

Abstract
Background
Immunotherapy has been shown to be a promising strategy against human cancers. A better understanding of the immune regulation in hepatocellular carcinoma (HCC) could help the development of immunotherapy against HCC. The epidermal growth factor receptor (EGFR) signaling is frequently activated in HCC and plays important roles in tumorigenesis. However, its role in HCC immunity is still largely unknown. This study aimed to investigate the impact of EGFR signaling on programmed death‐ligand 1 (PD‐L1) and human leukocyte antigen class‐I (HLA‐I) expression in HCC cells and its underlying mechanisms.
Methods
The expression of phosphorylated EGFR (p‐EGFR), PD‐L1, and HLA‐I (HLA‐ABC) in HCC specimens was detected by immunohistochemistry, and their correlations were analyzed. PD‐L1 and HLA‐ABC expression in EGFR‐activated HCC cells were detected by quantitative real‐time PCR, Western blotting, and flow cytometry, and T cell‐mediated lysis was performed to test the immunosuppressive effects of PD‐L1 and HLA‐ABC alterations in HCC cells. Furthermore, the underlying mechanisms of EGFR activation‐induced PD‐L1 up‐regulation and HLA‐ABC down‐regulation were explored by animal experiments, luciferase reporter assay, and gene gain‐ and loss‐of‐function studies.
Results
p‐EGFR was positively correlated with PD‐L1 and negatively correlated with HLA‐ABC expression in HCCs. EGFR activation by its ligand EGF up‐regulated PD‐L1 and down‐regulated HLA‐ABC in HCC cells, which was functionally important and could be abolished by the EGFR inhibitor, gefitinib, both in vitro and in vivo. Mechanistically, enhanced P38 mitogen‐activated protein kinase (MAPK) activation down‐regulated microRNA‐675‐5p (miR‐675‐5p) and up‐regulated glycolysis‐related enzyme hexokinase 2 (HK2); miR‐675‐5p down‐regulation enhanced the stability of PD‐L1 mRNA probably via the 3’‐untranslated region (3’‐UTR) of PD‐L1 and thereby caused PD‐L1 accumulation, and HK2 up‐regulation enhanced aerobic glycolysis and mediated a decrease in HLA‐ABC.
Conclusions
The EGFR‐P38 MAPK axis could up‐regulate PD‐L1 through miR‐675‐5p and down‐regulate HLA‐ABC via HK2 in HCC cells. Our study reveals a novel signaling network that may cause immune suppression in HCC and suggests that EGFR signaling can be targeted for HCC immunotherapy.
Keywords: EGFR signaling, P38 MAPK, PD‐L1, HLA‐ABC, miR‐675‐5p, Hexokinase‐2, Hepatocellular carcinoma
Abbreviations
- EGFR
epidermal growth factor receptor
- EGF
epidermal growth factor
- HCC
hepatocellular carcinoma
- HK2
hexokinase‐2
- HLA‐I
human leukocyte antigen class‐I
- PD‐1
programmed cell death protein 1
- PD‐L1
programmed death‐ligand 1
- 2‐DG
2‐Deoxy‐D‐glucose
- 3’‐UTR
3'‐untranslated region
- MAPK
mitogen‐activated protein kinase
- GAPDH
glyceraldehyde ‐3‐phosphate dehydrogenase
- DAPI, 4'
6‐diamidino‐2 ‐phenylindole
- 2‐NBDG
2‐(N‐(7‐Nitrobenz‐2‐oxa‐1, 3‐diazol‐4‐yl) Amino)‐2‐Deoxyglucose
- ERK 1/2
extracellular regulated protein kinases 1/2
- PI3K
phosphatidylinositol 3 kinase
- AKT
namely protein kinase B (PKB)
- STAT
signal‐transducing activator of transcription
- IFNγ
interferon‐gamma; miRNA, micro RNA
1. BACKGROUND
Hepatocellular carcinoma (HCC) is an aggressive malignancy that causes the fourth largest number of cancer deaths worldwide [1]. Curative treatment options are limited and only apply to patients in early stages of the disease [2]. Thus, novel therapeutic options are urgently required for this deadly disease. Recently, immunotherapies using immune checkpoint inhibitors and a combination of immunotherapeutic agents with chemotherapeutic drugs have been shown as a promising strategy against human cancers [3, 4, 5]. As a typical inflammation‐linked tumor, HCC represents a promising target for immune‐based therapeutics [6]. However, the current existing immunotherapies have only limited benefit in HCC patients [7]. This prompts us to explore the unique immune regulation in HCC patients to aid the development of novel or improved immunotherapies.
Epidermal growth factor receptor (EGFR) signaling activated by its ligand epidermal growth factor (EGF) plays important roles in cell proliferation, differentiation, and tumorigenesis [8, 9, 10]. EGFR signaling is frequently activated in HCC, promotes HCC occurrence and progression, and is associated with an aggressive phenotype, intrahepatic metastasis, and poor clinical outcomes [10, 11, 12, 13, 14]. Of note, blocking EGFR could promote anti‐tumor cellular immunity in head and neck cancer patients [15], and EGFR activation could regulate the expression of immune molecules in several types of tumor cells [16, 17, 18, 19], suggesting that EGFR signaling may be involved in the immune regulation of HCC.
Programmed death‐ligand 1 (PD‐L1) and human leukocyte antigen class‐I (HLA‐I) are two important immune regulatory molecules. PD‐L1 is widely expressed on the tumor cell surface and triggers negative co‐stimulatory signals [20]. It significantly inhibits the proliferation and function of T cells by binding with its ligand programmed cell death protein‐1 (PD‐1) on T cells [20]. On the contrary, the main HLA‐I molecules HLA‐A, HLA‐B, and HLA‐C promote immune response by presenting antigenic peptides to cytotoxic T cells [21]. Defective HLA‐I expression helps malignant cells escape from T cell‐specific lysis and attenuates the outcome of T‐cell‐based immunotherapy [22]. Aberrant PD‐L1 expression and HLA‐I deficiency frequently occur in HCC and are associated with impaired tumor immunity, aggressive disease phenotype, and poor prognosis of patients [23, 24, 25, 26, 27.]
In this study, we investigated the involvement of EGFR signaling in the regulation of PD‐L1 and HLA‐I in HCC cells. First, we analyzed the associations amongst EGFR activation, PD‐L1 expression, and HLA‐I (HLA‐ABC) expression in HCC tissues. Next, through in vitro and in vivo studies using HCC cell lines, we further explored the impact of EGFR signaling on the expression of PD‐L1 and HLA‐ABC, as well as the underlying mechanisms.
2. MATERIALS AND METHODS
2.1. Cell lines and cell culture
Human HCC cell lines SMMC‐7721 and HepG2 were obtained from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). The cells were maintained in DMEM culture medium (Gibco BRL, Gaithersburg, MD, USA) supplemented with heat‐inactivated endotoxin‐free 10% fetal bovine serum, 100 μg/mL streptomycin, and 100 units/mL penicillin under a humidified 5% CO2 atmosphere at 37°C.
2.2. Tumor tissues
To examine p‐EGFR, PD‐L1, and HLA‐ABC protein expression in HCC tissues, we obtained 152 specimens of HCC tissues from the Affiliated Cancer Hospital of Guangzhou Medical University (Guangzhou, Guangdong, China) which were collected between January 2014 and June 2018. All the tumor tissues were pathologically diagnosed as HCC according to the World Health Organization (WHO) classification criteria. This study was approved by the Ethics Committee of Guangzhou Medical University, and all methods were carried out in accordance with the approved guidelines for medical researches involving patient‐derived biological resources. HCC tissues were paraffin‐embedded and used to make tissue chips for immunohistochemistry studies.
2.3. Cell transfection
Cells were seeded on a 6‐well plate (2×105 cells/well) and cultured for 12 h. Then, the cells were transfected with 2 μg plasmids, 0.8 μmol microRNA (miRNA) mimics, or 1 μmol small interfering RNAs (siRNAs) mixed with lipofectamine 3000 reagent (#L3000150, Invitrogen, Carlsbad, CA, USA) in a complete medium with 10% fetal bovine serum (FBS) according to the manufacturer's instructions, and then incubated for indicated time before harvest. Mimics of miR‐675‐5p were purchased from GenePharma (Shanghai, China). siRNA against human hexokinase 2 (HK2) (siHK2) and control siRNA (siNC) were purchased from RiboBio (Guangzhou, Guangdong, China). The HLA‐B overexpression plasmid (pcDNA3.1‐HLA‐B) and its control vector (pcDNA3.1) were obtained from IGEbio (Guangzhou, Guangdong, China). pGL3‐Vector (#E1761) and pRL‐TK plasmid (#E2241) were purchased from Promega (Madison, WI, USA). Luciferase reporter containing a 1.3 kb fragment of the PD‐L1 3’‐UTR (pGL3‐PD‐L1 3’‐UTR, #107009) was obtained from Addgene (Cambridge, MA, USA).
2.4. Quantitative real‐time PCR
Total RNA was isolated from cells with RNAiso Plus reagent (#9108, TaKaRa E.Z.N.A® HP, Tokyo, Japan) according to the manufacturer's protocol. Total RNA (500 ng) was reversely transcribed using the PrimeScript RT Master Mix (#RR036A, TaKaRa E.Z.N.A® HP) in a final volume of 10 μL. Quantitative real‐time PCR (qRT‐PCR) was performed using the TB Green® Premix Ex Taq™ II (#RR820A, TaKaRa E.Z.N.A® HP) and analyzed using the ABI 7500 real‐time PCR system. The expression levels of miRNAs were measured using miDETECT A Track™ miRNA qRT‐PCR kits (#C10712‐1, RiboBio) according to the manufacturer's protocol. U6 and glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) were used as internal controls for miRNAs and mRNAs, respectively. Each sample was analyzed in triplicate. Expression levels for each target gene were calculated using the comparative threshold cycle (CT) method. The Δct values were calculated according to the formula Δct = ct (gene of interest) ‐ ct (GAPDH or U6), and the 2–ΔΔct was calculated according to the formula ΔΔct = Δct (control group) ‐ Δct (experimental group) for determination of relative expression. The primer sequences and the details of PCR products (%GC and Tm values) are listed in Supplementary Table S1.
2.5. Western blotting analysis
To analyze the protein levels of PD‐L1, HLA‐ABC, EGFR, p‐EGFR, P38, p‐P38, β‐actin, and HK2, Western blotting was performed, as previously described [28]. Primary antibodies against PD‐L1 (#13684), EGFR (#4267), p‐EGFR (#3777), P38 (#8690), p‐P38 (#4511), β‐actin (#4970), and HK2 (#2867) were obtained from Cell Signaling Technology (Danvers, MA, USA). Primary antibodies against HLA‐ABC (#ab70328) were obtained from Abcam Incorporated (Cambridge, MA, USA). All the above antibodies were used at 1:1000 dilutions.
2.6. Immunofluorescence assay
To detect the expression of p‐P38 mitogen‐activated protein kinase (MAPK) in cancer cells, the cells were seeded on confocal dishes (3×103 cells/well) overnight and treated with 20 ng/mL EGF (#AF‐100‐15, PeproTech, Rocky Hill, NJ, USA) for indicated time periods, then fixed with 4% paraformaldehyde for 20 min, and permeated by 1% Triton‐X100 for 15 min. Subsequently, the cells were blocked with 10% normal goat serum for 30 min at 37°C and incubated with antibodies against p‐P38 MAPK (1:100 dilution) overnight at 4°C. After washing with phosphate buffer solution (PBS), the cells were incubated with FITC‐conjugated secondary antibodies (#A32723; Invitrogen) and counterstained with 4',6‐diamidino‐2‐phenylindole (DAPI) (#D21490, Invitrogen) for 10 min. Representative images were taken with an inverted fluorescence microscope (Leica, Wetzlar, Germany) to examine the protein expression.
2.7. Flow cytometry
Flow cytometry was performed as previously described [29]. Briefly, cell suspensions were washed with PBS and then directly incubated with indicated fluorescence‐labeled antibodies (such as anti‐PD‐L1 or HLA‐ABC antibodies) or isotype controls for 1 h at 4°C. Subsequently, the cells were washed and resuspended with PBS, and then fluorescence data were collected on a flow cytometry machine (BD, San Diego, CA, USA). The data were analyzed using the FlowJo 7.6.1 software (TreeStar, Ashland, OR, USA). PE‐conjugated PD‐L1 antibody (#393608) and PE‐Cy5.5‐conjugated HLA‐ABC antibody (#311408) were purchased from Biolegend (San Diego, CA, USA).
2.8. Immunohistochemical examination
Immunohistochemical staining was performed as previously described [29]. Then, the stained sections were observed and images were captured by two pathologists under a light microscope at a magnification ×40 or ×400, and were blinded to the patients’ clinical information. The staining intensity was assessed using a modified quickscore method [30] on a scale of 0‐3 as negative (0), weak (1), moderate (2), or strong staining (3). The extent of staining, defined as the percentage of positively stained areas of cancer cells per the whole tumor area, was scored on a scale of 0 (0%), 1 (1%‐25%), 2 (26%‐50%), 3 (51%‐75%), and 4 (76%‐100%). An overall protein expression score (range, 0‐12) was calculated by multiplying the intensity and positivity scores according to the previous study [30].
2.9. Dual‐luciferase reporter assay
Cells cultured on 96‐well plates were transiently co‐transfected with firefly luciferase reporter plasmids (0.2 μg) and ranilla luciferase co‐reporter plasmids (pRL‐TK, 0.02 μg). Then, these cells were simultaneously or subsequently treated with EGF (20 ng/mL), P38 MAPK inhibitor SB203580 (10 μmol/L), or miR‐675‐5p mimics (20 μmol/L, add 0.4 μL into 100 μL medium per well) for the indicated time periods. Next, the cellular luciferase activity was measured using Dual‐luciferase® Reporter Assay System kit (#E1910, Promega) according to the manufacturer's instructions. Results were calculated as the ratios between the activity of firefly luciferase and ranilla luciferase. The recombinant human EGF (#AF‐100‐15) was purchased from PeproTech (Rocky Hill, NJ, USA), and P38 MAPK inhibitor SB203580 (#HY‐10256) was purchased from MedChem Express (Monmouth Junction, NJ, USA).
2.10. Establishment of SMMC‐7721pLV‐EGF cells with stable EGF overexpression
SMMC‐7721 cells were transfected using lentiviruses with EGF expression vector pLV‐EGF or the control vector pLV‐Null in the presence of 5 mg/mL of polybrene, then the transfected SMMC‐7721 cells were selected with puromycin (1 μg/mL) for 10‐14 days. The survived cells were then digested with trypsin and reseeded separately into 96‐well plates (almost a single cell per well) using a flow sorter (BD) for cell clone formation and expansion. The expanded monoclonal cell populations (SMMC‐7721pLV‐EGF and SMMC‐7721pLV‐Null) were collected and cryopreserved for further studies. Lentivirus vectors pLV‐EGF and pLV‐Null were purchased from VectorBuilder Inc. (Conroe, TA, USA).
2.11. Animal studies
Six‐week‐old female BALB/C nude mice were obtained from the Animal Experimental Center of Guangzhou Medical University (Guangzhou, China). The procedures for the handling and care of the mice were approved by the Animal Experimentation Ethics Committee of Guangzhou Medical University. Approximately 1 × 107 SMMC‐7721pLV‐EGF or SMMC‐7721pLV‐Null cells in matrigel were injected into the right flanks of nude mice to form xenograft tumors (at this stage, 7 mice per group and 28 mice in total were used). When the tumor volume reached ∼100 mm3, mice received either gefitinib (100 mg/kg) or 0.5% polysorbate vehicle, once daily by oral administration (0.1 mL per 10 g body weight) for one week. At the end of the whole treatment, tumors (5 mice per group) were collected, lysed, or digested for PD‐L1 and HLA‐ABC detection or CD8+ T cell‐mediated lysis assay.
2.12. Activation and isolation of CD8+ cytotoxic T cells
Cytotoxic T cells reactive to SMMC‐7721 cells were established as previously described [31]. Peripheral blood mononuclear cells (PBMCs) were collected from a healthy volunteer and co‐cultured with irradiated (40 Gy) SMMC‐7721 cells (as stimulators) at a ratio of 20:1 in RPMI‐1640 with 10% FBS and human recombinant interleukin‐2 (IL‐2) for 1 week. During this period, the IL‐2‐containing medium was replaced every 3 days. PBMCs were restimulated with irradiated SMMC‐7721 cells every10 days.
After pre‐stimulation with IL‐2, PBMCs were washed twice with PBS and resuspended in PBS prior to negative isolation of CD8+ T cells using CD8+ T cell isolation kits (#130‐096‐495, Miltenyi Biotech., Bergisch Gladbach, Germany) according to the manufacturer's instructions. Briefly, an antibody mix targeting the non‐CD8+ cells was added to the PBMC suspension and was allowed to bind to the cells. Dynabeads® were added and bound to the antibody‐labeled cells for 30 min on ice. The bead‐bound cells were quickly separated on a magnet and discarded. The remaining negatively isolated and untouched human CD8+ T cells were collected. The isolated CD8+ T cells were subsequently used as effector cells.
2.13. CD8+ T cell‐mediated lysis
Isolated human CD8+ T cells were used for cytotoxic T cell‐mediated lysis by co‐incubation with target tumor cells (SMMC‐7721 cells) at a ratio of 10:1 in 96‐well plates. The plates were centrifuged at 200 × g for 5 min and incubated at 37◦C in 5% CO2. After 6‐hour incubation, cytotoxicity was detected using the CytoTox 96® Non‐Radioactive Cytotoxicity Assay kit (#G1780, Promega) according to the manufacturer's instructions. The spontaneous release and maximum release were determined by incubating target cells without effector cells in medium alone or 0.5% NP‐40, respectively. The percent cytotoxicity was calculated as follows: (experimental release − spontaneous release) / (maximum release – spontaneous release) × 100%.
2.14. Glucose uptake assay
The glucose uptake assay was performed as previously described [32]. Cells were plated at 1 × 106/well in 6‐well plates and treated with 20 ng/mL EGF, 10 μmol/L SB203580, 10 μmol/L glycolysis inhibitor 2‐deoxyglucose (2‐DG) (#HY‐13966, MedChem Express, Monmouth Junction, NJ, USA) alone or in combinations for 24 h. Then, the cells were refreshed with serum‐starved (0.1% FBS) and glucose‐free DMEM, while continued with different treatments for 12 h. Next, cells were grown in the presence of 50 mmol/L 2‐(N‐(7‐Nitrobenz‐2‐oxa‐1,3‐diazol‐4‐yl)Amino)‐2‐Deoxyglucose (2‐NBDG) (#N13195, Thermo Fisher Scientific, Waltham, MA, USA) for 30 min, and then the cells were collected and the mean fluorescence intensity (MFI) indicating the cellular glucose uptake was measured on a flow cytometer. The glucose uptake in the experimental group was calculated by normalizing its MFI to the MFI in the control group.
2.15. Lactate production assay
In total, 1 × 106 cells were plated in 6‐well plates and treated with 20 ng/mL EGF, 10 μmol/L SB203580, 10 μmol/L 2‐DG alone or in combinations for 24 h, and the cell culture supernatants and cells were collected separately. Next, the lactate concentration in the culture supernatant was determined using the Lactate detection kit (#A019‐2‐1, Nanjing Jiancheng Bioengineering, Nanjing, Jiangsu, China) according to the manufacturer's protocol. Harvested cells were stained with trypan blue, and viable cells were counted using an automated cell counter (Bio‐Rad, Hercules, CA, USA). Then, the adjusted lactate concentration was calculated according to the following formula: adjusted lactate concentration = lactate concentration in experimental group / the ratio of cell number in the experimental group to that in the control group.
2.16. Statistical analysis
The data of continuous variables are expressed as mean ± standard deviation (SD) of three independent experiments, unless otherwise specified. Student's t‐test and one‐way analysis of variance (ANOVA) were performed to compare the differences between groups. The correlation between the expression levels of different proteins in tumor tissues was analyzed by Pearson's correlation coefficient. Statistical analyses were performed using the GraphPad Prism Software Version 5.0 (GraphPad Software Inc., La Jolla, CA, USA). P < 0.05 was considered statistically significant.
3. RESULTS
3.1. Correlations between EGFR activation and the expression of PD‐L1 and HLA‐ABC in HCC tissues
The clinicopathologic characteristics of the 152 patients, with a median age of 49 years (range, 18‐98 years), are summarized in Supplementary Table S2. To analyze the correlations between EGFR activation and the expression of PD‐L1 and HLA‐ABC in HCC tissues, we detected the expression of p‐EGFR, PD‐L1, and HLA‐ABC in 152 clinical HCC specimens by immunohistochemistry. The HCC specimens with high p‐EGFR expression had higher rates of high PD‐L1 expression and low HLA‐ABC expression than those with low p‐EGFR expression (Figure 1A). In addition, the expression of p‐EGFR was positively correlated with PD‐L1 expression and negatively correlated with HLA‐ABC expression (Figure 1B).
FIGURE 1.
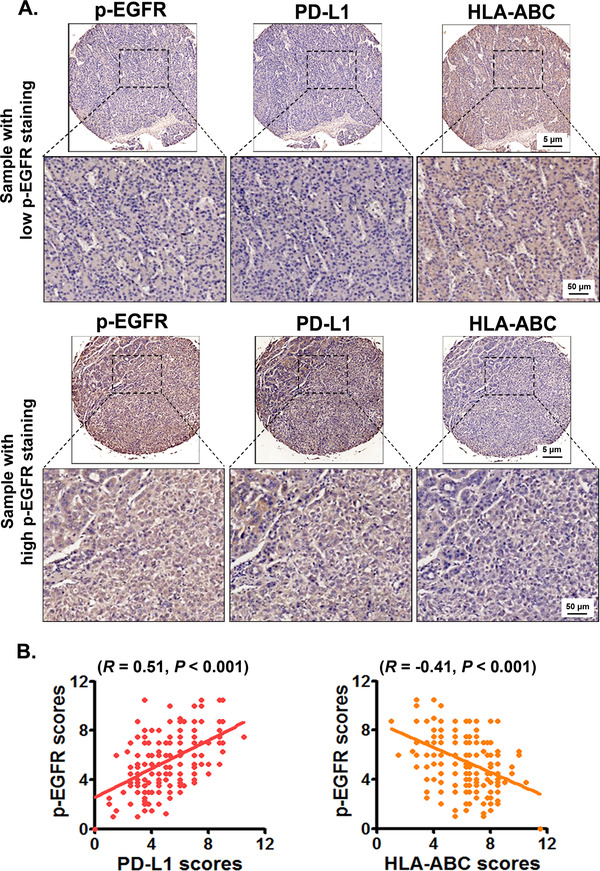
EGFR activation is positively correlated with PD‐L1 expression while negatively correlated with HLA‐ABC expression in HCC tissues. The expression of p‐EGFR, PD‐L1, and HLA‐ABC in HCC tissues was detected using immunohistochemistry. The average scores of p‐EGFR, PD‐L1, and HLA‐ABC expression in HCCs were analyzed by using a modified quickscore assessment method. (A) The representative images of PD‐L1 and HLA‐ABC expression in HCC samples with low and high p‐EGFR expression. (B) The correlation among p‐EGFR, PD‐L1, and HLA‐ABC expression in HCCs were analyzed by using Pearson's correlation coefficient. Abbreviations: EGFR, epidermal growth factor receptor; PD‐L1, programmed death‐ligand 1; HLA‐ABC, human leukocyte antigen class‐A, B, C; HCC, hepatocellular carcinoma.
3.2. Impact of EGFR activation on the expression of PD‐L1 and HLA‐ABC in HCC cells
Subsequently, we investigated the impact of EGFR signaling on the expression of PD‐L1 and HLA‐ABC in HCC cells. EGFR signaling activated by EGF significantly increased PD‐L1 expression and decreased HLA‐ABC expression in both SMMC‐7721 and HepG2 cells (Figure 2A‐C). EGFR activation promoted PD‐L1 expression and decreased HLA‐ABC expression in a time‐dependent manner, but not in a dose‐dependent manner (Figure 2D and Supplementary Figure S1).
FIGURE 2.
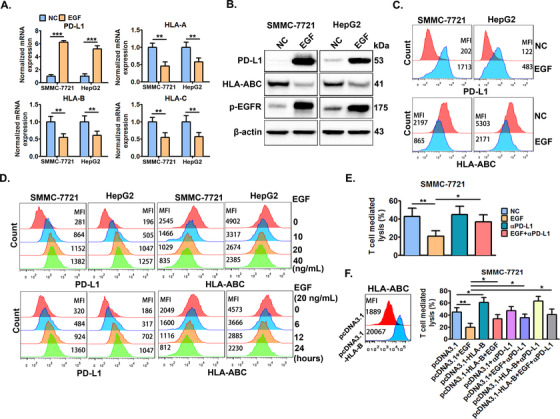
EGFR activation by EGF significantly induces PD‐L1 up‐regulation and HLA‐ABC down‐regulation. SMMC‐7721 and HepG2 cells were treated with or without EGF (20 ng/mL) for 24 h, then, the cells were collected to detect the expression of PD‐L1 and HLA‐ABC by (A) qRT‐PCR, (B) Western blotting, and (C) flow cytometry. (D) The cells were treated with the indicated doses of EGF (0‐40 ng/mL) for 24 h or treated with EGF (20 ng/mL) for the indicated time periods (0‐48 h), and then cellular PD‐L1 and HLA‐ABC expression was measured by flow cytometry. (E) SMMC‐7721 cells were pre‐treated with or without EGF (20 ng/mL) for 24 h, and then, the cells were collected and co‐incubated with CD8+ T cells. Next, the CD8+ T cell‐mediated lysis of tumor cells was detected 6 h later. (F) SMMC‐7721 cells were pre‐transfected with HLA‐B expression plasmids (pcDNA3.1‐HLA‐B) or control vector (pcDNA3.1) for 24 h, and a set of cells were collected for the detection of HLA‐ABC by flow cytometry. Another set of cells were further co‐stimulated with or without EGF (20 ng/mL) for another 24 h, and the cells were collected and co‐incubated with CD8+ T cells with or without the addition of anti‐PD‐L1 antibodies (αPD‐L1). Next, the CD8+ T cell‐mediated lysis of tumor cells was detected 6 h later. * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001. Abbreviations: EGFR, epidermal growth factor receptor; EGF, epidermal growth factor; PD‐L1, programmed death‐ligand 1; HLA‐ABC, human leukocyte antigen class‐A, B, C.
The in vitro CD8+ T cell‐mediated lysis assay demonstrated that EGFR activation by EGF significantly decreased CD8+ T cell‐mediated lysis of SMMC‐7721 cells, which could be significantly abolished by blocking cell surface PD‐L1 with PD‐L1 antibodies (Figure 2E). In addition, HLA‐B overexpression significantly reversed the EGFR activation‐caused decrease in the CD8+ T cell‐mediated lysis of SMMC‐7721, and HLA‐B overexpression combined with PD‐L1 blocking could maximize this effect (Figure 2F).
3.3. Impact of EGFR inhibitor gefitinib on EGFR activation‐induced PD‐L1 up‐regulation and HLA‐ABC down‐regulation in vitro and in vivo
We next studied if the clinically available EGFR inhibitor gefitinib could abolish the EGFR signaling‐induced PD‐L1 increase and HLA‐ABC decrease in HCC cells. It was found that gefitinib almost totally abrogated EGFR activation‐induced PD‐L1 up‐regulation and HLA‐ABC down‐regulation, but failed to significantly alter the basal expression of PD‐L1 and HLA‐B in both SMMC‐7721 and HepG2 cells (Figure 3A‐C).
FIGURE 3.
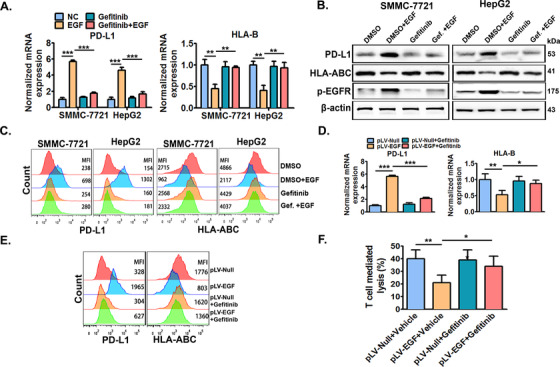
EGFR inhibitor gefitinib significantly abolishes EGFR activation‐induced PD‐L1 up‐regulation and HLA‐ABC down‐regulation in vitro and in vivo. SMMC‐7721 and HepG2 cells were pre‐treated with or without EGFR inhibitor gefitinib (10 μmol/L) for 6 h, and further co‐stimulated with or without EGF (20 ng/mL) for an additional 24 h. Then, cellular PD‐L1 and HLA‐ABC expression was measured by (A) qRT‐PCR, (B) Western blotting, and (C) flow cytometry. SMMC‐7721 cells with stable EGF expression (SMMC‐7721pLV‐EGF) and their control cells (SMMC‐7721pLV‐null) were previously established by infecting SMMC‐7721 cells using lentivirus with EGF expression vectors or empty vectors. Next, SMMC‐7721pLV‐EGF and SMMC‐7721pLV‐null cells were injected into the right flanks of nude mice to form xenograft tumors. When the tumor volume reached ∼100 mm3, tumor‐bearing mice were then treated with gefitinib (100 mg/kg) or 0.5% polysorbate vehicle once daily by oral administration (0.1 mL per 10 g body weight) for one week. Tumors were then collected, lysed, or digested for the detection of PD‐L1 and HLA‐ABC by (D) qRT‐PCR and (E) flow cytometry. SMMC‐7721pLV‐EGF and SMMC‐7721pLV‐null cells isolated from tumors treated with gefitinib or its vehicle were then co‐incubated with CD8+ T cells, and the specific lysis of tumor cells was detected 6 h later (F). * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001. Abbreviations: EGFR, epidermal growth factor receptor; EGF, epidermal growth factor; PD‐L1, programmed death‐ligand 1; HLA‐ABC, human leukocyte antigen class‐A, B, C.
Furthermore, the previously established SMMC‐7721pLV‐EGF cells with stable high EGF expression and the control SMMC‐7721pLV‐null cells (supplementary Figure S2) were used to test if gefitinib could abolish EGFR signaling‐induced PD‐L1 increase and HLA‐ABC decrease in mice bearing SMMC‐7721 tumors. It was demonstrated that PD‐L1 expression was significantly increased while HLA‐ABC expression was significantly inhibited in SMMC‐7721pLV‐EGF cell‐formed tumors compared with those in SMMC‐7721pLV‐Null cell‐formed tumors (Figure 3D and 3E). In addition, gefitinib treatment significantly abolished the PD‐L1 increase and HLA‐ABC decrease in SMMC‐7721pLV‐EGF cells (Figure 3D and 3E). Furthermore, tumor isolated SMMC‐7721pLV‐EGF cells had significantly lower CD8+ T cell‐mediated lysis rate compared with tumor isolated SMMC‐7721pLV‐Null cells in vitro, and gefitinib treatment significantly reversed the decrease in lysis rate of SMMC‐7721pLV‐EGF cells (Figure 3F).
3.4. The role of P38 MAPK in EGFR activation‐induced PD‐L1 up‐regulation and HLA‐ABC down‐regulation
Studies have revealed that EGFR activation mediates PD‐L1 expression mainly via the extracellular regulated protein kinases 1/2 (ERK 1/2)/c‐Jun, phosphatidylinositol 3 kinase (PI3K)/AKT (namely protein kinase B, PKB), or signal‐transducing activator of transcription (STAT) pathways in different cancer cell types, including NSCLC cells[33, 34] and esophageal squamous cell carcinoma cells [35]. In preliminary studies, we found that EGFR activation promoted c‐Jun signaling while inhibiting both PI3K/AKT and STAT signaling, and inhibition of these signaling pathways by their chemical inhibitors failed to abrogate the EGFR activation‐increased PD‐L1 transcription or ‐decreased HLA‐B transcription (Supplementary Figure S3A). Of note, EGFR activation significantly enhanced P38 MAPK signaling by increasing the phosphorylation of P38 MAPK, and inhibition of P38 MAPK signaling by its inhibitor SB203580 significantly abolished EGFR activation‐increased PD‐L1 transcription and ‐decreased HLA‐B transcription, suggesting a crucial regulatory role of P38 MAPK in EGFR activation‐induced PD‐L1 up‐regulation and HLA‐I down‐regulation (Supplementary Figure S3B). Immunofluorescence staining confirmed the increased phosphorylation of P38 MAPK in SMMC‐7721 cells after EGFR activation (Figure 4A). In addition, EGFR activation increased the phosphorylation of P38 MAPK in a time‐dependent manner within a short time (Figure 4B and Supplementary Figure S4), and gefitinib treatment almost totally abolished EGFR activation‐induced phosphorylation of P38 MAPK (Figure 4C). Next, it was confirmed that the inhibition of P38 MAPK activation by SB203580 almost totally reversed EGFR activation‐induced PD‐L1 up‐regulation and HLA‐ABC down‐regulation in both SMMC‐7721 and HepG2 cells (Figure 4D‐F).
FIGURE 4.
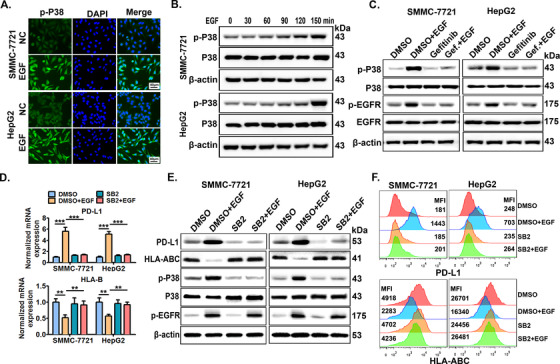
P38 MAPK mediates EGFR activation‐induced PD‐L1 up‐regulation and HLA‐ABC down‐regulation. (A) SMMC‐7721 cells were stimulated with EGF (20 ng/mL) for 12 h, then cells were collected and p‐P38 expression was detected by immunofluorescence. Scale bars: 40 μm. (B) SMMC‐7721 and HepG2 cells were stimulated with EGF (20 ng/mL) for indicated time periods (0‐150 min), then, the cells were collected to detect total P38 and phosphorylated P38 (p‐P38) expression by Western blotting. (C) Cells pre‐treated with EGFR inhibitor gefitinib for 6 h were further co‐stimulated with EGF (20 ng/mL) for an additional 4 h, then, the cells were collected to detect p‐P38, P38, p‐EGFR, and EGFR expression by Western blotting. Cells pre‐treated with P38 inhibitor SB203580 (SB2, 10 μmol/L) for 6 h were further stimulated with EGF (20 ng/mL) for 24 h, then PD‐L1 and HLA‐B transcription was detected by (D) qRT‐PCR, and PD‐L1 and HLA‐ABC protein levels were measured by (E) Western blotting and (F) flow cytometry. * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001. Abbreviations: EGFR, epidermal growth factor receptor; EGF, epidermal growth factor; PD‐L1, programmed death‐ligand 1; HLA‐ABC, human leukocyte antigen class‐A, B, C; MAPK, mitogen‐activated protein kinase.
3.5. The role of miR‐675‐5p‐enhanced PD‐L1 mRNA stability in the EGFR‐P38 MAPK axis‐induced PD‐L1 accumulation
PD‐L1 overexpression in multiple cancer cell types involves different regulatory mechanisms [36]. We demonstrated that PD‐L1 mRNA expression was increased in a time‐dependent manner after EGFR activation (Supplementary Figure S5). However, dual‐luciferase reporter assay demonstrated that EGFR activation failed to significantly alter the activity of the potential PD‐L1 promoter (Supplementary Figure S6). These findings suggested the involvement of a post‐transcriptional rather than a transcriptional regulatory mechanism. PD‐L1 expression can be enhanced by the increased stability of PD‐L1 mRNA [37]. Herein, we found that EGFR activation enhanced the stability of PD‐L1 mRNA in both SMMC‐7721 and HepG2 cells (Figure 5A), which could be significantly abrogated by the P38 MAPK inhibitor SB203580 (Figure 5B). These results demonstrated that the enhanced PD‐L1 mRNA stability mediated the EGFR‐P38 MAPK axis‐induced PD‐L1 up‐regulation in HCC cells.
FIGURE 5.
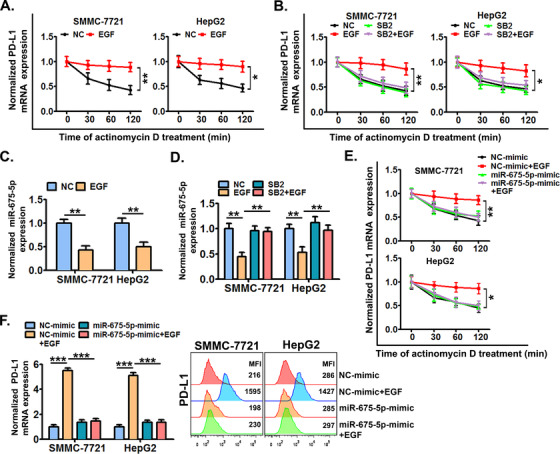
miR‐675‐5p‐enhanced PD‐L1 mRNA stability is crucial for the EGFR‐P38 MAPK axis‐induced PD‐L1 accumulation. (A) SMMC‐7721 and HepG2 cells were pre‐stimulated with or without EGF for 24 h and further treated with actinomycin D (5 μg/mL) for 0‐120 min, next cellular PD‐L1 mRNA expression was detected by qRT‐PCR. (B) Cells were pre‐treated with SB203580 (10 μmol/L) or dimethylsulfoxide (DMSO) for 6 h, and then co‐stimulated with or without EGF for an additional 24 h, next cells were further treated with actinomycin D (5 μg/mL) for 0‐120 min, and cellular PD‐L1 mRNA expression was detected by qRT‐PCR. (C) Cells were stimulated with or without EGF (20 ng/mL) for 24 h, and then the expression of miR‐675‐5p was detected by qRT‐PCR. (D) Cells pre‐treated with SB203580 (10 μmol/L) or DMSO for 6 h were further co‐treated with or without EGF (20 ng/mL) for an additional 24 h, and then, the expression of miR‐675‐5p was detected by qRT‐PCR. (E) Cells were pre‐treated with mimics of miR‐675‐5p or control mimics for 24 h, and then co‐stimulated with or without EGF for an additional 24 h. Next, the cells were treated with actinomycin D (5 μg/mL) for additional indicated time periods (0‐120 min), and the cellular PD‐L1 mRNA expression was detected by qRT‐PCR. (F) Cells were pre‐transfected with mimics of miR‐675‐5p or control mimics for 24 h, and then further co‐stimulated with or without EGF (20 ng/mL) for an additional 24 h.Next, the expression of PD‐L1 was detected by qRT‐PCR and flow cytometry. * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001. Abbreviations: EGFR, epidermal growth factor receptor; EGF, epidermal growth factor; PD‐L1, programmed death‐ligand 1; HLA‐ABC, human leukocyte antigen class‐A, B, C; MAPK, mitogen‐activated protein kinase.
miRNAs are a class of non‐coding RNAs with 19 to 25 nucleotides, which function as post‐transcriptional regulators in a variety of cellular processes associated with HCC progression [38]. Mechanistically, miRNAs can regulate the stability of target mRNAs by binding with them. In addition, miRNAs have been implicated in the regulation of PD‐L1 expression in cancer cells [39]. Thus we wondered if miRNAs were involved in the EGFR‐P38 MAPK axis‐induced PD‐L1 expression. RNA‐seq analysis showed that 84 miRNAs were down‐regulated in SMMC‐7721 cells after EGFR activation by EGF (supplementary Figure S7). Subsequently, the top 20 significantly down‐regulated miRNAs were chosen, and their potential roles were tested. By studying HCC cells transfected with mimics of these miRNAs, we found that miR‐675‐5p might be crucial for EGFR activation‐induced PD‐L1 expression in HCC cells (data not shown). qRT‐PCR confirmed that EGFR activation significantly inhibited the expression of miR‐675‐5p in both SMMC‐7721 and HepG2 cells (Figure 5C). In addition, EGFR activation‐inhibited miR‐675‐5p expression was significantly reversed by the inhibition of P38 MAPK by SB203580 (Figure 5D). Next, mimics of miR‐675‐5p almost totally reversed the EGFR‐P38 MAPK axis‐increased stability of PD‐L1 mRNA (Figure 5E) and PD‐L1 accumulation (Figure 5F).
Disruption of PD‐L1 3’‐UTR increases the stability of PD‐L1 mRNA in multiple cancers [40]. In addition, miR‐675‐5p regulates mRNA degradation of target genes by binding to their 3’‐UTR [41]. Thus, we next analyzed the role of PD‐L1 3’‐UTR in the miR‐675‐5p‐mediated increase in the stability of PD‐L1 mRNA induced by EGFR signaling. Using a luciferase reporter containing a fragment of 3’‐UTR of human PD‐L1 gene, we demonstrated that firefly luciferase activity in cells transfected with pGL3‐PD‐L1 3’‐UTR was significantly decreased compared with that in cells transfected with control vector pGL3‐Vector (Figure 6A). In addition, EGFR activation significantly increased the firefly luciferase activity in cells transfected with pGL3‐PD‐L1 3’‐UTR, but failed in cells transfected with pGL3‐Vector (Figure 6A). Next, we found that the inhibition of P38 MAPK by SB203580 and mimics of miR‐675‐5p both significantly abrogated EGFR activation‐increased firefly luciferase activity in cells transfected with pGL3‐PD‐L1 3’‐UTR (Figure 6B and 6C). These results collectively suggested that PD‐L1 3’‐UTR was probably required for the miR‐675‐5p‐mediated increase in the stability of PD‐L1 mRNA induced by the EGFR‐P38 MAPK axis.
FIGURE 6.
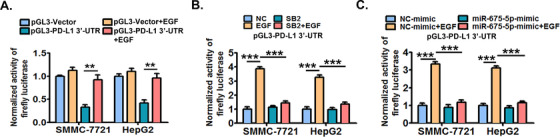
PD‐L1 3’‐UTR is probably required for the EGFR‐P38 MAPK‐miR‐675‐5p axis‐enhanced stability of PD‐L1 mRNA. (A) SMMC‐7721 and HepG2 cells were transfected with firefly luciferase reporter constructs containing the PD‐L1 3’‐UTR fragment (pGL3‐PD‐L1 3’‐UTR) or control vectors (pGL3‐Vector) for 24 h, and then co‐stimulated with or without EGF (20 ng/mL) for another 24 h. Next, the cellular luciferase activity was determined by dual‐luciferase reporter assay system. (B) Cells pre‐transfected with pGL3‐PD‐L1 3’‐UTR were further treated with SB203580 (10 μmol/L) or DMSO for 6 h, and then co‐stimulated with or without EGF (20 ng/mL) for an additional 24 h. Next, the cellular luciferase activity was determined by dual‐luciferase reporter assay system. (C) Cells pre‐transfected with pGL3‐PD‐L1 3’‐UTR were further treated with mimics of miR‐675‐5p or control mimics for 24 h, and then co‐stimulated with or without EGF (20 ng/mL) for an additional 24 h. Next, the cellular luciferase activity was determined by dual‐luciferase reporter assay system. * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001. Abbreviations: EGFR, epidermal growth factor receptor; EGF, epidermal growth factor; PD‐L1, programmed death‐ligand 1; HLA‐ABC, human leukocyte antigen class‐A, B, C; MAPK, mitogen‐activated protein kinase; 3’‐UTR, 3'‐untranslated region; DMSO, dimethylsulfoxide; miRNA, micro RNA.
3.6. The role of HK2‐enhanced aerobic glycolysis in the EGFR‐P38 MAPK axis‐induced HLA‐ABC down‐regulation
Enhanced aerobic glycolysis has been proposed to regulate tumor immunity and is characterized by enhanced glucose uptake and lactate production [42]. In addition, EGFR signaling is described as being able to induce aerobic glycolysis in cancer cells [43]. Thus, we examined if aerobic glycolysis was involved in EGFR signaling‐induced HLA‐ABC decrease in HCC cells. We demonstrated that EGFR activation significantly increased the cellular glucose uptake and lactate production in cell culture supernatants (Figure 7A), indicating that EGFR signaling could promote the aerobic glycolysis of HCC cells. Moreover, inhibition of P38 MAPK by SB203580 markedly abolished EGFR activation‐increased cellular glucose uptake and lactate production (Figure 7A). Next, it was confirmed that the glycolysis inhibitor 2‐DG significantly decreased both the basal and EGFR activation‐increased glucose uptake and lactate production (Figure 7B) and significantly increased both the basal and EGFR activation‐decreased HLA‐ABC expression in cancer cells (Figure 7C and 7D).
FIGURE 7.
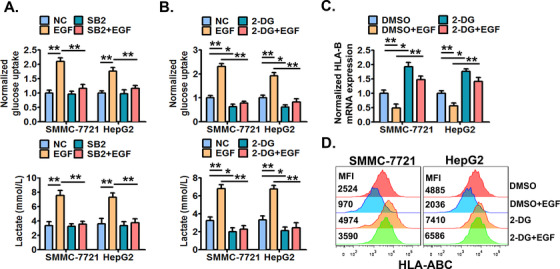
The EGFR‐P38 MAPK axis enhances the aerobic glycolysis which mediates HLA‐ABC down‐regulation in HCC cells. (A) SMMC‐7721 and HepG2 cells were pre‐treated with SB203580 (10 μmol/L) or DMSO for 6 h, and then stimulated with or without EGF (20 ng/mL) for an additional 24 h. Next, the cellular glucose uptake and lactate production were measured. Cells were pre‐treated with 2‐DG (10 μmol/L) or DMSO for 6 h, and then stimulated with or without EGF (20 ng/mL) for an additional 24 h. Next, (B) the cellular glucose uptake and lactate production were detected, (C) the cellular HLA‐B mRNA expression was detected by qRT‐PCR, and (D) the cell surface HLA‐ABC protein expression was detected by flow cytometry. * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001. Abbreviations: EGFR, epidermal growth factor receptor; EGF, epidermal growth factor; HLA‐ABC, human leukocyte antigen class‐A, B, C; MAPK, mitogen‐activated protein kinase; DMSO, dimethylsulfoxide; 2‐DG, 2‐Deoxy‐D‐glucose.
HK2 is a rate‐limiting enzyme that catalyzes the phosphorylation of hexose in the first step of glycolysis [44]. In addition, RNA‐seq analysis preliminarily showed that HK2 was up‐regulated in SMMC‐7721 cells with EGFR activation (Supplementary Figure S8). Thus, we wondered if HK2 was involved in the EGFR‐P38 MAPK axis‐enhanced aerobic glycolysis. It was demonstrated that EGFR activation significantly increased the expression of HK2 (Figure 8A). In addition, inhibition of P38 MAPK by SB203580 largely abrogated the EGFR activation‐increased HK2 expression (Figure 8B). Next, we demonstrated that EGFR activation‐increased glucose uptake and lactate production were significantly reversed when HK2 was successfully knocked down (Figure 8C and Supplementary Figure S9). Moreover, we showed that HK2 knockdown reversed EGFR signaling‐decreased HLA‐ABC expression (Figure 8D and 8E). The above results collectively demonstrated that HK2 enhanced aerobic glycolysis which was crucial for the EGFR‐P38 MAPK axis‐induced HLA‐ABC down‐regulation in HCC cells.
FIGURE 8.
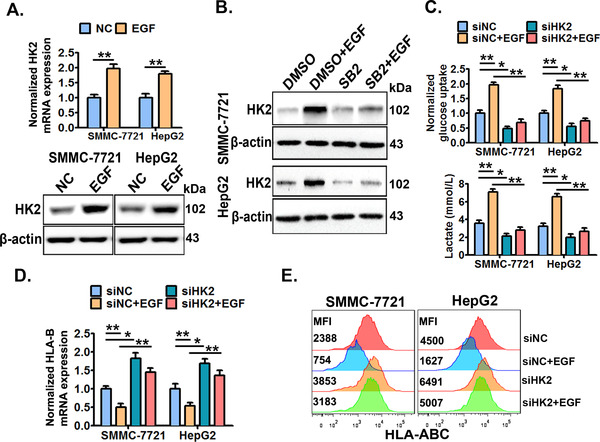
HK2 mediates the EGFR‐P38 MAPK axis‐enhanced aerobic glycolysis and HLA‐ABC down‐regulation. (A) SMMC‐7721 and HepG2 cells were treated with or without EGF (20 ng/mL) for 24 h, and then the cellular HK2 expression was detected by qRT‐PCR and Western blotting. (B) Cells were pre‐treated with SB203580 (10 μmol/L) or DMSO for 6 h, and then stimulated with or without EGF (20 ng/mL) for an additional 24 h, next the cellular HK2 expression was detected by Western blotting. Cells were pre‐transfected with HK2‐specific siRNAs (siHK2) or control siRNAs (siNC) for 24 h, and then treated with or without EGF (20 ng/mL) for an additional 24 h. Next, the cellular glucose uptake and lactate production were detected (C), the cellular HLA‐B mRNA expression was detected by qRT‐PCR (D), and the cell surface HLA‐ABC protein expression was detected by flow cytometry (E). * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001. Abbreviations: EGFR, epidermal growth factor receptor; EGF, epidermal growth factor; HLA‐ABC, human leukocyte antigen class‐A, B, C; MAPK, mitogen‐activated protein kinase; DMSO, dimethylsulfoxide; HK2, hexokinase‐2.
4. DISCUSSION
PD‐L1 and HLA‐I are important immune regulatory molecules. PD‐L1 inhibits the function of T cells, while HLA‐I deficiency causes failure in the presentation of tumor antigens, both impairing T cell‐mediated lysis of cancer cells [20, 22, 27]. Recently, EGFR signaling has been suggested to regulate PD‐L1 or HLA‐I expression in some cancer cell types [17, 18, 19]. Although a recent study suggested that MAPK might regulate PD‐L1 expression in liver cancer [45], the role of EGFR signaling in the regulation of PD‐L1 and HLA‐I in HCC still remains to be defined. In the present study, we illustrated that EGFR activation was positively correlated with PD‐L1 expression while negatively correlated with HLA‐ABC expression in HCC tissues. More importantly, EGFR activation by its ligand EGF significantly induced PD‐L1 up‐regulation and HLA‐ABC down‐regulation in HCC cells both in vitro and in vivo. Furthermore, we confirmed that EGFR activation‐induced PD‐L1 increase and HLA‐ABC decrease were functionally important in impairing the T cell‐mediated lysis of HCC cells, suggesting the involvement of EGFR signaling in the immune regulation of HCC by regulating both PD‐L1 and HLA‐ABC molecules. Furthermore, in the present study, the clinically available EGFR inhibitor gefitinib significantly abolished the PD‐L1 increase and HLA‐ABC decrease in vitro and in vivo, as well as the inhibition of T cell‐mediated lysis of HCC cells caused by EGFR activation. Clinically, anti‐PD‐1/PD‐L1 antibodies are mostly curative in treating tumors with high PD‐L1 expression, and interferon‐gamma (IFNγ) is used to stimulate HLA‐ABC expression in tumor cells [20, 26]. Thus, it is suggested that treatments with gefitinib, anti‐PD‐1/PD‐L1 antibodies, and/or IFNγ could be used to relieve the immunosuppression in HCC patients with aberrant EGFR activation.
PD‐L1 overexpression in multiple cancer types involves different regulatory mechanisms at the genetic, epigenetic, transcriptional, translational, post‐translational, or structural level [36]. In the present study, it appeared that the common modes of transcriptional regulation were not involved in EGFR signaling‐induced PD‐L1 expression since the potential PD‐L1 promoter activity was not significantly altered after EGFR activation. Instead, EGFR signaling endowed PD‐L1 mRNA with a longer half‐life and enhanced its stability, indicating the involvement of an epigenetic regulatory mechanism. miR‐675‐5p is a miRNA derived from its reservoir long non‐coding RNA H19 [46], and its expression levels are varied in different cancers [47, 48]. miR‐675‐5p plays different roles in cell proliferation, cell invasion, and metastasis in cancers as they respond to various stimuli [37, 42]. However, whether miR‐675‐5p is involved in immune regulation is still unknown. In this study, EGFR activation inhibited the expression of miR‐675‐5p, and mimics of miR‐675‐5p significantly abolished the increased stability of PD‐L1 mRNA and PD‐L1 accumulation induced by EGFR activation. This provides the evidence indicating the involvement of miR‐675‐5p in the regulation of immune molecules.
The 3’‐UTR plays an important role in the stability of mRNA [49, 50]. 3’‐UTR regulates the stability of mRNA mainly through binding with miRNAs on its miRNA‐binding sites to influence mRNA decay [50]. Disruption of the PD‐L1 3’‐UTR is able to increase the stability of PD‐L1 mRNA in multiple cancers [40]. In the current study, PD‐L1 3’‐UTR negatively regulated the expression of upstream linked firefly luciferase. More importantly, PD‐L1 3’‐UTR was required for EGFR activation‐induced expression of its upstream linked firefly luciferase. In addition, mimics of miR‐675‐5p significantly abolished EGFR activation‐induced expression of PD‐L1 3’‐UTR upstream linked firefly luciferase. Thus, we speculated that PD‐L1 3’‐UTR is probably required for the involvement of miR‐675‐5p in EGFR activation‐enhanced stability of PD‐L1 mRNA, although more direct evidence is still needed.
Aerobic glycolysis is the process of oxidation of glucose into pyruvate followed by lactate production under normoxic conditions. It frequently occurs in cancers and is known as the “Warburg effect”, and is characterized by increased glucose uptake and lactate production [51]. Previously, EGFR signaling has been shown to enhance the aerobic glycolysis of triple‐negative breast cancer cells [43]. In the present study, the EGFR‐P38 MAPK axis increased the aerobic glycolysis of HCC cells, as shown by the increased glucose uptake and lactate production. Enhanced tumor aerobic glycolysis is mainly known to support tumor cell growth and proliferation [52]. Recently, increasing evidence has revealed that it may contribute to tumor immunosuppression as well [42, 53]. Of interest, in the present study, we demonstrated that the enhanced aerobic glycolysis was crucial for the EGFR‐P38 MAPK axis down‐regulated HLA‐ABC expression in HCC cells. HK2 is a rate‐limiting enzyme that catalyzes the phosphorylation of hexose in the first step of glycolysis [40]. In the present study, the EGFR‐P38 MAPK axis promoted the expression of HK2 in HCC cells. In addition, knockdown of HK2 expression significantly reversed the EGFR activation‐induced aerobic glycolysis enhancement and HLA‐ABC down‐regulation. These collectively indicate that HK2 mediates EGFR activation‐enhanced aerobic glycolysis and thereby causes HLA‐ABC down‐regulation. The key byproducts and enzymes of aerobic glycolysis can function as signal regulators [53]. Thus, we speculated that some of the byproducts or enzymes of glycolysis may be involved in the EGFR‐P38 MAPK‐HK2 axis‐induced HLA‐ABC down‐regulation in HCC cells.
Of note, there are still some unresolved issues that need to be further studied. For example, how the EGFR‐P38 MAPK axis regulates miR‐675‐5p and HK2, what are the key sequences in the PD‐L1 promoter or factors contributing to miR‐675‐5p‐enhanced PD‐L1 mRNA stability, and whether the byproducts or enzymes of glycolysis were involved in the EGFR‐P38 MAPK‐HK2 axis‐induced HLA‐ABC decrease. Studying these issues will help to fully understand EGFR activation‐induced PD‐L1 increase and HLA‐ABC decrease, and provide more valuable potential targets for the immune intervention of HCC. In addition, to validate our findings from cell studies, using more animal models would provide more convincing in vivo evidence and help the clinical application of these results.
5. CONCLUSIONS
In summary, we demonstrated that EGFR activation was positively correlated with PD‐L1 expression while negatively correlated with HLA‐ABC expression in HCC tissues. EGFR signaling activation by EGF could up‐regulate PD‐L1 and down‐regulate HLA‐ABC in HCC cells, which was functionally important and could be significantly abolished by the EGFR inhibitor gefitinib. Mechanistically, enhanced P38 MAPK activation‐induced miR‐675‐5p down‐regulation and HK2 up‐regulation; miR‐675‐5p down‐regulation enhanced the PD‐L1 mRNA stability probably via 3’‐UTR and thereby caused PD‐L1 accumulation, and HK2 up‐regulation enhanced the aerobic glycolysis and then mediated the decrease in HLA‐ABC expression. The present study reveals a novel signaling network that may cause immune suppression in HCC cells and suggests that EGFR signaling could be potentially targeted for HCC immunotherapy.
DECLARATIONS
AUTHORS’ CONTRIBUTIONS
L. Liu, HF. Wang, ZC. Liu designed this study. ZC. Liu, F. Ning, YN. Cai, HY. Sheng, ZK. Lu, L. Su, XD. Chen conducted the relevant experiments. RD. Zheng, X. Yin collected the HCC specimens. HF. Wang, ZC. Liu performed the statistical analyses. ZC. Liu drafted the manuscript. HF. Wang, L. Liu, CH. Zeng provided critical comments, suggestions, and revised the manuscript. All authors read and approved the final version of the manuscript.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This study was approved by the Ethics Committee of Guangzhou Medical University, and all patients provided a signed informed consent. The procedures for the handling and care of the mice were approved by the Animal Experimentation Ethics Committee of Guangzhou Medical University.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
This work was supported by National Natural Science Foundation of China (No.81602493, No.81902926, No.31600746); Natural Science Foundation of Guangdong Frovince (No. 2018030310305); Fund from Guangzhou Women and Children's Medical Center/Internal Medicine Department (No. NKE‐PRE‐2019‐008).
CONFLICT OF INTEREST
The authors declared that they have no competing interests.
6.
Supporting information
Supporting Figure S1
Supporting Figure S2
Supporting Figure S3
Supporting Figure S4
Supporting Figure S5
Supporting Figure S6
Supporting Figure S7
Supporting Figure S8
Supporting Figure S9
Supporting Table S1
Supporting Table S2
ACKNOWLEDGMENT
We thank all the other members of the Laboratory of Endocrinology and Metabolism, and Guangzhou Institute of Pediatrics in Guangzhou Women and Children's Medical Center for sharing valuable materials and research support. We thank Hao Liu of the Affiliated Cancer Hospital of Guangzhou Medical University for sharing HCC samples. We thank Prof. Gendie Lash for the language editing.
Liu Z, Ning F, Cai Y, et al. The EGFR‐P38 MAPK axis up‐regulates PD‐L1 through miR‐675‐5p and down‐regulates HLA‐ABC via hexokinase‐2 in hepatocellular carcinoma cells. Cancer Commun. 2021;41:62–78. 10.1002/cac2.12117
Contributor Information
Haifang Wang, Email: wanghaifang@smu.edu.cn.
Li Liu, Email: liuli@gwcmc.org.
DATA AVAILABILITY STATEMENT
The supporting data of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018;68(6):394‐424. [DOI] [PubMed] [Google Scholar]
- 2. Zhu AX, Park JO, Ryoo B‐Y, et al. Ramucirumab versus placebo as second‐line treatment in patients with advanced hepatocellular carcinoma following first‐line therapy with sorafenib (reach): A randomised, double‐blind, multicentre, phase 3 trial. Lancet Oncol. 2015;16(7):859‐870. [DOI] [PubMed] [Google Scholar]
- 3. Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discovery 2015;14(8):561‐584. [DOI] [PubMed] [Google Scholar]
- 4. Houot R, Schultz LM, Marabelle A, et al. T‐cell–based immunotherapy: Adoptive cell transfer and checkpoint inhibition. Cancer Immunol. Res. 2015;3(10):1115. [DOI] [PubMed] [Google Scholar]
- 5. Yan Y, Kumar AB, Finnes H, et al. Combining immune checkpoint inhibitors with conventional cancer therapy. Front. Immunol. 2018;9(1739). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roth GS, Decaens T. Liver immunotolerance and hepatocellular carcinoma: Patho‐physiological mechanisms and therapeutic perspectives. Eur. J. Cancer 2017;87:101‐112. [DOI] [PubMed] [Google Scholar]
- 7. Ilan Y. Immune therapy for hepatocellular carcinoma. Hepatology International 2014;8(2):499‐504. [DOI] [PubMed] [Google Scholar]
- 8. Limaye PB, Bowen WC, Orr AV, et al. Mechanisms of hepatocyte growth factor–mediated and epidermal growth factor–mediated signaling in transdifferentiation of rat hepatocytes to biliary epithelium. Hepatology 2008;47(5):1702‐1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shiraga M, Komatsu N, Teshigawara K, et al. Epidermal growth factor stimulates proliferation of mouse uterine epithelial cells in primary culture. Zoolog. Sci. 2000;17(5):661‐666. [DOI] [PubMed] [Google Scholar]
- 10. Berasain C, Perugorria MJ, Latasa MU, et al. The epidermal growth factor receptor: A link between inflammation and liver cancer. Exp. Biol. Med. 2009;234(7):713‐725. [DOI] [PubMed] [Google Scholar]
- 11. Berasain C, Avila MA. The egfr signalling system in the liver: From hepatoprotection to hepatocarcinogenesis. J. Gastroenterol. 2014;49(1):9‐23. [DOI] [PubMed] [Google Scholar]
- 12. Daveau M, Scotte M, François A, et al. Hepatocyte growth factor, transforming growth factor α, and their receptors as combined markers of prognosis in hepatocellular carcinoma. Mol. Carcinog. 2003;36(3):130‐141. [DOI] [PubMed] [Google Scholar]
- 13. Blanc P, Etienne H, Daujat M, et al. Mitotic responsiveness of cultured adult human hepatocytes to epidermal growth factor, transforming growth factor α, and human serum. Gastroenterology 1992;102(4, Part 1):1340‐1350. [PubMed] [Google Scholar]
- 14. Tönjes RR, Löhler J, O'Sullivan JF, et al. Autocrine mitogen igegf cooperates with c‐myc or with the hcs locus during hepatocarcinogenesis in transgenic mice. Oncogene 1995;10(4):765‐768. [PubMed] [Google Scholar]
- 15. Luedke E, Jaime‐Ramirez AC, Bhave N, et al. Cetuximab therapy in head and neck cancer: Immune modulation with interleukin‐12 and other natural killer cell–activating cytokines. Surgery 2012;152(3):431‐440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Srivastava RM, Trivedi S, Concha‐Benavente F, et al. Cd137 stimulation enhances cetuximab‐induced natural killer: Dendritic cell priming of antitumor t‐cell immunity in patients with head and neck cancer. Clin. Cancer Res. 2017;23(3):707‐716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Okita R, Maeda A, Shimizu K, et al. Pd‐l1 overexpression is partially regulated by egfr/her2 signaling and associated with poor prognosis in patients with non‐small‐cell lung cancer. Cancer Immunol. Immunother. 2017;66(7):865‐876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang Y, Hu J, Ya Wang, et al. Egfr activation induced snail‐dependent emt and myc‐dependent pd‐l1 in human salivary adenoid cystic carcinoma cells. Cell Cycle 2018;17(12):1457‐1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen X‐H, Liu Z‐C, Zhang G, et al. Tgf‐β and egf induced hla‐i downregulation is associated with epithelial‐mesenchymal transition (emt) through upregulation of snail in prostate cancer cells. Mol. Immunol. 2015;65(1):34‐42. [DOI] [PubMed] [Google Scholar]
- 20. Zou W, Wolchok JD, Chen L. Pd‐l1 (b7‐h1) and pd‐1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016;8(328):rv4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Garcia‐Lora A, Algarra I, Garrido F. Mhc class i antigens, immune surveillance, and tumor immune escape. J. Cell. Physiol. 2003;195(3):346‐355. [DOI] [PubMed] [Google Scholar]
- 22. Seliger B, Cabrera T, Garrido F, et al. Hla class i antigen abnormalities and immune escape by malignant cells. Semin. Cancer Biol. 2002;12(1):3‐13. [DOI] [PubMed] [Google Scholar]
- 23. Shi F, Shi M, Zeng Z, et al. Pd‐1 and pd‐l1 upregulation promotes cd8+ t‐cell apoptosis and postoperative recurrence in hepatocellular carcinoma patients. Int. J. Cancer 2011;128(4):887‐896. [DOI] [PubMed] [Google Scholar]
- 24. Gao Q, Wang X‐Y, Qiu S‐J, et al. Overexpression of pd‐l1 significantly associates with tumor aggressiveness and postoperative recurrence in human hepatocellular carcinoma. Clin. Cancer Res. 2009;15(3):971. [DOI] [PubMed] [Google Scholar]
- 25. Calderaro J, Rousseau B, Amaddeo G, et al. Programmed death ligand 1 expression in hepatocellular carcinoma: Relationship with clinical and pathological features. Hepatology 2016;64(6):2038‐2046. [DOI] [PubMed] [Google Scholar]
- 26. Matsui M, Machida S, Itani‐Yohda T, et al. Downregulation of the proteasome subunits, transporter, and antigen presentation in hepatocellular carcinoma, and their restoration by interferon‐γ. J. Gastroenterol. Hepatol. 2002;17(8):897‐907. [DOI] [PubMed] [Google Scholar]
- 27. Kurokohchi K, Carrington M, Mann DL, et al. Expression of hla class i molecules and the transporter associated with antigen processing in hepatocellular carcinoma. Hepatology 1996;23(5):1181‐1188. [DOI] [PubMed] [Google Scholar]
- 28. Liu Z‐C, Ning F, Wang H‐F, et al. Epidermal growth factor and tumor necrosis factor α cooperatively promote the motility of hepatocellular carcinoma cell lines via synergistic induction of fibronectin by nf‐κb/p65. Biochimica et Biophysica Acta (BBA)‐General Subjects 2017;1861:2568‐2582. [DOI] [PubMed] [Google Scholar]
- 29. Wang H‐F, Ning F, Liu Z‐C, et al. Histone deacetylase inhibitors deplete myeloid‐derived suppressor cells induced by 4t1 mammary tumors in vivo and in vitro. Cancer Immunol. Immunother. 2017;66(3):355‐366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yao S, Zheng P, Wu H, et al. Erbin interacts with c‐Cbl and promotes tumourigenesis and tumour growth in colorectal cancer by preventing c‐Cbl‐mediated ubiquitination and down‐regulation of EGFR [J] .J Pathol 2015;236(1): 65‐77. [DOI] [PubMed] [Google Scholar]
- 31. Darrow TL, Slingluff CL, Seigler HF. The role of hla class i antigens in recognition of melanoma cells by tumor‐specific cytotoxic t lymphocytes. Evidence for shared tumor antigens. J. Immunol. 1989;142(9):3329. [PubMed] [Google Scholar]
- 32. Lei S, Yang J, Chen C, et al. Flipl is critical for aerobic glycolysis in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2016;35(1):79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen N, Fang W, Zhan J, et al. Upregulation of pd‐l1 by egfr activation mediates the immune escape in egfr‐driven nsclc: Implication for optional immune targeted therapy for nsclc patients with egfr mutation. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 2015;10(6):910‐923. [DOI] [PubMed] [Google Scholar]
- 34. Cheng C, Lin H, Tsai K, et al. Epidermal growth factor induces stat1 expression to exacerbate the ifnr‐mediated pd‐l1 axis in epidermal growth factor receptor‐positive cancers. Mol. Carcinog. 2018;57(11):1588‐1598. [DOI] [PubMed] [Google Scholar]
- 35.a) Zhang W, Pang Q, Yan C, et al. Induction of pd‐l1 expression by epidermal growth factor receptor‐mediated signaling in esophageal squamous cell carcinoma. OncoTargets and therapy 2017;10:763‐771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zerdes I, Matikas A, Bergh J, et al. Genetic, transcriptional and post‐translational regulation of the programmed death protein ligand 1 in cancer: Biology and clinical correlations. Oncogene 2018;37(34):4639‐4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Coelho MA, de Carné Trécesson S, Rana S, et al. Oncogenic ras signaling promotes tumor immunoresistance by stabilizing pd‐l1 mrna. Immunity 2017,47(6):1083‐1099.e 1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wong C, Tsang F, Ng I. Non‐coding rnas in hepatocellular carcinoma: Molecular functions and pathological implications. Nat. Rev. Gastroenterol. Hepatol. 2018;15(3):137‐151. [DOI] [PubMed] [Google Scholar]
- 39. Wang Q, Lin W, Tang X, et al. The roles of micrornas in regulating the expression of pd‐1/pd‐l1 immune checkpoint. Int. J. Mol. Sci. 2017;18(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kataoka K, Shiraishi Y, Takeda Y, et al. Aberrant pd‐l1 expression through 3′‐utr disruption in multiple cancers. Nature 2016;534(7607):402‐406. [DOI] [PubMed] [Google Scholar]
- 41. Zhu M, Chen Q, Liu X, et al. Lncrna h19/mir‐675 axis represses prostate cancer metastasis by targeting tgfbi. FEBS J. 2014;281(16):3766‐3775. [DOI] [PubMed] [Google Scholar]
- 42. Cascone T, McKenzie JA, Mbofung RM, et al. Increased tumor glycolysis characterizes immune resistance to adoptive t cell therapy. Cell Metab. 2018;27(5):977‐987.e974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lim S‐O, Li C‐W, Xia W, et al. Egfr signaling enhances aerobic glycolysis in triple‐negative breast cancer cells to promote tumor growth and immune escape. Cancer Res. 2016;76(5):1284‐1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. DeWaal D, Nogueira V, Terry A, et al. Hexokinase‐2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat. Commun. 2018;9(1):446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xing S‐J, Chen S‐L, Yang X‐L et al. Role of MAPK activity in PD‐L1 expression in hepatocellular carcinoma cells. Journal of BUON 2020;25(4): 1875‐1882. [PubMed] [Google Scholar]
- 46. Keniry A, Oxley D, Monnier P, et al. The h19 lincrna is a developmental reservoir of mir‐675 that suppresses growth and igf1r. Nat. Cell Biol. 2012;14(7):659‐665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. He D, Wang J, Zhang C, et al. Down‐regulation of mir‐675‐5p contributes to tumor progression and development by targeting pro‐tumorigenic gpr55 in non‐small cell lung cancer. Mol. Cancer 2015;14(1):73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schmitz KJ, Helwig J, Bertram S, et al. Differential expression of microrna‐675, microrna‐139‐3p and microrna‐335 in benign and malignant adrenocortical tumours. J. Clin. Pathol. 2011;64(6):529‐535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tushev G, Glock C, Heumüller M, et al. Alternative 3' utrs modify the localization, regulatory potential, stability, and plasticity of mrnas in neuronal compartments. Neuron 2018;98(3):495‐511.e496. [DOI] [PubMed] [Google Scholar]
- 50. Chatterjee S, Pal JK. Role of 5′‐ and 3′‐untranslated regions of mrnas in human diseases. Biol. Cell 2009;101(5):251‐262. [DOI] [PubMed] [Google Scholar]
- 51. Ganapathy‐Kanniappan S. Molecular intricacies of aerobic glycolysis in cancer: Current insights into the classic metabolic phenotype. Crit. Rev. Biochem. Mol. Biol. 2018;53(6):667‐682. [DOI] [PubMed] [Google Scholar]
- 52. Chen D‐P, Ning W‐R, Jiang Z‐Z, et al. Glycolytic activation of peritumoral monocytes fosters immune privilege via the pfkfb3‐pd‐l1 axis in human hepatocellular carcinoma. J. Hepatol. 2019;71(2):333‐343. [DOI] [PubMed] [Google Scholar]
- 53. Li W, Tanikawa T, Kryczek I, et al. Aerobic glycolysis controls myeloid‐derived suppressor cells and tumor immunity via a specific cebpb isoform in triple‐negative breast cancer. Cell Metab. 2018;28(1):87‐103.e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Figure S1
Supporting Figure S2
Supporting Figure S3
Supporting Figure S4
Supporting Figure S5
Supporting Figure S6
Supporting Figure S7
Supporting Figure S8
Supporting Figure S9
Supporting Table S1
Supporting Table S2
Data Availability Statement
The supporting data of this study are available from the corresponding author upon reasonable request.