
Figure 1.
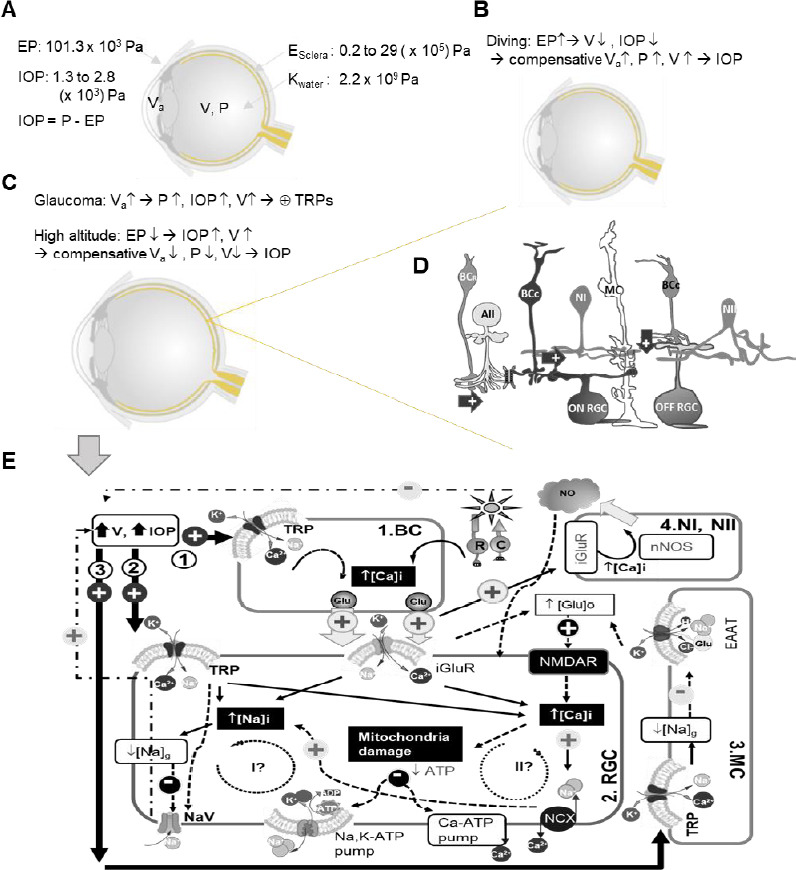
Diagram of potential routes for TRPs to mediate RGC pathophysiology in glaucoma.
Ocular physical parameters are depicted in A–C. E: Young’s modulus; EP: external pressure; IOP: intraocular pressure; K: bulk modulus; P: pressure; V: eyeball volume; Va: volume of aqueous humor. In D, arrow: excitatory glutamate synapse; AII: AII amacrine cell; and butterfly shape: gap junction. In D and E, BCR: rod bipolar cell (BC); BCC: cone BC; MC: Müller cell; Type I and II nitroxidergic amacrine cell (NI and NII) and RGC: retinal ganglion cell. E displays the possible (arrow) and presumable (dashed arrow) effect of TRP activation upon increasing V and IOP, and three potential pathophysiological routes are illustrated involved by BC (1), RGC (2), and MC (3). TRP activation is known to cause influxes of Na+ and Ca2+, higher [Ca2+]i, and the reduction of Na+ electrochemical gradien ([Na]g)/membrane depolarization, which are to be superimposed on light-evoked activities driven by the photoreceptor rod (R) and cone (C) and mediated by iGluRs in RGCs. Higher [Ca2+]i presumably increases glutamate (Glu) release in BCs, and in Müller cell (MC) TRP-mediated decrease of [Na]g would reduce Glu transportation by the excitatory amino acid transporter (EAAT) to increase [Glu]o. The increased [Glu]o can activate N-methyl-D-aspartate receptor (NMDAR) in RGCs, damaging RGC mitochondria via Ca2+ and other excitotoxic mechanisms. The increase of [Ca2+]i and [Na+]i needs to consume more ATPs to restore their normal electrochemical gradients. Two potential pathophysiological cycles (I and II) are further proposed for RGCs. Cycle I anticipates that an extra amount of Na+ influx would consume more ATPs to cause or enhance ATP shortage, and a dramatic reduction of [Na]g could lead to the deactivation of the voltage-gated Na+ channel (NaV) and the loss of RGC function. Cycle II proposes that an extra amount of Ca2+ influx via TRPs would increase [Ca2+]i to further damage the mitochondria, causing or enhancing ATP shortage to further increase [Ca2+]i and exacerbate the cellular damage.