

Abstract
Background and Aims
Hepatic sinusoidal cells are known actors in the fibrogenic response to injury. Activated hepatic stellate cells (HSCs), liver sinusoidal endothelial cells, and Kupffer cells are responsible for sinusoidal capillarization and perisinusoidal matrix deposition, impairing vascular exchange and heightening the risk of advanced fibrosis. While the overall pathogenesis is well understood, functional relations between cellular transitions during fibrogenesis are only beginning to be resolved. At single‐cell resolution, we here explored the heterogeneity of individual cell types and dissected their transitions and crosstalk during fibrogenesis.
Approach and Results
We applied single‐cell transcriptomics to map the heterogeneity of sinusoid‐associated cells in healthy and injured livers and reconstructed the single‐lineage HSC trajectory from pericyte to myofibroblast. Stratifying each sinusoidal cell population by activation state, we projected shifts in sinusoidal communication upon injury. Weighted gene correlation network analysis of the HSC trajectory led to the identification of core genes whose expression proved highly predictive of advanced fibrosis in patients with nonalcoholic steatohepatitis (NASH). Among the core members of the injury‐repressed gene module, we identified plasmalemma vesicle–associated protein (PLVAP) as a protein amply expressed by mouse and human HSCs. PLVAP expression was suppressed in activated HSCs upon injury and may hence define hitherto unknown roles for HSCs in the regulation of microcirculatory exchange and its breakdown in chronic liver disease.
Conclusions
Our study offers a single‐cell resolved account of drug‐induced injury of the mammalian liver and identifies key genes that may serve important roles in sinusoidal integrity and as markers of advanced fibrosis in human NASH.
Abbreviations
- Adgrg6
adhesion G protein–coupled receptor G6
- Angptl6
angiopoietin‐like 6
- ASH
alcoholic steatohepatitis
- AUROC
area under the receiver operating characteristics
- Bmp5
bone morphogenetic protein 5
- Ccr2
chemokine (C‐C motif) receptor 2
- CD
cluster of differentiation
- Clec4f
C‐type lectin domain family 4 member F
- COL1A1
collagen type 1 alpha 1 chain
- CV
central vein
- Cxcl13
chemokine (C‐X‐C motif) ligand 13
- Cx3cr1
chemokine (C‐X3‐C motif) receptor 1
- DILI
drug‐induced liver injury
- Dpep1
dipeptidase 1
- Dpt
dermatopontin
- ECM
extracellular matrix
- Ecm1
ECM protein 1
- Ednrb
endothelin receptor type B
- Esm1
endothelial cell–specific molecule 1
- Fabp4
fatty acid binding protein 4
- Gas6
growth arrest–specific 6
- GO
gene ontology
- Gpnmb
glycoprotein Nmb
- HSC
hepatic stellate cell
- KC
Kupffer cell
- LEC
liver endothelial cell
- LSEC
liver sinusoidal endothelial cell
- MDM
monocyte‐derived macrophage
- Mfap4
microfibril‐associated protein 4
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- Pcdh17
protocadherin 17
- PE
phycoerythrin
- PLVAP
plasmalemma vesicle–associated protein
- PN
portal node
- Rspo3
R‐spondin 3
- scRNAseq
single‐cell RNA sequencing
- aSMA
alpha‐smooth muscle actin
- Stab2
stabilin 2
- Trem2
triggering receptor expressed on myeloid cells 2
- Tsc22d1
TSC22 domain family member 1
- UMAP
uniform manifold approximation and projection
- VEGF
vascular endothelial growth factor
- Veh
vehicle
- WGCNA
weighted gene correlation network analysis
The hepatic sinusoid is a vascular matrix defined by liver sinusoidal endothelial cells (LSECs), intrasinusoidal Kupffer cells (KCs), natural killer cells, perisinusoidal hepatic stellate cells (HSCs), and extracellular matrix (ECM) components.( 6 ) In particular, LSECs and pericytic HSCs are intimately associated along the porous sinusoid,( 7 ) where they manage blood flow, the trans‐sinusoidal migration of cells, and exchange of lipoprotein particles.( 8 , 9 , 10 ) Hepatic injury and inflammatory cues trigger sinusoidal cell activation, causing sinusoidal capillarization and fibrogenesis. Cytokines and growth factors from the microenvironment directly activate HSCs, which transdifferentiate into fibrogenic myofibroblasts.( 11 ) Similarly, LSECs are transformed by angiogenic cytokines and growth factors, which act in concert with hypoxic and mechanical stress from altered blood flow and tissue stiffness.( 12 ) LSEC activation leads to their dedifferentiation and loss of fenestration, resulting in endothelial dysfunction.( 13 ) If the underlying insults persist, capillarization and widespread fibrosis increase portal vascular resistance, resulting in portal hypertension and aberrant angiogenesis.( 8 ) Despite these insights and the urgency of decoding the concerted response to hepatic injury, the molecular events that forego advanced liver disease remain poorly understood. Hepatic tissue complexity, cellular heterogeneity of the sinusoids, and dynamic sinusoidal communication demand systems‐level approaches for their analysis.
Single‐cell transcriptomics has reshaped our view and understanding of complex biological systems. Cell‐resolved transcriptomics offers high‐dimensional information about tissues and unprecedented insight into cellular composition and response to perturbation. Although, single‐cell RNA‐sequencing (scRNAseq) has been conducted on livers from mice and humans, only few studies have investigated hepatic injury, and the yield of HSCs in particular has been modest. Recent studies include one establishing HSC zonation as a determinant of the fibrotic response,( 14 ) one detailing cellular crosstalk in murine nonalcoholic steatohepatitis (NASH) resolving endothelial and macrophage populations( 15 ) and one demonstrating HSC heterogeneity in the injured liver but offering limited resolution and analytical depth.( 16 )
We set out to characterize the major cell types associated with the hepatic sinusoid, HSCs, KCs/monocyte‐derived macrophages (MDMs), and liver endothelial cells (LECs), by single‐cell transcriptomics after drug‐induced liver injury (DILI) and early fibrogenesis. We identified discrete subpopulations of each cell type and recorded their dynamic transformations and interactions. Specifically, we show how HSC activation and transdifferentiation proceed through discrete activation states defined by unique gene expression networks, which may shape the biology of HSCs and interacting cell types. Intriguingly, we identify PLVAP as an HSC‐enriched protein in mouse and human liver and repression of HSC Plvap expression as a key event during HSC activation. Conversely, robust induction of modules of highly correlated, HSC‐specific genes signified late‐stage HSC activation and may serve as a composite diagnostic marker of advanced hepatic fibrosis in humans.
Materials and Methods
Animal Experiments
Female C57BL6/J mice were housed and treated for 2‐4 weeks either with carbon tetrachloride (CCl4) to induce liver injury or vehicle as described( 1 ) and detailed in the Supporting Information. Animal experiments were approved by the Danish Animal Experiments Inspectorate and adhered to the ARRIVE guidelines.
Isolation of Hepatic Macrophages, Stellate Cells, and Endothelial Cells
Retrograde perfusion and enzymatic dissociation of mouse livers was performed as described.( 2 ) Dissociated livers were strained using a 100‐μm cell strainer and labeled with anti–cluster of differentiation 105 (CD105)‐phycoerythrin (PE) and anti‐F4/80‐PE. Live, PE‐positive singlets were sorted by flow cytometry for single‐cell RNA sequencing. Liver dissociation, antibody labeling, and reagents are detailed in the Supporting Information.
Single‐Cell RNA Sequencing and Data Analysis
Sorted cells were loaded onto a 10x Genomics Chromium controller (10x Genomics, Pleasanton, CA) and libraries prepared according to the manufacturer's instructions. Libraries were paired‐end sequenced with a mean coverage of >50,000 raw reads/cell. Alignment, aggregation, and analysis of the single‐cell data sets were performed in R and Python as detailed in the Supporting Information.
Whole‐Liver/Bulk RNA Purification, Sequencing, and Analysis
Whole‐liver RNA was extracted from snap‐frozen livers from C57BL6/J female mice treated as above and paired‐end sequenced. RNA purification and data analyses are detailed in the Supporting Information.
Human Expression Array Analysis
Expression data from two studies of 72 and 55 patients with nonalcoholic fatty liver disease (NAFLD) staged for hepatic fibrosis, respectively, were retrieved from Gene Expression Omnibus repositories GSE49541 ( 3 ) and GSE89632 ( 4 ) and analyzed in R. Statistical analyses are detailed in the Supporting Information.
Human Liver Biopsies and Sections
The study of liver biopsies from patients with alcoholic steatohepatitis (ASH) has been described.( 5 ) The study of human liver sections (histologically normal tissue obtained from surgical specimens from patients undergoing resection of colorectal liver metastases) was approved by the Ethics Committee of the Region of Southern Denmark and informed consent was collected.
RNA In Situ Hybridization and Immunohistochemical Staining of Formalin‐Fixed Paraffin‐Embedded Tissues
Fixation of liver tissue in 4% paraformaldehyde (PFA), paraffin embedding, and processing for immunohistochemistry and RNA in situ hybridization are detailed in the Supporting Information.
Immunofluorescence
Fluorescence double‐stainings of CD34/alpha‐smooth muscle actin (αSMA) and plasmalemma vesicle–associated protein (PLVAP)/mCherry were performed on mouse cryosections prepared form 4‐hour PFA‐fixed livers as described.( 1 ) Confocal imaging and deconvolution are detailed in the Supporting Information.
Results
Single‐Cell Analysis of Sinusoid‐Associated Cells From Healthy and Fibrotic Mice
DILI may be recapitulated in mice by the administration of CCl4. To get an overview of the transcriptional response to acute hepatic injury, we sequenced whole‐liver mRNA from mice treated with CCl4 for 2 weeks (n = 3) or 4 weeks (n = 3) as well as controls treated with vehicle (veh, n = 2). Clustering and gene ontology (GO) analyses of the genes that changed significantly between groups (4 weeks versus vehicle; P adj < 0.05) pointed to processes mainly driven by nonparenchymal cells including inflammation, wound healing, and vasculogenesis (Fig. 1A; Supporting Fig. S1A). These categories suggested major changes in sinusoidal gene expression, testifying to a profound transcriptional plasticity. We validated fibrogenesis by picrosirius red and verified the appearance of αSMA‐positive myofibroblasts and induction of CD34, marking sinusoidal capillarization( 17 ) (Fig. 1B; Supporting Fig. S1B). Indicative of ongoing inflammation, we also observed redistribution of F4/80hi KCs after only 2 weeks of treatment.
Fig. 1.
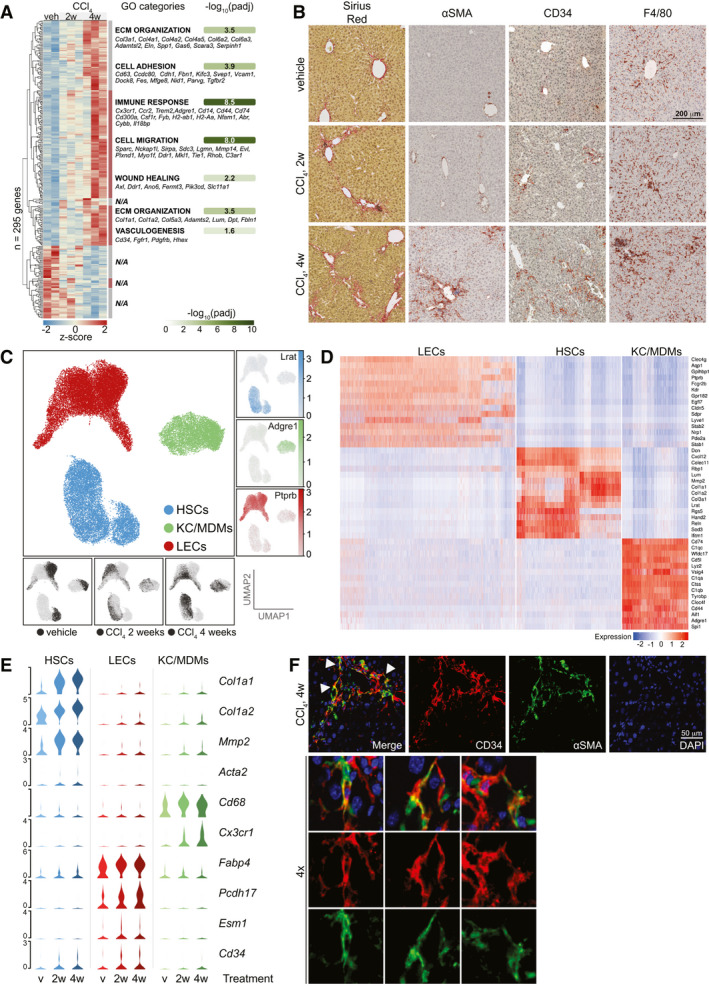
Single‐cell analysis of sinusoidal cells from healthy and fibrotic mouse livers. (A) Whole‐liver RNA‐sequencing of mice treated with CCl4 or veh (n = 2‐3). Z‐scores of 295 differentially expressed genes (4w CCl4 vs. veh, Padj < 0.05, DESeq2). Seven hierarchical clusters are indicated with enriched GO‐categories (Padj < 0.05) and exemplary genes of each category (N/A: No significantly enriched category for cluster). (B) Sirius red, αSMA, CD34 and F4/80 IHC of representative livers from mice treated as indicated. (C) Centre; UMAP of >35K single cells colored according to cell type. Right; UMAPs showing log2‐expression of marker genes. Bottom; UMAPs indicating treatment group of each cell. (D) Scaled log2‐expression of marker genes in all cells grouped by cell type. (E) Scaled, log2‐expression of select cell type‐specific and injury‐responsive genes stratified by cell type and treatment group (v = vehicle). (F) IF of CD34 (red) and αSMA (green) in livers of 4w CCl4‐treated mice. Arrows indicate double‐positive cells shown at 4x magnification.
We then enriched for HSCs, LECs, and KCs/MDMs from CCl4‐treated and vehicle‐treated mice for single‐cell RNA sequencing. Similar to LECs, HSCs express the surface receptor CD105 (Endoglin [Eng]),( 18 ) allowing us to enrich for these two cell types by flow cytometry using anti‐CD105 antibodies. We independently confirmed CD105 expression on quiescent and activated HSCs (not shown). KCs and MDMs were enriched using the surface receptor F4/80. We furthermore gated for singlets and viability (Supporting Fig. S1C). Cells (2x 10,000) from three biological replicates for each treatment group were sorted for scRNAseq and bulk RNA sequencing. We recorded transcriptomes for >56,000 cells, confidently mapping 90% of reads to the mouse genome (Supporting Fig. S1D) and resolved individual cell populations using the uniform manifold approximation and projection (UMAP) technique for dimensionality reduction of our data. We removed doublets and one biological replicate (4‐week CCl4) containing high levels of erythrocyte‐derived transcripts (Supporting Fig. S1E). The cells separated into three well‐defined louvain clusters corresponding to HSCs, LECs, and KCs/MDMs (Fig. 1C; Supporting Figs. S1F‐H). Expression of marker genes and a comparison to the well‐annotated Tabula Muris( 19 ) data set confirmed the identities of the clusters (Fig. 1D; Supporting Fig. S1I). We further confirmed assay specificity by analyzing the expression of Eng and adhesion G protein–coupled receptor E1 (encoding CD105 and F4/80, respectively) and cell type–specific markers (Supporting Figs. S1J,K and S3G). To focus our analyses, minor clusters of other cell types were omitted from further analysis. A survey of changes within individual cell populations showed that the activation markers collagen type 1 alpha 1 chain (Col1a1), Col1a2, matrix metalloproteinase 2 and the myofibroblast marker actin alpha 2 smooth muscle were induced in the HSC population (Fig. 1E). In the KC/MDM population, induction of the leukocytic markers Cd68 and chemokine (C‐X3‐C motif) receptor 1 (Cx3cr1) reflected transcriptional changes in KCs and recruitment of MDMs. Fatty acid binding protein 4 (Fabp4) and protocadherin 17 (Pcdh17) levels were increased in LECs. FABP4 was previously linked to vascular injury and vascular endothelial growth factor A (VEGFA) signaling and PCDH17 to cell–cell connections. Also, the observed induction of endothelial cell–specific molecule 1 (Esm1) and Cd34 indicates endothelial dysfunction and sinusoidal capillarization.( 17 , 20 ) Cd34 was also modestly induced in HSCs. We confirmed partial, lobular, and periportal coexpression of CD34 and αSMA by double‐immunofluorescence on livers from CCl4‐treated mice (Fig. 1F; Supporting Fig. S1L). CD34 is an adhesion molecule also expressed in progenitor cells and fibroblasts, but the significance of Cd34 induction in HSCs is unclear.
Stellate Cell Dynamics in Healthy and Fibrotic Livers
Having recapitulated the sinusoidal response to injury at the single‐cell level and ascribed key transcriptional changes to specific cell populations, we next focused on the dynamics within these populations. Louvain clustering helped us identify four HSC subclusters. These aligned well with treatment groups ranging from <1% HSCs from CCl4‐treated mice in subcluster 1 to >99% in subcluster 4 (Fig. 2A). A near complete shift in the HSC population from subcluster 1 to the contiguous subclusters 2‐4 was hence seen already after 2 weeks of CCl4 treatment. The overall changes in gene expression between subclusters were calculated (Supporting Fig. S2A).
Fig. 2.
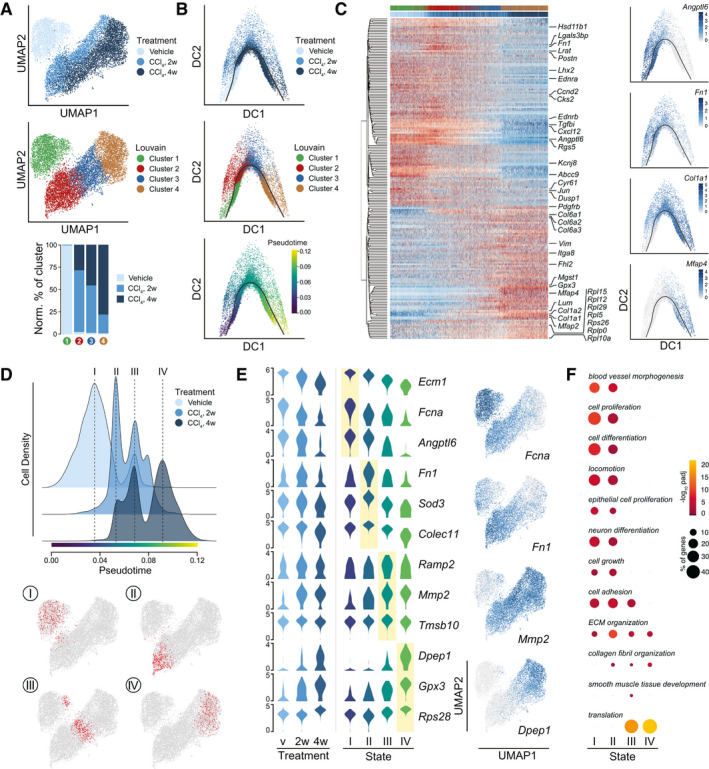
Hepatic stellate cell dynamics and functionality in healthy and fibrotic livers. (A) UMAPs of 10582 HSCs with indication of treatment groups and louvain clustering. Lower panel shows the representation of each treatment group in the louvain clusters 1‐4. Counts normalized by total HSC counts in each treatment group. (B) Diffusion maps of HSCs colored by treatment group, louvain clustering, and pseudotime. Black curve indicates the single‐lineage HSC activation trajectory. (C) Scaled, log2‐expression of top‐200 highly variable genes in HSCs over pseudotime. Select genes are highlighted and louvain cluster and treatment group of each cell are indicated. Right; diffusion maps showing log2‐expression of characteristic genes. (D) Cellular densities of HSCs along pseudotime with indication of four discrete, high‐density states I‐IV. HSCs are stratified by treatment group. Bottom; UMAPs highlighting the 500 HSCs around each density maximum. (E) Log2‐expression of top, unique signature genes of each state shown in all HSCs stratified by treatment group (left) and in the 500 HSCs around the density maximum of each HSC state (middle). Right: UMAPs of HSCs showing log2‐expression levels of select signature genes. (F) Enriched (Padj < 0.05) GO‐categories associated with each HSC state. Colors indicate significance of enrichment and circle sizes indicate number of genes falling into respective categories.
To further resolve the HSC activation process and transdifferentiation, we generated diffusion maps and calculated a single‐lineage trajectory of the HSC‐to‐myofibroblast transition for pseudotemporal ordering of the HSCs. In keeping with the cell distribution across subclusters, the unbranched trajectory proceeded from louvain subcluster 1 to 4 (Fig. 2B). We analyzed HSC gene expression in pseudotime along this trajectory and plotted top variable genes in a heatmap (Fig. 2C). Select genes are indicated, and expression of representative genes is shown in diffusion maps. Putative promoters of sinusoidal homeostasis, including angiopoietin‐like 6 (Angptl6), bone morphogenetic protein 5 (Bmp5), and ECM protein 1 (Ecm1), were repressed upon HSC activation and superseded by fibrosis‐associated genes such as Fn1, Col1a1/2, and microfibril‐associated protein 4 (Mfap4) as well as the induction of ribosomal subunit genes.
The segmented appearance of the expression profiles in pseudotime led us to examine cell densities over pseudotime for each treatment group (Fig. 2D). HSCs from the different treatment groups distributed among four main activation states refining the four louvain clusters: state I corresponding to nonactivated HSCs, state IV populated by the most activated HSCs from CCl4‐treated animals, and two intermediate states (II and III) also with HSCs derived mainly from CCl4‐treated animals. Expression of top state‐defining genes across treatment groups, activation states, and louvain clusters is presented in violin plots and in UMAP space (Fig. 2E; Supporting Fig. S2B and Table S2). Accommodating the transition from sinusoidal pericyte to fibrogenic myofibroblast, HSC transcriptomes shifted from supporting blood vessel morphogenesis and cell proliferation and differentiation, over genes affecting cell adhesion and ECM organization, to smooth muscle tissue development and protein translation (Fig. 2F).
Sinusoidal Dynamics and Interactions in Healthy and Fibrotic Livers
The emergence of HSC subpopulations occurred in the context of a wider sinusoidal reaction to DILI. We therefore turned our attention to the six subpopulations of LECs (Fig. 3A). Upon CCl4 treatment, we observed the progression of LECs through the central subclusters 1‐3, ranging from 7% LECs derived from CCl4‐treated animals in subcluster 1 to 94% in subcluster 3. Expression profiles identified these central clusters as stabilin 2 (Stab2)‐expressing LSECs as defined by Geraud et al.,( 21 ) although LSEC marker genes were reduced in the cells of subcluster 3 upon injury (Fig. 3B). In contrast, LEC subclusters 4 and 5 appeared unaffected by injury, containing comparable cell numbers from each treatment group. The relative position to either the portal and centrolobular areas results in subpopulations of LECs with unique gene expression signatures. Based on expression profiles from studies of lobular zonation,( 22 ) we classified subcluster 4 and 5 LECs as portal node (PN)‐proximal and central vein (CV)‐proximal LECs, respectively. Adhesion G protein–coupled receptor G6 (Adgrg6) encodes a developmentally important adhesion G protein–coupled receptor and is a highly specific marker of PN‐derived LECs. Differentially expressed genes across LEC subclusters are shown in Fig. 3B and Supporting Fig. S3A and summarized in Supporting Fig. S3B.
Fig. 3.
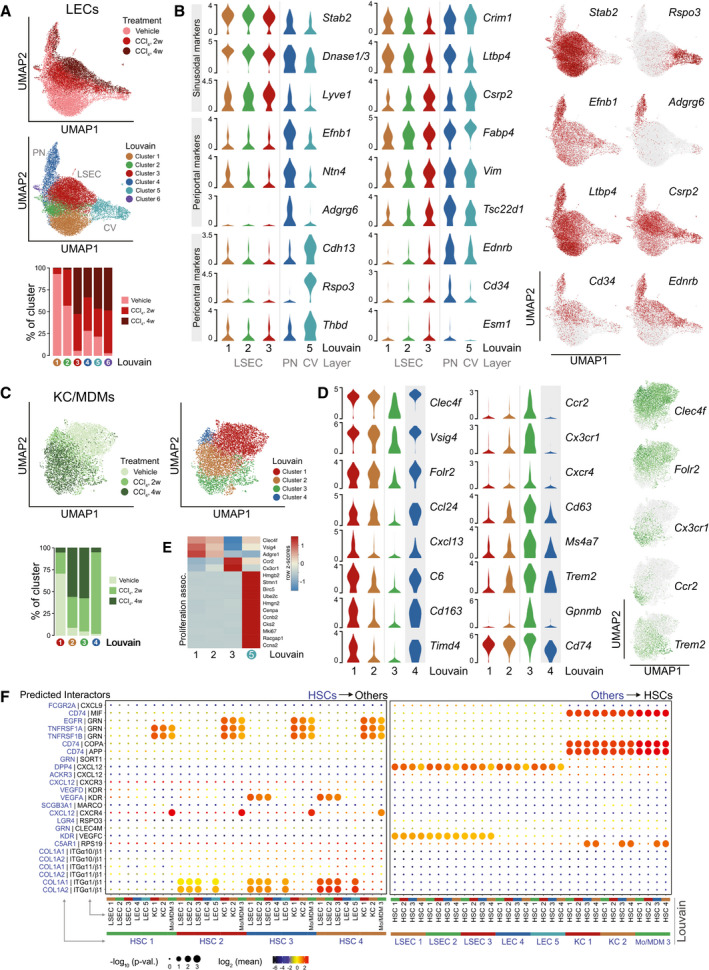
Sinusoidal dynamics and interactions in healthy and fibrotic livers. (A) UMAPs of 17748 LECs with indication of treatment groups and louvain clustering (PN: portal node‐proximal LECs; CV: central vein‐proximal LECs). Lower panel shows the representation of each treatment group in the louvain clusters 1‐6. Counts normalized by total LEC counts in each treatment group. (B) Log2‐expression of cluster‐selective marker genes and select injury‐responsive genes across LEC louvain clusters 1‐5. Right; UMAPs of LECs showing log2‐expression levels of select genes. (C) UMAPs of 6611 KC/MDMs with indication of treatment groups and louvain clustering. Lower panel showing the representation of each treatment group in the louvain clusters 1‐6. Counts normalized by total KC/MDM counts in each treatment group. (D) Log2‐expression of cluster‐selective marker genes and select injury‐responsive genes across KC/MDM louvain clusters 1‐4. Right; UMAPs of KC/MDMs showing log2‐expression of select genes. (E) Row‐normalized z‐scores of select marker genes and proliferation‐associated genes in KC/MDM louvain clusters 1‐3 and 5. (F) Directional cell‐cell interactions inferred from ligand‐receptor pairs and stratified by louvain clusters. Left; HSC‐to‐LEC/KC/MDM interactions. Right; LEC/KC/MDM‐to‐HSC interactions. Circle size indicates enrichment of expression of interacting partners in the interacting cluster pairs and color indicates mean expression of interacting partner subunits. Applied mean expression cut‐off is ≥3 for at least one interacting cluster pair.
Having annotated our subclusters, we found that LSECs from the CCl4‐treated animals had induced expression of genes otherwise expressed in nonsinusoidal LECs, supporting the notion of LSEC activation and dedifferentiation. In addition to Fabp4, Pcdh17, Esm1, and Cd34, injury‐induced genes include cysteine and glycine rich protein 2 (Csrp2), vimentin (Vim), TSC22 domain family member 1 (Tsc22d1), endothelin receptor type B (Ednrb), and mesoderm‐specific transcript (Mest), several of which are known to be transforming growth factor beta–responsive. Mest is an early marker of the mesodermal lineage linked to angiogenesis. The endothelin receptor gene Ednrb was expressed in LSECs and CV‐derived LECs only after injury, while being constitutively expressed in PN‐derived LECs (UMAP; Fig. 3B). Ednrb induction occurred in parallel to loss of Ednrb expression in activated HSCs (see Fig. 2C) and suggests another coupling of HSC and LSEC biology. Also, Ednra expression was repressed in HSCs (Fig. 2C) but not detected in LECs. LEC subcluster 6 consisted of baculoviral IAP repeat containing 5, cyclin B2, stathmin 1, and ubiquitin conjugating enzyme E2 C–positive LECs derived largely from CCl4‐treated animals and indicative of injury‐associated LSEC proliferation (Supporting Fig. S3C). Indeed, this subpopulation expressed Stab2 but neither Adgrg6 nor R‐spondin 3 (Rspo3). Notably, they showed highly elevated expression of vimentin and hepatoma‐derived growth factor (Hdgf) but expressed only low levels of hepatocyte growth factor, arguing against bone marrow origin.( 23 ) A minor subcluster of 12 LECs with elevated levels of hepatocyte‐specific transcripts was excluded from the data set.
Subclustering of the KC/MDM population resulted in five subclusters (Fig. 3C; Supporting Fig. S3D). Similar to HSCs and LSECs, the KC/MDM population shifted upon injury (Fig. 3C). Subcluster 1 cells were mainly from healthy animals and cells in subclusters 2 and 3 predominantly from CCl4‐treated animals. In line with such transition, the expression of KC marker genes C‐type lectin domain family 4 member F (Clec4f), V‐set and immunoglobulin domain containing 4 (Vsig4), and Folr2 established subclusters 1 and 2 as KCs,( 24 ) while the chemokine (C‐C motif) receptor 2 (Ccr2 hi)/Cx3cr1 hi cells of subcluster 3 were of monocytic origin (Fig. 3D). In KCs from CCl4‐treated mice, genes associated with scavenging and clearance were repressed including chemokine (C‐X‐C motif) ligand 13 (Cxcl13), C6, Cd163, T‐cell immunoglobulin and mucin domain containing 4 (Timd4), and macrophage receptor with collagenous structure (Marco), while inflammation‐associated genes such as Cx3cr1, Cd63, triggering receptor expressed on myeloid cells 2 (Trem2), and glycoprotein Nmb (Gpnmb) were induced. We summarized changes in gene expression between KC subclusters 1 and 2 (Supporting Fig. S3E). The minor KC/MDM subcluster 4 consisted of KCs from 2‐week CCl4‐treated mice and was very similar to subcluster 1. A fifth cluster of 92 KC/MDMs emerged but was for visualization purposes initially omitted. This subcluster expressed KC markers Clec4f/Vsig4 and high levels of proliferation‐associated genes but was not enriched for cells from any treatment group in particular (Fig. 3E; Supporting Fig. S3D).
All subclusters aligned closely with injury status and allowed for an unbiased review of specifically expressed ligand–receptor pairs, which could predict sinusoidal communication. We tapped into a recently compiled and curated repository of ligand–receptor interaction pairs, CellPhoneDB,( 25 ) taking into account the subunit architecture of receptors and ligands. We focused our predictions on interactions involving HSCs and omitted LEC subcluster 6 and KC/MDM subclusters 4 and 5 due to their similarity to the remaining subclusters. Figure 3F shows significant (P < 0.05) ligand–receptor pairs with a mean expression ≥3 for at least one interaction. The most significant, specific interactions were predicted to originate from the HSCs expressing ligands potentially affecting either LSECs (VEGFA, VEGFC), LSECs/PN‐derived LECs (COL1A1, COL1A2), KC/MDM populations (macrophage migration inhibitory factor [MIF], amyloid beta precursor protein, coatomer subunit alpha [COPA]), or both LECs and macrophages (CXCL12). Together with Col1a1 and Col1a2, expression of Vegfa and Mif was induced in subsets of HSCs upon activation (Supporting Fig. S3F), emphasizing the proinflammatory and proliferative roles activated HSCs may play at the site of injury. High expression of the trophogenic chemokine Cxcl12 in HSCs raises the possibility of chemokine (C‐X‐C motif) receptor 4 (CXCR4) signaling by KC/MDMs or atypical chemokine receptor 3 (ACKR3) signaling by subpopulations of LECs and HSCs (Fig. S3F). The CXCL12–ACKR3/CXCR4 signaling axes were previously implicated in liver repair( 26 ); however, the functionality of such an interaction is unclear as Cxcl12 expression drops upon HSC activation and the CXCL12 modulator dipeptidyl peptidase 4 (Dpp4) is highly expressed in LECs. KCs and MDMs may also tune HSC function by secreting progranulin and in turn modulate epidermal growth factor receptor (EGFR) and tumor necrosis factor receptor superfamily members 1A/1B (TNFRSF1A/1B) signaling. Progranulin could function as an HSC survival signal by stimulating EGFR signaling through ephrin receptor A2( 27 ) (Supporting Fig. S3F). Progranulin inhibition of tumor necrosis factor alpha receptor signaling remains contested. An extended ligand–receptor table with a lower cutoff (mean subunit expression ≥1) is found in Supporting Fig. S3H.
Weighted Gene Correlation Network Analysis Identifies a Highly Predictive Fibrosis Marker and Putative HSC Functions
Having found discrete HSC subpopulations appearing upon liver injury and genes marking each state, we wanted to identify the transcriptional networks most tightly associated with the HSC trajectory. To identify networks correlating with HSC activation in pseudotime, we performed weighted gene correlation network analysis (WGCNA)( 28 ) on the combined HSC population. The analysis returned three nonredundant modules including 38 top member genes that met our criteria for module membership (score >0.65) and gene significance (>0.20) (Supporting Figs. S4A,B). Nineteen genes correlated positively with HSC activation and 19 genes correlated negatively as represented by their module eigengenes (Fig. 4A). Reassuringly, the identified top members were also found among the top‐ranking genes defining HSC subclusters 1‐4 (Fig. 4A, middle) and HSC activation states. Modules I and II were negatively correlated (Pearson r = −0.85), indicative of mutually exclusive transcriptional networks instructing pericyte and myofibroblast functions, respectively. Module‐II top member genes signify the myofibroblastic state and include Col1a1, Col1a2, dermatopontin (Dpt), growth arrest–specific 6 (Gas6), and Mfap4, encoding secreted proteins linked to hepatic fibrosis. Circulating GAS6 and MFAP4 proteins have been proposed as biomarkers of advanced liver fibrosis in patients.( 29 , 30 ) Whereas activated HSCs have been identified as a source of GAS6 stimulating MER proto‐oncogene tyrosine kinase and AXL receptor tyrosine kinase receptors on LECs and KC/MDMs (Supporting Fig. S3H), we also demonstrated Mfap4 induction in HSCs. Similarly, module‐II top member Dpep1 encoding dipeptidase‐1 has not been linked to HSC biology, but intriguingly, DPEP1 was recently linked to neutrophil recruitment.( 31 ) Concurrent with the transcriptional rewiring, induction of ribosomal protein genes of module III may ensure the translational capacity to accommodate HSC transdifferentiation and ECM production. Both the switch in core transcriptional networks and the increased translational capacity align with the previously identified GO categories (Fig. 2F).
Fig. 4.
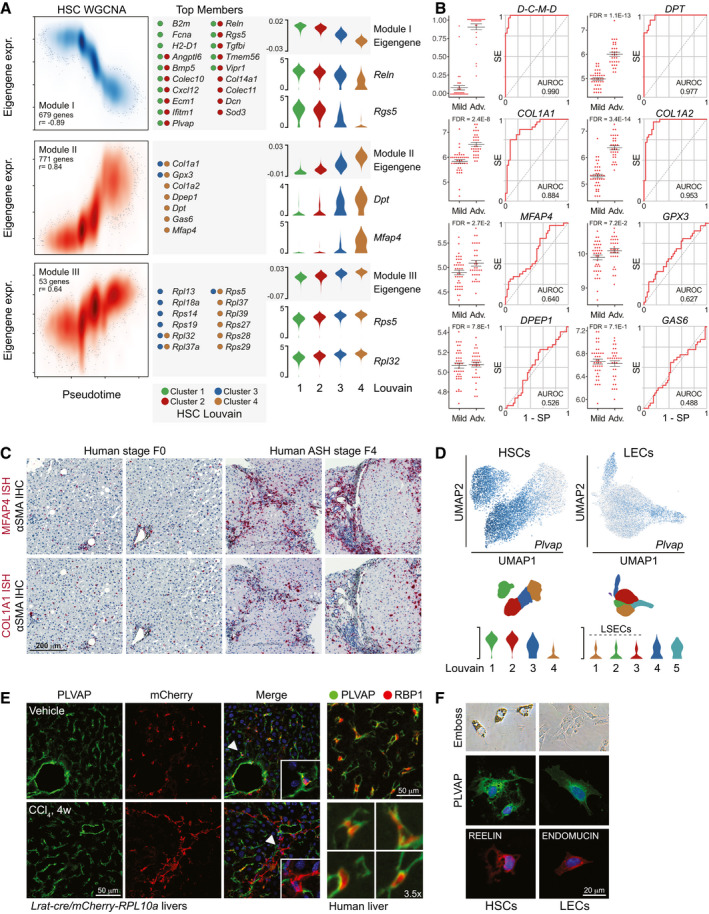
WGCNA identifies highly predictive fibrosis marker and putative HSC functionality. (A) Left; smoothened scatterplot showing WGCNA module I‐III eigengene expression and densities of HSCs over pseudotime. Module member gene count and eigengene‐pseudotime correlation (Pearson) indicated for each module. Center; top module member genes (membership score > 0.65; gene significance > 0.20) with indication of louvain cluster‐association. Right; log2‐expression levels of module eigengenes and select top module member genes. (B) Differential expression of module II top member genes in human biopsies from patients with mild (F0‐F1; n = 40) or severe (F3‐F4; n = 32) NAFLD. Average expression, standard error, and FDR‐values from DE analysis are shown. For the combination of DPT, COL1A2, MFAP4 and DPEP1 (D‐C‐M‐D), the probabilities that the expression of the combination correctly predicts fibrosis classifications are shown. ROC curves indicate the discriminatory performance of each transcript and of the D‐C‐M‐D combination. SE: Sensitivity, SP: Specificity. (C) MFAP4 and COL1A1 in situ hybridization (red) combined with αSMA IHC (black) of human liver biopsies, fibrosis stage F0 and F4, respectively. (D) Top; UMAPs of HSCs and LECs showing log2‐expression of Plvap. Bottom; log2‐expression of Plvap across HSC and LEC louvain clusters. (E) Left; confocal IF analysis of PLVAP (green) in vehicle‐ or 4w CCl4‐treated Lrat‐cre/mCherry‐Rpl10aA knock‐in mice. mCherry‐RPL10A+ HSCs and HSC‐derived MFBs are red, DAPI‐stained nuclei are blue. Right; IF analysis of PLVAP (green) and RBP1 (red) in normal human liver. (F) Emboss contrast microscopy of primary mouse HSCs and LECs and confocal IF analysis of PLVAP (green), REELIN (red), and ENDOMUCIN (red).
As WGCNA module II follows HSC activation, top member genes are potential diagnostic markers for advanced fibrosis where conventional clinical parameters generally show suboptimal sensitivity. Publicly available microarray data of liver biopsies from a cross‐sectional study of patients with NAFLD( 3 ) was used for our validation. Expression of DPT, COL1A1, COL1A2, and MFAP4 was significantly different (false discovery rate < 0.05) between patients with mild NAFLD (fibrosis stage F0/F1, n = 40) and severe NAFLD (F3/F4, n = 32) (Fig. 4B). We next evaluated the diagnostic accuracy of the individual genes and a minimal combination of four genes, DPT, COL1A2, MFAP4, and DPEP1 (D‐C‐M‐D), identified among the seven top member genes by logistic regression. We focused on combinations of HSC‐specific genes specifically signifying louvain cluster 4 and therefore omitted GAS6, broadly expressed in the injured liver. Indeed, the D‐C‐M‐D gene combination very effectively detected advanced fibrosis with an area under the receiver operating characteristic curve (AUROC) of 0.990 (95% confidence interval [CI], 0.899‐1.000), selectivity of 0.990, and specificity of 1.00, resulting in 100% detection of patients with advanced fibrosis (Fig. 4B; Supporting Figs. S4C‐E). Among individual genes, DPT expression best predicted advanced fibrosis with an AUROC of 0.977 (95% CI, 0.828‐0.972), selectivity of 0.900, and specificity of 0.938; COL1A1 showed the highest sensitivity (selectivity, 0.925); and COL1A2 showed the highest specificity (0.969). Testing of the four‐gene combination on a second NAFLD cohort( 4 ) resulted in an AUROC of 0.880 for the prediction of significant fibrosis (stage F2‐F4; Supporting Fig. S4F). These findings demonstrate that our four‐gene combination derived from the HSC WGCNA translated into human fibrogenesis and accurately predicted disease severity. We validated the induction of MFAP4 and COL1A1 by RNA in situ hybridization on liver biopsies from patients with ASH, showing highly increased lobular and periportal expression of both genes in cirrhotic liver compared to nonfibrotic liver (Fig. 4C; Supporting Fig. S4G).
While module II mirrored disease progression, module‐I top member genes may give insight into HSC functionality in healthy liver. Despite being recognized as an endothelium‐specific gene, Plvap encoding the PLVAP protein was highly expressed in nonactivated HSCs and lost upon their activation (Fig. 4A,D). Plvap was of similar abundance in nonsinusoidal LECs but sparsely expressed in LSECs regardless of treatment (Fig. 4D). PLVAP plays a prominent role in vascular integrity and permeability,( 10 , 32 ) and our findings allude to hitherto unknown functions of HSCs in sinusoidal vascular integrity, which is lost as activated HSCs disengage. The likely importance of HSC Plvap repression in sinusoidal remodeling led us to validate our findings at the protein level. In mCherry knock‐in mice crossed with the Lrat‐cre driver line,( 33 ) coexpression of mCherry and PLVAP in HSCs indeed confirmed our findings (Fig. 4E left; Supporting Fig. S4H). Moreover, CCl4 treatment of the Lrat‐cre/mCherry knock‐in mice led to mCherry‐positive cells devoid of PLVAP protein. Outside HSCs, PLVAP protein was detected in both LSECs and nonsinusoidal LECs near bigger vessels. Importantly, also in human liver, retinol‐binding protein 1 (Rbp1)‐positive HSCs clearly expressed PLVAP protein (Fig. 4E, right; Supporting Figs. S4I‐J). Probing of isolated murine HSCs and LECs confirmed PLVAP protein expression in HSCs ex vivo (Fig. 4F).
Discussion
Sinusoidal remodeling is a defining event during the development of hepatic fibrosis. HSCs and LSECs associated along the healthy sinusoids undergo phenotypic transitions, leading to the loss of sinusoidal fenestrations, basal lamina formation, and perisinusoidal fibrosis.( 13 ) This perturbs microvascular exchange between the sinusoid and hepatic parenchyma, increases vascular resistance, and raises portal pressure.
In the current study we applied single‐cell RNA sequencing to determine the transcriptional dynamics of sinusoidal cell populations during DILI in mice and validated key findings using patient data and liver tissue. Experimental DILI led to global activation of all HSCs, followed by stepwise progression through discrete activation states from Angptl6‐, Bmp5‐, Ecm1‐, and Plvap‐expressing pericytes to myofibroblastic cells expressing ECM proteins and profibrotic mediators. The acute repression of genes upon HSC activation, causing an immediate decline in pericyte function, may directly implicate HSC activation in LSEC capillarization. The loss of PLVAP in particular could have major implications for microvascular dynamics. In fetal and neonatal livers, PLVAP is confined to the diaphragmed fenestra of LSECs, forming a physical sieve that regulates cellular transmigration and passage of lipoprotein particles across the sinusoid.( 10 , 32 ) Our findings indicate that PLVAP expression partly shifts to HSCs in the adult sinusoid, displaying open fenestra without diaphragm. The expression of PLVAP by HSCs highlights HSCs and LSECs as a functional entity of the sinusoidal barrier, where suppression of PLVAP upon injury may affect permeability and parenchymal accessibility for immune cells. Notably, Plvap‐null livers are marked by inflammation and diffuse perisinusoidal fibrosis.( 32 )
The hepatic fibrosis score is the most robust predictor of mortality in patients with ASH or NASH,( 34 , 35 ) and genes that mirror HSC activation trajectory would have obvious diagnostic value. Using the powerful WGCNA approach on the HSC population resolved in pseudotime, we identified three core gene modules that reflected the fundamental shift in HSC function. The induced module II correlated best with HSC activation and led to seven top member genes proving very effective in the prediction of advanced liver fibrosis in patients with NAFLD from biopsy gene expression data. The four‐gene combination, DPT, COL1A2, MFAP4, and DPEP1, in particular, predicted fibrosis severity with high accuracy. Other four‐gene combinations of the seven top member genes gave similar or slightly lower prediction power. Adding more genes only marginally improved accuracy. Combining the signature genes improved robustness over individual genes and correctly classified 100% of patients with advanced fibrosis in the study by Moylan et al.( 3 ) Three genes (DPT, COL1A2, MFAP4) encode secreted proteins, and future studies should evaluate serum levels of transcripts and proteins. Importantly, MFAP4 protein was previously detected in serum from patients with cirrhosis,( 29 ) and hepatic DPT expression was, as part of a 24‐gene panel, shown to reflect histological improvements in patients with NAFLD after gastric bypass.( 36 )
Alongside global HSC activation, we saw profound shifts of the LSEC and KC populations after 2 weeks of treatment, with few cells remaining unaffected. Proliferating Stab2+ LSECs also appeared after 2 weeks, possibly reflecting angiogenesis. LSEC proliferation has mainly been studied ex vivo, and the source of LSEC recruitment upon acute injury is unclear. The proliferative LSECs identified here expressed high levels of Hdgf and vimentin. Vim expression gradually increased also in nonproliferative LSECs after CCl4 treatment, making them a likely source. Microvascular regeneration is essential for hepatic recovery, and further insights into LSEC recruitment, proliferation, and inactivation are needed. Based on our results, a better understanding of the role of HSCs in these processes should be prioritized.
We inferred the sinusoidal communication between the identified subpopulations by their expression of ligand–receptor pairs and found both known and unknown interactions. Our distinction between individual subpopulations allowed us to investigate injury‐associated changes and supported the idea of injury‐induced diversion of HSCs from actively preserving sinusoidal integrity to promoting regeneration. This is in contrast to the notion of HSCs as merely quiescent cells becoming active. Recent predictions of secretomes of nonparenchymal hepatic cell types in murine NASH supports this concept, pointing to putative interactions.( 15 ) While aspects of HSC signaling are uncovered in both studies (e.g., Bmp5, Cxcl12, and Gas6 expression by HSCs), we failed to see indications of others (e.g., chemokine [C‐C motif] ligand 2, CXCL10, CXCL16, and interleukin‐11 signaling by HSCs). Whether this discrepancy is due to the analytical stringency, misassignment of transcripts to the HSC population by Xiong et al.,( 15 ) or differences between fibrosis models remains to be tested. Ligand and receptor subunit architecture were considered in our predictions, but protein expression, ligand secretion, and localization of receptors need to be validated. Our analyses hinge on the capture of sinusoidal cells using the surface markers CD105 and F4/80. Expression of both markers showed only subtle differences across treatment groups and subclusters and was unlikely to introduce significant bias (Supporting Figs. S1J and S3G). This conclusion is backed by the overall concordance between the LEC and KC/MDM subpopulations identified in our study and in the study by Xiong et al.( 15 ) A curious difference between our studies is the likely origin of Trem2 hi macrophages. We observe Trem2 induction in Clec4f hi KCs as well as the emergence of a Trem2 hi/Ccr2 hi/Clec4f lo subpopulation, while NASH‐associated Trem2 hi macrophages were all of Ccr2 lo KC origin. This may be explained by different severities and durations of inflammation. Importantly, Trem2 hi macrophages seem involved in a general response to hepatocyte damage, not only in NASH.
In conclusion, our study offers a comprehensive molecular account of drug‐induced injury of the mammalian liver and highlights the power of single‐cell resolution in elucidating complex biological systems. By dissecting sinusoidal cell populations, we mapped transitions of individual cell types upon injury and interactions between their subpopulations. Combining single‐cell transcriptomics with WGCNA, we identified key HSC genes that may play important roles in human sinusoidal integrity and serve as composite markers of advanced fibrosis in patients.
Author Contributions
M.K.T. and K.R. conceptualized the study. M.K.T., S.M.B., D.H., R.N., A.S., T.L.A., and K.R. developed the study methodology and conducted the investigation. M.K.T., E.A.H.S., A.F.M., K.N.N., and K.R. conducted the formal NGS data analysis. G.L.S., A.K., K.N.N., S.D., and H.D. provided resources for the study and made significant intellectual contributions to the study design or data interpretation. K.R. supervised, managed, and coordinated the study. Funding for the study was mainly acquired by S.M. and K.R.
Supporting information
Supplementary Material
Acknowledgment
We thank Tenna P. Mortensen, Maibrith Wishoff, Naja L. Lorenzen, and Inger Nissen for expert technical assistance. We thank Jonathan J. Thompson and Tune H. Pers (CBMR, Copenhagen University) for valuable discussion of the WGCNA, Robert F. Schwabe (Columbia University) for the Lrat‐cre mouse strain, and Maja Thiele (SDU) for advice on the human biopsies. Bioimaging was partly performed at DaMBIC, a bioimaging research core facility at the University of Southern Denmark (SDU), established by an equipment grant from the Danish Agency for Science, Technology and Innovation and internal funding from SDU.
Supported by the National Danish Research Foundation and the University of Southern Denmark (ATLAS; M.K.T., S.M.B., R.N., S.M., A.K., K.R.), the Villum Foundation (K.N.N.), the Danish Institute for Advanced Study (K.N.N.), the Fuhrmann Foundation (D.H.), the Villum Foundation (K.N.N.), the Danish Institute for Advanced Study (K.N.N.), the Independent Research Fund Denmark (H.D., K.R.), the European Commission's Marie Skłodowska‐Curie Action (K.R.), and the European Foundation for the Study of Diabetes (K.R.).
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Marcher AB, Bendixen SM, Terkelsen MK, Hohmann SS, Hansen MH, Larsen BD, et al. Transcriptional regulation of hepatic stellate cell activation in NASH. Sci Rep 2019;9:2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mederacke I, Dapito DH, Affo S, Uchinami H, Schwabe RF. High‐yield and high‐purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat Protoc 2015;10:305‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moylan CA, Pang H, Dellinger A, Suzuki A, Garrett ME, Guy CD, et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology 2014;59:471‐482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Arendt BM, Comelli EM, Ma DW, Lou W, Teterina A, Kim T, et al. Altered hepatic gene expression in nonalcoholic fatty liver disease is associated with lower hepatic n‐3 and n‐6 polyunsaturated fatty acids. Hepatology 2015;61:1565‐1578. [DOI] [PubMed] [Google Scholar]
- 5. Thiele M, Madsen BS, Hansen JF, Detlefsen S, Antonsen S, Krag A. Accuracy of the enhanced liver fibrosis test vs fibrotest, elastography, and indirect markers in detection of advanced fibrosis in patients with alcoholic liver disease. Gastroenterology 2018;154:1369‐1379. [DOI] [PubMed] [Google Scholar]
- 6. Wake K, Sato T. “The sinusoid” in the liver: lessons learned from the original definition by Charles Sedgwick Minot (1900). Anat Rec (Hoboken) 2015;298:2071‐2080. [DOI] [PubMed] [Google Scholar]
- 7. Wisse E, de Zanger RB, Charels K, van der Smissen P, McCuskey RS. The liver sieve: considerations concerning the structure and function of endothelial fenestrae, the sinusoidal wall and the space of disse. Hepatology 1985;5:683‐692. [DOI] [PubMed] [Google Scholar]
- 8. Gracia‐Sancho J, Marrone G, Fernandez‐Iglesias A. Hepatic microcirculation and mechanisms of portal hypertension. Nat Rev Gastroenterol Hepatol 2019;16:221‐234. [DOI] [PubMed] [Google Scholar]
- 9. Carpenter B, Lin Y, Stoll S, Raffai RL, McCuskey R, Wang R. VEGF is crucial for the hepatic vascular development required for lipoprotein uptake. Development 2005;132:3293‐3303. [DOI] [PubMed] [Google Scholar]
- 10. Rantakari P, Jappinen N, Lokka E, Mokkala E, Gerke H, Peuhu E, et al. Fetal liver endothelium regulates the seeding of tissue‐resident macrophages. Nature 2016;538:392‐396. [DOI] [PubMed] [Google Scholar]
- 11. Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol 2014;14:181‐194. [DOI] [PubMed] [Google Scholar]
- 12. Marrone G, Shah VH, Gracia‐Sancho J. Sinusoidal communication in liver fibrosis and regeneration. J Hepatol 2016;65:608‐617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Poisson J, Lemoinne S, Boulanger C, Durand F, Moreau R, Valla D, et al. Liver sinusoidal endothelial cells: physiology and role in liver diseases. J Hepatol 2017;66:212‐227. [DOI] [PubMed] [Google Scholar]
- 14. Dobie R, Wilson‐Kanamori JR, Henderson BEP, Smith JR, Matchett KP, Portman JR, et al. Single‐cell transcriptomics uncovers zonation of function in the mesenchyme during liver fibrosis. Cell Rep 2019;29:1832‐1847.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S, et al. Landscape of intercellular crosstalk in healthy and NASH liver revealed by single‐cell secretome gene analysis. Mol Cell 2019;75:644‐660.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Krenkel O, Hundertmark J, Ritz TP, Weiskirchen R, Single TF. Cell RNA sequencing identifies subsets of hepatic stellate cells and myofibroblasts in liver fibrosis. Cells 2019;8:503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Narita M, Oussoultzoglou E, Chenard MP, Fuchshuber P, Rather M, Rosso E, et al. Liver injury due to chemotherapy‐induced sinusoidal obstruction syndrome is associated with sinusoidal capillarization. Ann Surg Oncol 2012;19:2230‐2237. [DOI] [PubMed] [Google Scholar]
- 18. Meurer SK, Tihaa L, Lahme B, Gressner AM, Weiskirchen R. Identification of endoglin in rat hepatic stellate cells: new insights into transforming growth factor beta receptor signaling. J Biol Chem 2005;280:3078‐3087. [DOI] [PubMed] [Google Scholar]
- 19. Tabula Muris Consortium , Overall Coordination , Logistical Coordination , Organ Collection and Processing , Library Preparation and Sequencing , Computational Data Analysis , et al. Single‐cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018;562:367‐372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Elsheikh E, Younoszai Z, Otgonsuren M, Hunt S, Raybuck B, Younossi ZM. Markers of endothelial dysfunction in patients with non‐alcoholic fatty liver disease and coronary artery disease. J Gastroenterol Hepatol 2014;29:1528‐1534. [DOI] [PubMed] [Google Scholar]
- 21. Geraud C, Koch PS, Zierow J, Klapproth K, Busch K, Olsavszky V, et al. GATA4‐dependent organ‐specific endothelial differentiation controls liver development and embryonic hematopoiesis. J Clin Invest 2017;127:1099‐1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Halpern KB, Shenhav R, Massalha H, Toth B, Egozi A, Massasa EE, et al. Paired‐cell sequencing enables spatial gene expression mapping of liver endothelial cells. Nat Biotechnol 2018;36:962‐970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang L, Wang X, Xie G, Wang L, Hill CK, DeLeve LD. Liver sinusoidal endothelial cell progenitor cells promote liver regeneration in rats. J Clin Invest 2012;122:1567‐1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Scott CL, T'Jonck W, Martens L, Todorov H, Sichien D, Soen B, et al. The transcription factor ZEB2 is required to maintain the tissue‐specific identities of macrophages. Immunity 2018;49:312‐325.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vento‐Tormo R, Efremova M, Botting RA, Turco MY, Vento‐Tormo M, Meyer KB, et al. Single‐cell reconstruction of the early maternal–fetal interface in humans. Nature 2018;563:347‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ding BS, Cao Z, Lis R, Nolan DJ, Guo P, Simons M, et al. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature 2014;505:97‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Neill T, Buraschi S, Goyal A, Sharpe C, Natkanski E, Schaefer L, et al. EphA2 is a functional receptor for the growth factor progranulin. J Cell Biol 2016;215:687‐703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang B, Horvath S. A general framework for weighted gene co‐expression network analysis. Stat Appl Genet Mol Biol 2005;4:Article17. [DOI] [PubMed] [Google Scholar]
- 29. Molleken C, Sitek B, Henkel C, Poschmann G, Sipos B, Wiese S, et al. Detection of novel biomarkers of liver cirrhosis by proteomic analysis. Hepatology 2009;49:1257‐1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Barcena C, Stefanovic M, Tutusaus A, Joannas L, Menendez A, Garcia‐Ruiz C, et al. Gas6/Axl pathway is activated in chronic liver disease and its targeting reduces fibrosis via hepatic stellate cell inactivation. J Hepatol 2015;63:670‐678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Choudhury SR, Babes L, Rahn JJ, Ahn BY, Goring KR, King JC, et al. Dipeptidase‐1 is an adhesion receptor for neutrophil recruitment in lungs and liver. Cell 2019;178:1205‐1221.e17. [DOI] [PubMed] [Google Scholar]
- 32. Herrnberger L, Hennig R, Kremer W, Hellerbrand C, Goepferich A, Kalbitzer HR, et al. Formation of fenestrae in murine liver sinusoids depends on plasmalemma vesicle‐associated protein and is required for lipoprotein passage. PLoS One 2014;9:e115005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun 2013;4:2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dulai PS, Singh S, Patel J, Soni M, Prokop LJ, Younossi Z, et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: systematic review and meta‐analysis. Hepatology 2017;65:1557‐1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lackner C, Spindelboeck W, Haybaeck J, Douschan P, Rainer F, Terracciano L, et al. Histological parameters and alcohol abstinence determine long‐term prognosis in patients with alcoholic liver disease. J Hepatol 2017;66:610‐618. [DOI] [PubMed] [Google Scholar]
- 36. Lefebvre P, Lalloyer F, Bauge E, Pawlak M, Gheeraert C, Dehondt H, et al. Interspecies NASH disease activity whole‐genome profiling identifies a fibrogenic role of PPARalpha‐regulated dermatopontin. JCI Insight 2017;2:e92264. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material