. 2017, 176, 116. [DOI] [PubMed] [Google Scholar]
- 80. Ring H. C., Mogensen M., Hussain A. A., Steadman N., Banzhaf C., Themstrup L., Jemec G. B., J. Eur. Acad. Dermatol. Venereol. 2015, 29, 890. [DOI] [PubMed] [Google Scholar]
- 81. Robinson D. R., Corbett E., Hamilton T. R., Surg. Forum 1951, 505. [PubMed] [Google Scholar]
- 82. Slemp A. E., Kirschner R. E., Curr. Opin. Pediatr. 2006, 18, 396. [DOI] [PubMed] [Google Scholar]
- 83. Murray J. C., Pollack S. V., Pinnell S. R., J. Am. Acad. Dermatol. 1981, 4, 461. [DOI] [PubMed] [Google Scholar]
- 84. Song C., Ann. Plast. Surg. 2014, 73, S108. [DOI] [PubMed] [Google Scholar]
- 85. Thomas D. W., Hopkinson I., Harding K. G., Shepherd J. P., Int. J. Oral Maxillofac. Surg. 1994, 23, 232. [DOI] [PubMed] [Google Scholar]
- 86. Davies D. M., Plast. Reconstr. Surg. 1985, 290, 1056. [Google Scholar]
- 87. Brissett A. E., Sherris D. A., Facial Plast. Surg. 2001, 17, 263. [DOI] [PubMed] [Google Scholar]
- 88. McGrouther D. A., Eye 1994, 8, 200. [DOI] [PubMed] [Google Scholar]
- 89. Ladin D. A., Garner W. L., Smith D. J., Wound Repair Regen. 1995, 3, 6. [DOI] [PubMed] [Google Scholar]
- 90. Huang C., Murphy G. F., Akaishi S., Ogawa R., Plast. Reconstr. Surg. Glob. Open 2013, 1, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. English R. S., Shenefelt P. D., Dermatol. Surg. 1999, 25, 631. [DOI] [PubMed] [Google Scholar]
- 92. Canady J., Karrer S., Fleck M., Bosserhoff A. K., J. Dermatol. Sci. 2013, 70, 151. [DOI] [PubMed] [Google Scholar]
- 93. Bran G. M., Goessler U. R., Hormann K., Riedel F., Sadick H., Int. J. Mol. Med. 2009, 24, 283. [DOI] [PubMed] [Google Scholar]
- 94. Trusler H. M., Bauer T. B., Arch. Surg. 1948, 57, 539. [DOI] [PubMed] [Google Scholar]
- 95. Gold M. H., McGuire M., Mustoe T. A., Pusic A., Sachdev M., Waibel J., Murcia C., International Advisory Panel on Scar Management , Dermatol. Surg. 2014, 40, 825. [DOI] [PubMed] [Google Scholar]
- 96. Jumper N., Hodgkinson T., Paus R., Bayat A., PLoS One 2017, 12, e0172955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lee Y. S., Liang Y. C., Wu P., Kulber D. A., Tanabe K., Chuong C. M., Widelitz R., Tuan T. L., Exp. Dermatol. 2019, 28, 480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Russell S. B., Russell J. D., Trupin K. M., Gayden A. E., Opalenik S. R., Nanney L. B., Broquist A. H., Raju L., Williams S. M., J. Invest. Dermatol. 2010, 130, 2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Khorshid F. A., Med. Sci. Monit. 2005, 11, BR212. [PubMed] [Google Scholar]
- 100. Morris D. E., Wu L., Zhao L. L., Bolton L., Roth S. I., Ladin D. A., Mustoe T. A., Plast. Reconstr. Surg. 1997, 100, 674. [DOI] [PubMed] [Google Scholar]
- 101. Zhang Q., Yamaza T., Kelly A. P., Shi S., Wang S., Brown J., Wang L., French S. W., Shi S., Le A. D., PLoS One 2009, 4, e7798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Lee Y.‐S., Hsu T., Chiu W.‐C., Sarkozy H., Kulber D. A., Choi A., Kim E. W., Benya P. D., Tuan T. L., Wound Repair Regen. 2015, 24, 302. [DOI] [PubMed] [Google Scholar]
- 103. Supp D. M., Hahn J. M., Glaser K., McFarland K. L., Boyce S. T., Plast. Reconstr. Surg. 2012, 129, 1259. [DOI] [PubMed] [Google Scholar]
- 104. Supp D. M., Glaser K., Hahn J. M., McFarland K. L., Boyce S. T., Eplasty 2012, 12, 184. [PMC free article] [PubMed] [Google Scholar]
- 105. Momtazi M., Kwan P., Ding J., Anderson C. C., Honardoust D., Goekjian S., Tredget E. E., Wound Repair Regen. 2013, 21, 77. [DOI] [PubMed] [Google Scholar]
- 106. Yang D. Y., Li S. R., Wu J. L., Chen Y. Q., Li G., Bi S., Dai X., Plast. Reconstr. Surg. 2007, 119, 104. [DOI] [PubMed] [Google Scholar]
- 107. Aarabi S., Bhatt K. A., Shi Y., Paterno J., Chang E. I., Loh S. A., Holmes J. W., Longaker M. T., Yee H., Gurtner G. C., FASEB J. 2007, 21, 3250. [DOI] [PubMed] [Google Scholar]
- 108. Derderian C. A., Bastidas N., Lerman O. Z., Bhatt K. A., Lin S.‐E., Voss J., Holmes J. W., Levine J. P., Gurtner G. C., Ann. Plast. Surg. 2005, 55, 69. [DOI] [PubMed] [Google Scholar]
- 109. Teofoli P., Barduagni S., Ribuffo M., Campanella A., De Pita’ O., Puddu P., J. Dermatol. Sci. 1999, 22, 31. [DOI] [PubMed] [Google Scholar]
- 110. Abdou A. G., Maraee A. H., Al‐Bara A. M., Diab W. M., Am. J. Dermatopathol. 2011, 33, 84. [DOI] [PubMed] [Google Scholar]
- 111. Zhao J., Zhong A., Friedrich E. E., Jia S., Xie P., Galiano R. D., Mustoe T. A., Hong S. J., J. Invest. Dermatol. 2017, 137, 650. [DOI] [PubMed] [Google Scholar]
- 112. Akasaka Y., Fujita K., Ishikawa Y., Asuwa N., Inuzuka K., Ishihara M., Ito M., Masuda T., Akishima Y., Zhang L., Ito K., Ishii T., Wound Repair Regen. 2001, 9, 501. [DOI] [PubMed] [Google Scholar]
- 113. Tanriverdi‐Akhisaroglu S., Menderes A., Oktay G., Cell Biochem. Funct. 2009, 27, 81. [DOI] [PubMed] [Google Scholar]
- 114. Zhou H.‐M., Wang J., Elliott C., Wen W., Hamilton D. W., Conway S. J., J. Cell Commun. Signal. 2010, 4, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Bertheim U., Hellström S., Br. J. Plast. Surg. 1994, 47, 483. [DOI] [PubMed] [Google Scholar]
- 116. Knapp T. R., Daniels R. J., Kaplan E. N., Am. J. Pathol. 1977, 86, 47. [PMC free article] [PubMed] [Google Scholar]
- 117. Milsom J. P., Craig R. D. P., Br. J. Dermatol. 1973, 89, 635. [DOI] [PubMed] [Google Scholar]
- 118. Neely A. N., Clendening C. E., Gardner J., Greenhalgh D. G., Warden G. D., Wound Repair Regen. 1999, 7, 166. [DOI] [PubMed] [Google Scholar]
- 119. Ulrich D., Ulrich F., Unglaub F., Piatkowski A., Pallua N., J. Plast. Reconstr. Aesthetic Surg. 2010, 63, 1015. [DOI] [PubMed] [Google Scholar]
- 120. Ueda K., Furuya E., Yasuda Y., Oba S., Tajima S., Plast. Reconstr. Surg. 1999, 104, 694. [DOI] [PubMed] [Google Scholar]
- 121. Kiya K., Kubo T., Kawai K., Matsuzaki S., Maeda D., Fujiwara T., Nishibayashi A., Kanazawa S., Yano K., Amano G., Katayama T., Hosokawa K. O., Exp. Dermatol. 2016, 26, 705. [DOI] [PubMed] [Google Scholar]
- 122. Touchi R., Ueda K., Kurokawa N., Tsuji M., J. Plast. Reconstr. Aesthetic Surg. 2016, 69, e35. [DOI] [PubMed] [Google Scholar]
- 123. Beer T. W., Baldwin H., West L., Gallagher P. J., Wright D. H., Br. J. Opthalmol. 1998, 82, 691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Aiba S., Tabata N., Ishii H., Ootani H., Tagami H., Br. J. Dermatol. 1992, 127, 79. [DOI] [PubMed] [Google Scholar]
- 125. Onodera M., Ueno M., Ito O., Suzuki S., Igawa H. H., Sakamoto H., Pathol. Int. 2007, 57, 337. [DOI] [PubMed] [Google Scholar]
- 126. Chen D., Wang Q., Bao W., Xu S., Tang Y., Chin. Med. J. 2003, 116, 314. [PubMed] [Google Scholar]
- 127. Akasaka Y., Ishikawa Y., Ono I., Fujita K., Masuda T., Asuwa N., Inuzuka K., Kiguchi H., Ishii T., Lab. Investig. 2000, 80, 345. [DOI] [PubMed] [Google Scholar]
- 128. Akasaka Y., Ito K., Fujita K., Komiyama K., Ono I., Ishikawa Y., Akishima Y., Sato H., Ishii T., Wound Repair Regen. 2005, 13, 373. [DOI] [PubMed] [Google Scholar]
- 129. Yang E., Qipa Z., Hengshu Z., Wounds 2014, 26, 139‐146. [PubMed] [Google Scholar]
- 130. Kössi J., Laato M., Pathobiology 2000, 68, 29. [DOI] [PubMed] [Google Scholar]
- 131. Kössi J., Peltonen J., Uotila P., Laato M., Arch. Dermatol. Res. 2001, 293, 126. [DOI] [PubMed] [Google Scholar]
- 132. Kössi J., Vähä‐Kreula M., Peltonen J., Risteli J., Laato M., World J. Surg. 2001, 25, 142. [DOI] [PubMed] [Google Scholar]
- 133. Wong V. W., You F., Januszyk M., Kuang A. A., J. Surg. Res. 2013, 184, 678. [DOI] [PubMed] [Google Scholar]
- 134. Zhu W., Wu X., Yang B. O., Yao X., Cui X., Xu P., Chen X., Acta Biochim. Biophys. Sin. 2019, 51, 185. [DOI] [PubMed] [Google Scholar]
- 135. Sato M., Ishikawa O., Miyachi Y., Br. J. Dermatol. 1998, 138, 938. [DOI] [PubMed] [Google Scholar]
- 136. van der Slot A. J., Zuurmond A.‐M., van den Bogaerdt A. J., Ulrich M. M. W., Middelkoop E., Boers W., Karel Ronday H., DeGroot J., Huizinga T. W. J., Bank R. A., Matrix Biol. 2004, 23, 251. [DOI] [PubMed] [Google Scholar]
- 137. Drummond P. D., Dawson L. F., Wood F. M., Fear M. W., Burns 2017, 44, 582. [DOI] [PubMed] [Google Scholar]
- 138. Mendoza‐Garcia J., Sebastian A., Alonso‐Rasgado T., Bayat A., Photodermatol. Photoimmunol. Photomed. 2015, 31, 239. [DOI] [PubMed] [Google Scholar]
- 139. Phan T. T., Lim I. J., Aalami O., Lorget F., Khoo A., Tan E. K., Mukhopadhyay A., Longaker M. T., J. Pathol. 2005, 207, 232. [DOI] [PubMed] [Google Scholar]
- 140. Pavelecini M., Zettler C. G., Fernandes M. C., Ely P. B., Plast. Reconstr. Surg. Glob. Open 2019, 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Abdou A. G., Maraee A. H., Abd‐Elsattar Saif H. F., Am. J. Dermatopathol. 2014, 36, 311. [DOI] [PubMed] [Google Scholar]
- 142. Sideek M. A., Teia A., Kopecki Z., Cowin A. J., Gibson M. A., J. Mol. Histol. 2016, 47, 35. [DOI] [PubMed] [Google Scholar]
- 143. Aiba S., Tagami H., J. Cutan. Pathol. 1997, 24, 65. [DOI] [PubMed] [Google Scholar]
- 144. Andreoli A., Ruf M. T., Itin P., Pluschke G., Schmid P., Br. J. Dermatol. 2015, 172, 1415. [DOI] [PubMed] [Google Scholar]
- 145. Tanaka A., Hatoko M., Tada H., Iioka H., Niitsuma K., Miyagawa S., J. Dermatol. Sci. 2004, 34, 17. [DOI] [PubMed] [Google Scholar]
- 146. Cobbold C. A., Sherratt J. A., J. Theor. Biol. 2000, 204, 257. [DOI] [PubMed] [Google Scholar]
- 147. Smith C. J., Smith J. C., Finn M. C., J. Burn Care Rehabilit. 1987, 8, 126. [DOI] [PubMed] [Google Scholar]
- 148. Prathiba V., Rao K. S., Gupta P. D., Cytobios 2001, 104, 43‐51. [PubMed] [Google Scholar]
- 149. Kim J.‐E., Lee J.‐H., Jeong K.‐H., Kim G. M., Kang H., Ann. Dermatol. 2014, 26, 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Hu Z.‐C., Tang B., Guo D., Zhang J., Liang Y.‐Y., Ma D., Zhu J.‐Y., Clin. Exp. Dermatol. 2014, 39, 822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Nirodi C. S., Devalaraja R., Nanney L. B., Arrindell S., Russell S., Trupin J., Richmond A., Wound Repair Regen. 2000, 8, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Liao W.‐T., Yu H.‐S., Arbiser J. L., Hong C.‐H., Govindarajan B., Chai C.‐Y., Shan W.‐J., Lin Y.‐F., Chen G.‐S., Lee C.‐H., Exp. Dermatol. 2010, 19, e142. [DOI] [PubMed] [Google Scholar]
- 153. Hsu C., Lin H., Harn H. I., Ogawa R., Wang Y. K., Ho Y. T., Chen W. R., Lee Y. C., Lee J. Y., Shieh S. J., Cheng C. M., McGrath J. A., Tang M. J., J. Invest. Dermatol. 2018, 138, 208. [DOI] [PubMed] [Google Scholar]
- 154. De Felice B., Ciarmiello L. F., Mondola P., Damiano S., Seru R., Argenziano C., Nacca M., Santoriello M., Garbi C., DNA Cell Biol. 2007, 26, 541. [DOI] [PubMed] [Google Scholar]
- 155. Lu F., Gao J. H., Ogawa R., Hyakusoku H., Plast. Reconstr. Surg. 2007, 119, 844. [DOI] [PubMed] [Google Scholar]
- 156. Sahara K., Kucukcelebi A., Ko F., Phillips L., Robson M., Wound Repair Regen. 1993, 1, 22. [DOI] [PubMed] [Google Scholar]
- 157. Younai S., Nichter L. S., Wellisz T., Reinisch J., Nimni M. E., Tuan T.‐L., Ann. Plast. Surg. 1994, 33, 148. [DOI] [PubMed] [Google Scholar]
- 158. Beer T. W., Baldwin H. C., Goddard J. R., Gallagher P. J., Wright D. H., Hum. Pathol. 1998, 29, 1273. [DOI] [PubMed] [Google Scholar]
- 159. Ueda K., Yasuda Y., Furuya E., Oba S., Scand. J. Plast. Reconstr. Surg. Hand Surg. 2004, 38, 267. [DOI] [PubMed] [Google Scholar]
- 160. Kurokawa N., Ueda K., Tsuji M., J. Plast. Surg. Hand Surg. 2010, 44, 272. [DOI] [PubMed] [Google Scholar]
- 161. Janssen de Limpens A. M., Cormane R. H., Aesthetic Plast. Surg. 1982, 6, 149. [DOI] [PubMed] [Google Scholar]
- 162. Laurentaci G., Dioguardi D., Arch. Dermatol. 1977, 113, 1726. [PubMed] [Google Scholar]
- 163. Dong X., Zhang C., Ma S., Wen H., Int. J. Clin. Exp. Pathol. 2014, 7, 3596. [PMC free article] [PubMed] [Google Scholar]
- 164. Mitts T. F., Bunda S., Wang Y., Hinek A., J. Invest. Dermatol. 2010, 130, 2396. [DOI] [PubMed] [Google Scholar]
- 165. Dohi T., Padmanabhan J., Akaishi S., Than P. A., Terashima M., Matsumoto N. N., Ogawa R., Gurtner G. C., Plast. Reconstr. Surg. 2019, 144, 58. [DOI] [PubMed] [Google Scholar]
- 166. Elrefaie A. M., Salem R. M., Faheem M. H.. Lasers Med. Sci. 2020, 35, 379. [DOI] [PubMed] [Google Scholar]
- 167. Ghazizadeh M., Miyata N., Sasaki Y., Arai K., Aihara K., Am. J. Dermatopathol. 1997, 19, 468. [DOI] [PubMed] [Google Scholar]
- 168. Seleit I., Bakry O. A., Samaka R. M., Tawfik A. S., Appl. Immunohistochem. Mol. Morphol. 2016, 24, 296. [DOI] [PubMed] [Google Scholar]