

Abstract
High‐throughput sequencing analysis represented both a medical diagnosis and technological revolution. Gene panel analysis is now routinely performed in the exploration of hereditary predisposition to cancer, which is becoming increasingly heterogeneous, both clinically and molecularly. We present 1530 patients with suspicion of hereditary predisposition to cancer, for which two types of analyses were performed: a) oriented according to the clinical presentation (n = 417), or b) extended to genes involved in hereditary predisposition to adult cancer (n = 1113). Extended panel analysis had a higher detection rate compared to oriented analysis in hereditary predisposition to breast / ovarian cancer (P < .001) and in digestive cancers (P < .094) (respectively 15% vs 5% and 19.3%, vs 12.5%). This higher detection is explained by the inclusion of moderate penetrance genes, as well as the identification of incident mutations and double mutations. Our study underscores the utility of proposing extended gene panel analysis to patients with suspicion of hereditary predisposition to adult cancer.
Keywords: double mutation, HBOC, HNPCC, incidental findings ATM, panel sequencing, predisposition to cancer

1. INTRODUCTION
Hereditary predisposition to cancer is caused by constitutional deleterious mutations in a tumor suppressor gene or in an oncogene and is characterized by an increased risk of specific types of cancer compared to that of the general population. More than 80 genes have been identified as predisposing to different cancers. 1 Many of the high‐penetrance risk genes were identified using the Sanger sequencing and linkage analysis in the 1990's. 2
Next‐generation‐sequencing (NGS), which appeared in 2004 in the wake of the Human Genome Project, represented a technological and medical revolution. The impact of high‐throughput sequencing has been considerable for the molecular exploration of genetic disease, whether hereditary diseases with Mendelian transmission, or acquired genetic diseases such as cancer. 1
In hereditary predisposition to cancer, whole‐exome‐sequencing and whole‐genome‐sequencing have identified new genes implicated in colorectal cancer (POLE, POLD1, NTHL1, GREM1, RNF43), 3 uveal melanoma (BAP1) or pheochromocytoma (MAX). 4 NGS has also been useful in better defining the penetrance of cancer predisposition genes and the spectrum of associated tumors. Several elements have led to the establishment of gene panel analysis as the reference technique for molecular genetic diagnostics, notably the multiplication of genes identified as being associated with cancer risk, the molecular heterogeneity observed in different predisposing syndromes, as well as the superior sensitivity and falling cost of high‐throughput sequencing. 1
At least 15 genes are currently known to be involved in hereditary predisposition to breast and / or ovarian cancer (HBOC), 2 and at least 10 genes in colon cancer predisposition. 3 Tumor spectra overlap for different familial syndromes, making extended panel (EP) analysis medically appropriate. Here we present the results of cancer panel analysis of 1530 probands with an indication for oncogenetic analysis. Two types of analyses were proposed: the first was comprehensive analysis of 38–46 actionable genes involved in the hereditary predisposition to cancer, regardless of the syndrome of hereditary cancer suspected in the patient; the objective of this approach was to determine the interest of carrying out an EP to identify hereditary risk that would not have been identified by an oriented analysis in population. Alternatively, analysis of an oriented panel (OP) focused on genes directly involved in the hereditary predisposition to cancer suspected in the patient. EP or OP analysis was decided by the medical geneticist; patients not consenting to EP were also then proposed OP analysis.
2. MATERIALS AND METHODS
2.1. Samples
Between 2016 and 2018, 1530 patients consulting in our Oncogenetics Department underwent panel analysis for hereditary predisposition to cancer according to national or international recommendations if available, or according to data from the literature. Indications included HBOC, digestive, endocrine, renal, cutaneous and syndromic cancers (Figure 1). The syndromic cancer indication includes syndromes in which the clinical presentation, associated with particular disorders, is suggestive of a single gene, as well as patients with multiple cancers. An indication “other” includes patients who cannot be integrated into other indications. For the rare families with multiple indications, only the indication best describing the patient analyzed was considered in this study. Each patient signed informed consent for genetic diagnosis of hereditary disease. Two initial samples were collected for each patient: peripheral blood on EDTA, and a cheek swab transferred to FTA paper. DNA was extracted from peripheral blood using QIAamp DNA Blood maxikit (Qiagen). Additional samples for the confirmation of variants were requested as necessary.
FIGURE 1.
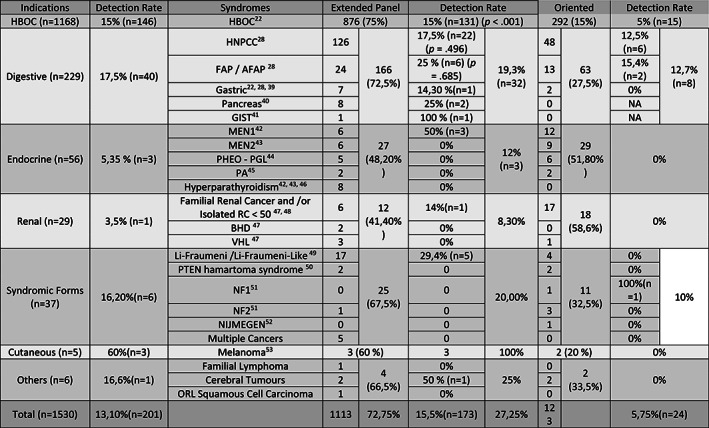
Indications of analysis, panel performed and detection rates. The detection rate is significantly higher in HBOC indication (P<.001), and higher in digestive indications. HBOC: Hereditary Breast and Ovarian Cancer, HNPCC: Hereditary Non Polyposis Colorectal Cancer, FAP / AFAP: Familial adenomatous polyposis / Attenuated Familial Adenomatous Polyposis, GIST: Gastro‐Intestinal Stromal Tumor, MEN1: Multiple endocrine neoplasia type 1, MEN2: Multiple endocrine neoplasia type 2, PHEO – PGL: Pheochromocytoma – Paraganglioma, PA: Pituitary Adenoma, BHD: Birt‐Hogg‐Dubé, VHL: Von Hippel Lindau, NF1: Neurobromatosis Type 1, NF2: Neurobromatosis Type 2
2.2. Panel sequencing
The 1113 patients were analyzed for a panel of diagnostic genes for which medical management can be proposed: 1037 for a first version of 38 genes, and 76 for a second version including 46 genes (Table 1). The 417 patients underwent analysis of a limited number of genes directly involved in the familial pathology (oriented panel, Table 2). The HBOC oriented panel (OP) did not include genes for which the cancer risk levels are controversial and not clearly associated with a relative risk >3, such as ATM or CHEK2. OP were designed by our laboratory according to the literature. All patients with a class 4 or 5 variant were given recommendations for health management.
TABLE 1.
Diagnostic genes
| Diagnostic genes in panel V1 | Additional genes in panel V2 | ||||
|---|---|---|---|---|---|
| APC | (LRG_130tl & t2) | PALB2 | (LRG_308) | AIP | (LRG_460) |
| ATM | (LRG_135) | PMS2 | (LRG_161) | CASR | (NM_000388.3) |
| BAP1 | (LRG_529) | POLD1 | (LRG_785 t1) | CDC73 | (LRG_507) |
| BMPR1A | (LRG_298) | POLE | (LRG_789) | CDK4 | (LRG_490) |
| BRCA1 | (LRG_292) | PTEN | (LRG_311) | FH | (LRG_504) |
| BRCA2 | (LRG_293) | RAD51C | (LRG_314) | MET | (LRG_662) |
| BRIP1 | (LRG_300) | RAD51D | (LRG_516) | MITF | (LRG_776) |
| CDH1 | (LRG_301) | RET | (LRG_518 t1) | NF1 | (LRG_214t1 & 2) |
| CDKN2A | (LRG_11t1 & t2) | SDHA | (NM_004168.3) | ||
| CHEK2 | (NM_007194.3) | SDHAF2 | (LRG_519) | ||
| EPCAM | (LRG_215) | SDHB | (LRG_316) | ||
| FLCN | (LRG_325) | SDHC | (LRG_317) | ||
| MAX | (LRG_530) | SDHD | (NM_003002.3) | ||
| MEN1 | (LRG_509 t2) | SMAD4 | (LRG_318) | ||
| MLH1 | (LRG_216) | STK11 | (LRG_319) | ||
| MSH2 | (LRG_218) | TMEM127 | (LRG_528) | ||
| MSH6 | (LRG_219) | TP53 | (LRG_321 t1) | ||
| MUTYH | (NM_001048171.1) | VHL | (LRG_322) | ||
| NBN | (LRG_158) | ||||
| NF2 | (LRG_S11 t1) | ||||
Note: The EP included 38 genes in the first version, and 46 genes in the second version. All genes were analyzed regardless of the pedigree presentation suggesting hereditary predisposition to cancer.
TABLE 2.
Oriented panels
| HBOC panel | Colorectal panel | PGL/PHC panel | Renal panel |
|---|---|---|---|
| BRCA1 | APC | MAX | FH |
| BRCA2 | EPCAM | RET | FLCN |
| PALB2 | MLH1 | SDHA | MET |
| MLH1 | MSH2 | SDHAF2 | SDHB |
| MSH2 | MSH6 | 5DHB | VHL |
| MSH6 | MUTYH | SDHC | MITF |
| PMS2 | PMS2 | SDHD | |
| EPCAM | POLD1 | TMEM127 | |
| RAD51C | POLE | VHL | |
| RAD51D | |||
| BRIP1 | |||
| PTEN | |||
| TP53 | |||
| CDH1 |
Note: Four OP were defined. The genes below the line were optional, and were analyzed according to the patient's personal and family history. For the HBOC panel, analysis of genes in green was performed if ovarian cancer was present. Genes in blue were analyzed for specific syndromes (PTEN hamartoma tumor syndrome, Li‐Fraumeni syndrome and diffuse gastric cancer syndrome). For the colorectal panel, genes involved in polyposis and/or HNPCC were analyzed according to the clinical presentation. MITF was analyzed if renal cancer was associated with melanoma or pancreatic cancer.
Whether tested for the full panel, or for an oriented subset of genes, all samples underwent the same fragmentation, capture, and sequencing protocol: data for unrequested genes were masked before bio‐informatic processing and interpretation.
Sonic fragmentation of DNA from peripheral blood was performed on a Bioruptor instrument (Diagenode). Kapa HTP library preparation and SeqCap EZ Choice probes and reagents (Roche) were used for library preparation and capture. Quality of fragmentation, library and capture were controlled using a Bioanalyzer 2100 instrument (Agilent). Sequencing was performed using Miseq v2 kit (300 cycles) on Miseq Instrument (Illumina). Each run included 21 patients. All steps were performed following providers' guidelines. Analysis of exons 11 to 15 of PMS2 and exons 1, 13, and 14 of SDHA, and quantitative analysis of WRN exon 10 was not performed, due to high identity with paralogs genes.
2.3. Bio‐informatic analysis
De‐multiplexing was performed using bcl2fastq2 Conversion Software (Illumina). Alignment was performed on University of California Santa Cruz human genome reference build 19 using the Burrows‐Wheeler Aligner. Genome Analysis Toolkit (GATK) and PICARD tools were used for base quality score recalibration (BaseRecalibrator) and realignment (RealignerTargetCreator, IndelRealigner), as recommended by Eurogentest guidelines (Matthijs et al., 2016). Variant calling was performed using GATK HaplotypeCaller and annotated using EnsemblVariantEffectPredictor. Copy number variation analysis was performed using ExomeDef. Variants were filtered for quality score ≥ 30, depth ≥ 50x, and present in ≥20% of reads.
2.4. Variant interpretation
Variant interpretation was aided using ALAMUT (Interactive BioSoftware), which includes splice site analysis tools (SpliceSiteFinder, MaxEntScan), protein‐function prediction tools (SIFT, Polyphen 2.0), and links to relevant databases (ClinVar, Leiden Open Variation Database [LOVD], other syndrome‐specific databases). Variants were classified according to the American College of Medical Genetics (ACMG) recommendations, 5 aided by the French National Database of variants (Groupe Génétique et Cancer). Only pathogenic or probably pathogenic variants (classes 4 and 5) were considered in this study.
2.5. Complementary analyses
All class 5 (pathogenic) or 4 (likely pathogenic) variants were confirmed on a second patient sample using techniques appropriate for the variant. Sanger sequencing was performed using a 3500xl instrument and BigDye terminator kit 3.1 (Applied Biosystems). Interpretation was performed using Seqman software (DNASTAR). copy number variation (CNV) were confirmed using QMPSF (quantitative multiplex PCR of short fragments). At least one probe within the CNV and if possible at least one probe in the same gene but outside the CNV, as well as control probes on other chromosomes were used in each multiplex mix. Interpretation was performed using GeneMapper software (Applied Biosystems). If the breakpoints of large deletions were identified in the NGS data, allele‐specific PCR and Sanger sequencing were used to confirm the CNV.
Variants likely to affect splicing were confirmed by RT‐PCR unless the classes 4 and 5 character of the variant was already well established in the literature or national databases. Blood drawn on PaxGene tubes was used for RNA extraction using PAXgene Blood RNA Kit (Qiagen). RNA was reverse‐transcribed using High Capacity RNA to cDNA kit (Applied Biosystems) and amplified for the region of interest. PCR products were gel‐purified if appropriate before sequencing.
Any exons with insufficient coverage depth (< 50x) in genes pertinent for the clinical presentation were analyzed for point mutations by Sanger sequencing and for CNV by QMPSF as described above.
2.6. Statistical analyses
For the HBOC and hereditary nonpolyposis colorectal cancer (HNPCC) cases, differences in the mutation detection rates between the EP and the OP were tested using Fisher's exact test. These differences in mutational detection rates between the two panels were also measured on the common genes in order to look for a potential confounding effect. The mutation rates of the ATM and BRCA2 genes observed in our study were compared to those of the general population using the exact binomial test. Statistical analyses were conducted with R software, version 3.6.1 (R‐Project, http://cran.r-project.org/). All statistical tests were two‐sided and with 5% significance level.
3. RESULTS
The indications for genetic testing were dominated by hereditary breast and ovarian cancer syndrome (HBOC) (n = 1168, 76.3%) and predisposition to digestive cancers (n = 229, 15%), followed by endocrine tumor predisposition (n = 56, 3.6%), syndromic predisposition to cancer (n = 37, 2.4%) and renal cancer predisposition (n = 29, 1.9%). Other indications were rare (Figure 1).
EP analysis was performed in 72.8% of cases overall, ranging from 41 to 75% depending on the indication for referral. OP were more frequently prescribed for rare or syndromic presentations, with as few as a single gene to be analyzed, except for Li‐Fraumeni and Li‐Fraumeni‐Like syndrome.
Two hundred one class 4 and 5 variants were identified and confirmed in 195 cases (Supplemental File 1). These variants were all observed in a heterozygous state, except for one homozygous mutation in MUTYH. The detection rate was 13.1% overall, with 15.5% for EP and 5.75% for OP.
3.1. Identification of class 4 and 5 variants by predisposition syndrome
3.1.1. HBOC cases
One hundred forty‐six class 4 and 5 variants were identified in HBOC patients (Figure 2, Supplemental File 1). Mutations in the major HBOC genes were the most common, including BRCA1 (n = 26, 17.8%), BRCA2 (n = 37, 25.4%), followed by ATM (n = 16, 11%), PALB2 (n = 11, 7.5%) and CHEK2 (n = 10, 6.8%).
FIGURE 2.
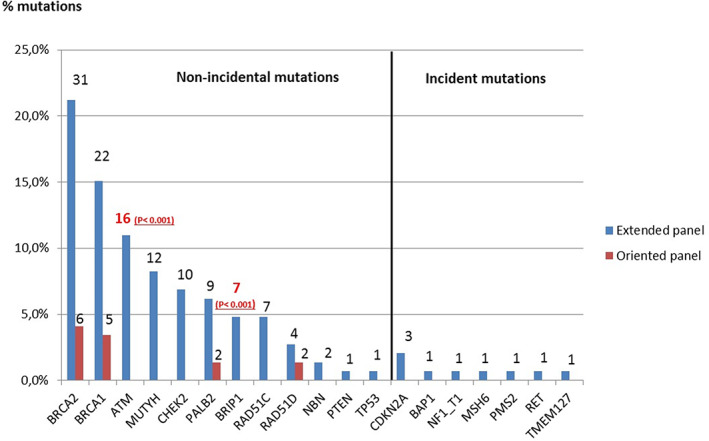
Genes with a pathogenic or probably pathogenic mutation identified in HBOC cases. Class 4 and 5 variants were identified using EP (blue) and OP (red) according to clinical presentation. ATM and BRIP1 have been associated significantly to HBOC patients in comparison to general population. Incidental findings include high penetrance genes CDKN2A, BAP1, NF1, MSH6, PMS2, RET and TMEM127. The heterozygous variants in MUTYH are not presented [Colour figure can be viewed at wileyonlinelibrary.com]
For the 1168 patients referred for HBOC, the EP was analyzed in 876 (75%), and all or part of the HBOC OP was analyzed in 292 (25%). Overall, the detection rate for class 4 and 5 variants in HBOC cases was 12.5%. The detection rate was 15% for EP analysis and 5% for OP, with a significant statistical difference (P < .001). The discovery of mutations in the major HBOC genes was comparable (8.5% in EP, 5% in OP, P = .074), while the EP more frequently uncovered mutations in other genes involved in hereditary predisposition to breast cancer (12.4%). The EP identified nine incidental mutations in BAP1, CDKN2A, MSH6, NF1, PMS2, RET, and TMEM127, and 12 heterozygous mutations in MUTYH.
3.2. Digestive cases
For the 229 patients referred for predisposition to digestive cancers, the EP was analyzed in 166 cases (72.5%), and an OP in 63 cases (27.5%). Forty class 4 and 5 variants were found (Figure 3, Supplemental File 2).
FIGURE 3.
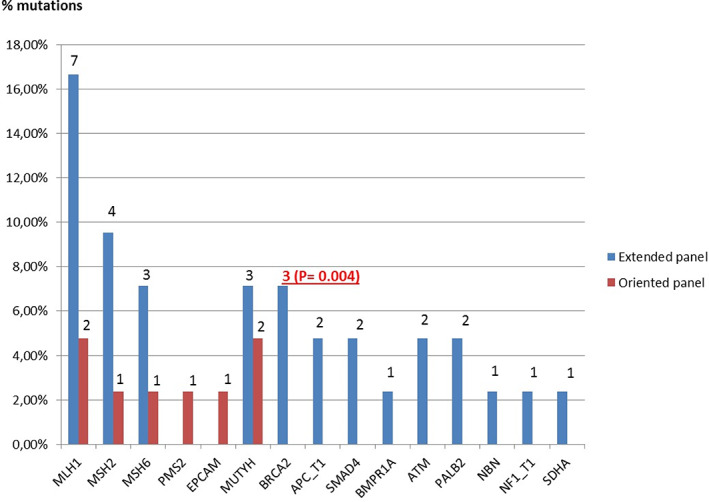
Genes with a pathogenic or probably pathogenic mutation identified in digestive cases. Class 4 and 5 variants were identified using EP (blue) and OP (red) according to clinical presentation. BRCA2 represents the third most frequently mutated gene. It is significantly associated to digestive indication. Others incidental findings included ATM, PALB2, NBN, NF1, and SDHA [Colour figure can be viewed at wileyonlinelibrary.com]
Overall, the mutation detection rate was 17.5%. The detection rate was 19.6% with the EP and 11.3% with an OP (P < .094). The difference is not significant, probably because the number of patients was not large enough here, unlike HBOC cases. The most common mutations were found in mismatch repair genes (MMR) (n = 20, 50%), including MLH1 (n = 9, 22.5%), MSH2 (n = 5, 12.5%), MSH6 (n = 4, 10%) PMS2 (n = 1, 2.5%) and EPCAM (n = 1, 2.5%). Beyond MLH1 and MSH2, the most frequently mutated genes were MUTYH (n = 5, with 1 homozygous mutation, 12.5%) and BRCA2 (n = 3, 7.5%). Mutations were also found in other known digestive cancer genes (APC, BMPR1A and SMAD4). Nine incidental findings were observed in BRCA2, PALB2, NF1, SDHA, and heterozygous NBN (22.5% of mutations in cases at risk of digestive cancer).
For HNPCC cases, the detection rate was 17.5% with the EP and 12.7% with an OP, but the difference was not significant (P = .496). The detection rate of mutations in MMR genes was comparable (13.5% and 12.5% respectively, P = 1). The difference of the overall mutation detection rate was thus due to incidental findings in ATM, BRCA2, PALB2, NBN, SDHA, and mutations in MUTYH (including one homozygote).
For PAF cases, the detection rate was 25% with the EP and 15.4% with an OP, with two heterozygous mutations in MUTYH (P = .685). EP analysis identified mutations in APC, BRCA2, BMPR1A, and SMAD4.
Other class 4 and 5 variants were identified in cases with stomach cancer (APC), pancreatic cancer (ATM, PALB2) and familial GIST (NF1), all by EP analysis.
3.3. Other cases
The proportion of OP analysis for other indications was 51.8% (n = 29) for neuroendocrine tumors, 58.6% (n = 18) for renal tumors, and 40% (n = 11) for syndromic indications. All mutations but one were found using EP. The detection rate was 12% for endocrine indications, with two mutations in MEN1 and one heterozygous mutation in MUTYH found in MEN1 patients. One mutation in ATM was found in a patient with renal carcinoma (Supplemental File 3).
Among syndromic indications, the detection rates were about 16.2%, with mutations found in TP53, BRCA1, and NBN using EP in Li‐Fraumeni patients, while one mutation was identified in NF1 using OP analysis. Mutations were also found in CDKN2A in patients with melanoma and in RAD51D in a patient with a brain tumor.
3.4. Incidental pathogenic mutations
The EP analysis identified class 4 and 5 variants in some genes unrelated to the clinical presentation. Excluding 15 cases with mono‐allelic pathogenic variants in MUTYH, incidental findings were found in 19 cases using EP, representing 10.9% of all mutations identified. Incidental findings in high penetrance genes concerned BRCA1, BRCA2, BAP1, CDKN2A, PALB2, PMS2, NF1, RET, and RAD51D. We observed bi‐allelic mutations in BRCA2 in a case with colorectal polyposis. Additional incidental findings concerned genes of indeterminate or moderate penetrance, including ATM, NBN, SDHA, and TMEM127.
3.5. Identification of double mutations
EP sequencing identified four patients with pathogenic variants in two distinct genes (Supplemental File 4). These double mutations including the following combinations: ATM / NBN, BRCA1 / NBN, BRCA2 / RAD51D and CHEK2 / MSH6. The first three were associated with a classic HBOC phenotype. The carrier of CHEK2 and MSH6 mutations presented with breast cancer at 40 years of age. Her familial branch with the CHEK2 mutation presented an HBOC phenotype, while the familial branch with the MSH6 mutation presented an HNPCC phenotype.
4. DISCUSSION
In the present study, 1530 consecutive patients for whom hereditary predisposition to cancer was suspected were analyzed by capture and sequencing of relevant gene regions. Regardless of personal and familial history, similar proportions of patients accepted testing of an EP in HBOC and digestive indications. Thus, if the choice is offered to them, most patients with these indications agree to an extensive analysis of actionable genes, for which medical management can be proposed in the event of the identification of a pathogenic variant, even if these variants are incidental. This proportion was stable over the recruitment period, but was somewhat dependent on the prescribing physician (data not shown), underlining the importance of the presentation by the physicians of the various choices proposed and their individual and familial consequences.
Our mutation detection rate, including class 4 and 5 variants, was 15.5% for EP analysis, which is comparable to similar studies of inherited cancer risk (8 to 15%). 1 , 6 , 7 , 8 , 9 Most mutations were identified in genes known to be associated with the personal and familial clinical presentation of patients. The BRCA1 and BRCA2 genes thus accounted for 40% of mutations observed in HBOC cases. For these patients, mutations were also observed in the ovarian cancer risk genes BRIP1, RAD51C, and RAD51D, as well as the breast cancer risk genes PALB2, TP53, PTEN, ATM, CHEK2 and NBN. While PALB2, PTEN, TP53, RAD51C, and RAD51D were included in targeted HBOC analysis, ATM, BRIP1, CHEK2, and NBN were only analyzed with the EP. The mutations identified in these latter four genes explain the higher detection rate using EP (15%) to OP (5%).
ATM emerged as a major contributor to hereditary breast cancer risk in our study. Frequent in the general population (0.3 to 0.6%), 10 mono‐allelic mutation carriers have a moderate risk of breast cancer (odds ratio [OR] 2.8). 11 , 12 Of the 19 heterozygous ATM mutations observed, 16 occurred in HBOC cases, representing the third most frequently mutated gene in HBOC, with a statistically higher prevalence than the general population 10 (1.8% in our study, P < .001). One ATM mutation was found in a familial pancreatic indication, and an increased risk of pancreatic cancer has been attributed to ATM. 13
NBN and CHEK2 mutations also confer moderate risk of breast cancer (relative risk [RR] 2 to 3). 12 , 14 , 15 , 16 NBN is less well‐studied, and specific rare mutations are known to cause Nijmegen Breakage Syndrome (NBS) when bi‐allelic. 17 Four heterozygous cases with null mutations not seen in NBS were observed: two in HBOC cases, one in a sarcoma patient, and one associated with colorectal cancer. The contribution of NBN to cancer risk and associated tumor spectrum needs further study. Mutations in CHEK2 were more frequent, with 10 HBOC cases presenting a mutation. No CHEK2 mutations were observed in colorectal or thyroid cancer cases, though increased risk of these cancers has been described for CHEK2. 18 , 19 The majority of mutations were the northern‐European c.1100delC allele, though we have previously observed a variety of stop, frameshift, and splicing mutations of this gene in HBOC families. 20 The prevalence of mutations in CHEK2 was not statistically higher than the general population in our study 16 (1.1% vs 0.7%, P = .148).
ATM, CHEK2, and NBN were not in our EP because of them penetrance. Indeed, a heterozygous mutation in these genes confers a moderate risk comparable to that induced by some first‐degree relative with breast cancer (RR of 3.3 if one first degree relative with breast cancer before the age of 50, RR of 2.9 if two first‐degree relatives with breast cancer). 6 , 21 Thus, their portion attributable in the occurrence of breast cancer in families at risk remains unclear.
Specific health management measures may proposed to patients with a mutation in one of these moderate risk genes, such as an annual MRI from the age of 40, 22 without interrupting the surveillance offered to first‐degree relatives of a patient with breast cancer, based on family history.
Moreover, ATM is causal for ataxia‐telangiectasia, a severe autosomal recessive hereditary disease involving progressive cerebellar degeneration, immunodeficiency, very high cancer risk, and extreme radiosensitivity. 23 The identification of heterozygous mutation carriers, which is frequent, could identify families at risk of ataxia‐telangiectasia, and represent an opportunity to provide appropriate genetic counseling.
Concerning ovarian cancer risk, BRIP1 mutations confer a moderate risk of ovarian cancer (RR = 3.41) 24 that is comparable to a first‐degree relative risk (RR = 2.56 to 3.83). 25 Thus, the management of ovarian cancer risk for the first‐degree relatives of a patient with an ovarian cancer should be based on family history but not on BRIP1 mutational status. Specific management could be proposed to BRIP1 mutation carriers without a first‐degree relative with ovarian cancer. The prevalence of mutations in BRIP1 was statistically higher than the general population in HBOC cases in our study 24 (0.8% vs 0.1%, P < .001).
Six out of seven mutations in BRIP1 were identified in our study in patients with breast cancer, with no personal or familial history of ovarian cancer. Thus, the involvement of BRIP1 in breast cancer risk needs further investigation.
The significantly higher detection rate for EP analysis of HBOC cases is also partly explained by incidental findings (6.8% of mutations of the EP in HBOC) and heterozygous mutation of MUTYH (8.1% of mutations of the EP in HBOC).
The frequency of mono‐allelic mutation carrier in MUTYH in the general population is high, 1 to 2%, comparable that the frequency in our study. 26 It represents the fourth most frequent mutated gene in HBOC. Digestive cancer risk is well described for persons with bi‐allelic mutations of this gene, while the significance of a single mutated allele is less clear. 27 The interest of the identification of a mono‐allelic mutation would be to propose to the carriers with a first‐degree related with colon cancer, an adapted screening of this cancer from 40 years old using colonoscopy every 5 years, 28 and to carry out a genetics investigation at the spouse in order to offer appropriate family genetic counseling. However, according to the Hardy–Weinberg model, 29 the probability of a mono‐allelic carrier of MUTYH mutation giving birth to a child with a bi‐allelic mutation in this gene is rare, estimated at 0.4%. Moreover, the only bi‐allelic mutation in MUTYH in our study was identified in an HNPCC indication. Thus, the interest of the inclusion of this gene outside a “colon cancer” indication remains low.
Using EP, incidental mutations were found in 1% of patients presenting HBOC, representing 7% of mutations identified in this context. Thus, the identification of an incidental finding in HBOC indication is rare. However, these incidental findings were all observed in high penetrance genes (BAP1, CDKN2A, MSH6, NF1, PMS2, RET, TMEM127), which have a significant medical impact, and for which appropriate medical care must be offered. These mutations would never have been identified by OP.
As genetic testing for cancer predisposition becomes more widespread, and panel‐based testing becomes the norm, the tumor spectra associated with different predisposition genes has appeared to broaden. Thus, some mutations have been identified in known ovarian cancer genes as RAD51C, RAD51D or BRIP1 2 in breast cancer families. In addition, a mutation in the TP53 gene was found in a 28‐year‐old female patient with isolated breast cancer. Despite analysis of TP53 now being standard for women with breast cancer occurring before 31 years old, 30 the frequency of TP53 mutation without personal or familial history of Li‐Fraumeni syndrome tumors is <1%. 31
This phenotype ‐ genotype dissociation was also observed in patients referred for predisposition to cancers of the digestive tract, where BRCA2 was the most frequently mutated gene after MLH1 and MSH2. Two brothers presenting attenuated familial adenomatous polyposis where shown to have bi‐allelic mutations in BRCA2 (one pathogenic, one probably pathogenic). No signs of Fanconi Anemia (FA) were observed on retrospective analysis of these patients. 32 A pathogenic variant in BRCA2 was also found in an HNPCC patient who developed colon cancer at 39 years of age, in association with a variant of unknown significance on the other allele of BRCA2.
These finding are consistent with reports in the literature of bi‐allelic BRCA2 mutations associated not with FA but with colorectal cancer, 33 and some indications that support BRCA2 as contributing to familial colorectal cancer type X (FCC‐X), a hereditary nonpolyposis colorectal cancer syndrome. 34 On the other hand, a meta‐analysis of 18 studies of BRCA‐associated cancers did not identify significant risk of colorectal cancer for BRCA2 mutations. 35
As expected, mutations for digestive indications were most frequently found in the MMR genes. However, incidental findings represented 25% of mutations found in these cases, including moderate penetrance genes (ATM, NBN and SDHA) and high penetrance genes (BRCA2, PALB2, NF1), contributing to a higher detection rate in the expanded panel (19.3%) in comparison to OP analysis (12.7%). The identification of mutations in genes involved in rare syndromes (SMAD4, BMPR1A), or in a digestive gene with an unexpected clinical presentation (one bi‐allelic mutation in MUTYH in a HNPCC patient without polyposis) also increased the detection rate for the EP. The mutation rate of BRCA2 in digestive cases was statistically higher than that observed in the general population 36 (1.9% vs 0.2%, P = .004), suggesting that BRCA2 mutations were associated with digestive cases in our study.
Nine percent of patients presented with indications other than HBOC or digestive cancers. EP identified some mutations in unexpected genes, including high penetrance (BRCA1, RAD51C) and moderate penetrance genes (ATM). As the numbers of each indication are small, no conclusion can be proposed. However, it is surprising that no mutations were found in patients with medullary thyroid carcinoma (n = 15), for whom a mutation in the RET gene is classically described in 25% of cases, 37 or in patients with paraganglioma ‐ pheochromocytoma (n = 11), for whom a hereditary genetic cause is identified in 40% of cases. 38
The EP identified double mutations in four patients. Rare (0.5% of individuals analyzed using EP), these identifications report phenotypic presentation of some double mutations, and offer to propose an adapted genetic counseling to the two parental branches of the proband concerned by a hereditary risk of cancer, which could not have been identified by an OP.
5. CONCLUSION
Our study demonstrates that an EP of 38–46 genes improved the detection rate of class 4 and 5 variants in patients suspected to have a genetic predisposition to adult cancer, significantly in HBOC indication, compared to a panel analysis of 1 to 13 genes oriented according to personal and family history.
The inclusion of moderate penetrance genes, which is somewhat controversial, contributed to the significant high discovery rate in HBOC patients, in particular ATM (P < .001), CHEK2, BRIP1 and NBN which represented 27.3% of identified mutation. Being associated with a moderate risk of cancer, a specific management can be offered to the mono‐allelic mutation carriers, without however interrupting the care of the first degree relatives with a RR of cancer ≥3 based on familial history.
More extensive testing increases the likelihood of identifying incidental findings, in particular in digestive indications. In our series, 10.9% of mutations identified (25% in digestive indications) using the expanded panel (not including heterozygous MUTYH mutations) were not directly linked to the patient's pathology or associated with atypical presentation. Because these mutations are most often present in high penetrance genes, their identification has a strong medical impact for taking care of patients and their families. Moreover, almost all of the identification of classes 4 and 5 variants in a gene with moderate penetrance to modify the management of patients.
Routine analysis of EP such as ours could refine the tumor spectrum associated with a gene, such as digestive cancer with BRCA2 mutations, or breast cancer with BRIP1, RAD51C or RAD51D mutations. The impact of double mutations on cancer risk and type may also become clearer as more cases are described.
Finally, an improvement in the detection rate by extensive panel analysis, whether through the inclusion of moderately penetrated genes, rare genes, or the identification of incident mutations, determines the hereditary cancer to which the patient and their families are exposed, based on their history and genetics as part of personalized medicine.
CONFLICT OF INTEREST
The authors declare non conflict of interest. The study has been approved by personal protection committee (IRB: 2020 / CE57)
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/cge.13864.
Supporting information
Supplemental File 1: Variants identified in hereditary breast and ovarian cancer syndrome indication
Supplemental File 2: Variants identified in digestive indications
Supplemental File 3: Variants identified in others indications than hereditary breast and ovarian cancer syndrome and digestive indications
Supplemental File 4: Double mutations identified in our study
ACKNOWLEDGEMENTS
The authors thank Centre Jean Perrin. The samples used in this study were conserved in the Biological Resource Center of Jean Perrin Comprehensive Cancer Center, identified under No. BB‐0033‐00075 (Clermont‐ Ferrand, France)
Cavaillé M, Uhrhammer N, Privat M, et al. Feedback of extended panel sequencing in 1530 patients referred for suspicion of hereditary predisposition to adult cancers. Clinical Genetics. 2021;99:166–175. 10.1111/cge.13864
DATA AVAILABILITY STATEMENT
All variants of interest identified in this study are in supplemental file. Other data are available on request from the authors upon reasonable request.
REFERENCES
- 1. Kamps R et al. Next‐generation sequencing in oncology: genetic diagnosis, risk prediction and cancer classification. Int J Mol Sci. 2017;18(2):308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nielsen FC, van Overeem Hansen T, Sørensen CS. Hereditary breast and ovarian cancer: new genes in confined pathways. Nat. Rev Cancer. 2016;16:599‐612. [DOI] [PubMed] [Google Scholar]
- 3. Valle L. Recent discoveries in the genetics of familial colorectal cancer and polyposis. Clin. Gastroenterol. Hepatol. 2017;15:809‐819. [DOI] [PubMed] [Google Scholar]
- 4. Sokolenko AP et al. Identification of novel hereditary cancer genes by whole exome sequencing. Cancer Lett. 2015;369:274‐288. [DOI] [PubMed] [Google Scholar]
- 5. Richards S et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17:405‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Susswein LR et al. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next‐generation cancer panel testing. Genetics Med. 2016;18:823‐832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baert‐Desurmont S et al. Optimization of the diagnosis of inherited colorectal cancer using NGS and capture of exonic and intronic sequences of panel genes. Eur. J. Hum. Genet. 2018;26:1597‐1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Neben CL et al. Multi‐gene panel testing of 23,179 individuals for hereditary cancer risk identifies pathogenic variant carriers missed by current genetic testing guidelines. J Mol Diagnostics. 2019;21:646‐657. [DOI] [PubMed] [Google Scholar]
- 9. LaDuca H et al. A clinical guide to hereditary cancer panel testing: evaluation of gene‐specific cancer associations and sensitivity of genetic testing criteria in a cohort of 165,000 high‐risk patients. Genet Med. 2020;22:407‐415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Paglia LL et al. ATM germline mutations in women with familial breast cancer and a relative with haematological malignancy. Breast Cancer Res Treat. 2010;119:443‐452. [DOI] [PubMed] [Google Scholar]
- 11. Easton DF et al. Gene‐panel sequencing and the prediction of breast‐cancer risk. N Engl J Med. 2015;372:2243‐2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Couch FJ et al. Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol. 2017;3:1190‐1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Roberts NJ et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2012;2:41‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Australia, C. Risk factors for breast cancer: A review of the evidence 2018. Available at: https://canceraustralia.gov.au/publications-and-resources/cancer-australia-publications/risk-factors-breast-cancer-review-evidence-2018
- 15. Bogdanova N et al. Nijmegen breakage syndrome mutations and risk of breast cancer. Int. J. Cancer. 2008;122:802‐806. [DOI] [PubMed] [Google Scholar]
- 16. Zhang B, Beeghly‐Fadiel A, Long J, Zheng W. Genetic variants associated with breast‐cancer risk: comprehensive research synopsis, meta‐analysis, and epidemiological evidence. Lancet Oncol. 2011;12:477‐488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pastorczak A, Szczepanski T, Mlynarski W. Clinical course and therapeutic implications for lymphoid malignancies in Nijmegen breakage syndrome. Eur J Med Genet. 2016;59:126‐132. [DOI] [PubMed] [Google Scholar]
- 18. Gronwald J et al. Cancer risks in first‐degree relatives of CHEK2 mutation carriers: effects of mutation type and cancer site in proband. Br. J. Cancer. 2009;100:1508‐1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Siołek M et al. CHEK2 mutations and the risk of papillary thyroid cancer. Int. J. Cancer. 2015;137:548‐552. [DOI] [PubMed] [Google Scholar]
- 20. Desrichard A, Bidet Y, Uhrhammer N, Bignon Y‐J. CHEK2 contribution to hereditary breast cancer in non‐BRCA families. Breast Cancer Res. 2011;13:R119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Collaborative Group on Hormonal Factors in Breast Cancer . Familial breast cancer: collaborative reanalysis of individual data from 52 epidemiological studies including 58,209 women with breast cancer and 101,986 women without the disease. Lancet. 2001;358:1389‐1399. [DOI] [PubMed] [Google Scholar]
- 22. NCCN Guidelines Insights . Genetic/familial high‐risk assessment: breast and ovarian. NCCN Clinic Practice Guidelines Oncol. 2019;3:2019. [Google Scholar]
- 23. Rothblum‐Oviatt C et al. Ataxia telangiectasia: a review. Orphanet J Rare Dis. 2016;11(1):159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ramus SJ et al. Germline mutations in the BRIP1, BARD1, PALB2, and NBN genes in women with ovarian cancer. J. Natl. Cancer Inst. 2015;107(11):djv214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jervis S et al. Ovarian cancer familial relative risks by tumour subtypes and by known ovarian cancer genetic susceptibility variants. J. Med. Genet. 2014;51:108‐113. [DOI] [PubMed] [Google Scholar]
- 26. Hegde M et al. ACMG technical standards and guidelines for genetic testing for inherited colorectal cancer (lynch syndrome, familial adenomatous polyposis, and MYH‐associated polyposis). Genetics in Medicine. 2014;16:101‐116. [DOI] [PubMed] [Google Scholar]
- 27. Win AK et al. Risk of colorectal cancer for carriers of mutations in MUTYH, with and without a family history of cancer. Gastroenterology. 2014;146:1208‐1211.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gupta S et al. NCCN guidelines insights: genetic/familial high‐risk assessment: colorectal, version 3.2017. J Natl Compr Canc Netw. 2017;15:1465‐1475. [DOI] [PubMed] [Google Scholar]
- 29. Zhou JJ, Lange K, Papp JC, Sinsheimer JS. A heterozygote‐homozygote test of hardy‐Weinberg equilibrium. Eur J Hum Genet. 2009;17:1495‐1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kratz CP et al. Cancer screening recommendations for individuals with li‐Fraumeni syndrome. Clin. Cancer Res. 2017;23:e38‐e45. [DOI] [PubMed] [Google Scholar]
- 31. Bakhuizen JJ et al. TP53 germline mutation testing in early‐onset breast cancer: findings from a nationwide cohort. Fam. Cancer. 2019;18(2):273–280. 10.1007/s10689-018-00118-0. [DOI] [PubMed] [Google Scholar]
- 32. Gay‐Bellile M et al. Is BRCA2 involved in early onset colorectal cancer risk. Clin Genet. 2019;97(4):668–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Degrolard‐Courcet E et al. Development of primary early‐onset colorectal cancers due to biallelic mutations of the FANCD1/BRCA2 gene. Eur. J. Hum. Genet. 2014;22:979‐987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Garre P et al. BRCA2 gene: a candidate for clinical testing in familial colorectal cancer type X. Clinical Genetics. 2015;87:582‐587. [DOI] [PubMed] [Google Scholar]
- 35. Oh M et al. BRCA1 and BRCA2 gene mutations and colorectal cancer risk: systematic review and meta‐analysis. J Natl Cancer Inst. 2018;110:1178‐1189. [DOI] [PubMed] [Google Scholar]
- 36. Balmaña J, Díez O, Rubio IT, Cardoso F. BRCA in breast cancer: ESMO clinical practice guidelines. Ann Oncol. 2011;22:vi31‐vi34. [DOI] [PubMed] [Google Scholar]
- 37. Accardo G et al. Genetics of medullary thyroid cancer: an overview. International Journal of Surgery. 2017;41:S2‐S6. [DOI] [PubMed] [Google Scholar]
- 38. Turchini J, Cheung VKY, Tischler AS, De Krijger RR, Gill AJ. Pathology and genetics of phaeochromocytoma and paraganglioma. Histopathology. 2018;72:97‐105. [DOI] [PubMed] [Google Scholar]
- 39. Oliveira C, Pinheiro H, Figueiredo J, Seruca R, Carneiro F. Familial gastric cancer: genetic susceptibility, pathology. And Implications for Management. Lancet Oncol. 2015;16:e60‐e70. [DOI] [PubMed] [Google Scholar]
- 40. Matsubayashi H et al. Familial pancreatic cancer: concept, management and issues. World J Gastroenterol. 2017;23:935‐948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Niinuma T, Suzuki H, Sugai T. Molecular characterization and pathogenesis of gastrointestinal stromal tumor. Transl Gastroenterol Hepatol. 2018;3:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Thakker RV et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J. Clin. Endocrinol. Metab. 2012;97:2990‐3011. [DOI] [PubMed] [Google Scholar]
- 43. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid. 2015;6:567‐610. https://pubmed-ncbi-nlm-nih-gov.proxy.insermbiblio.inist.fr/25810047/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lenders JWM et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014;99:1915‐1942. [DOI] [PubMed] [Google Scholar]
- 45. Beckers A, Aaltonen LA, Daly AF, Karhu A. Familial isolated pituitary adenomas (FIPA) and the pituitary adenoma predisposition due to mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene. Endocr Rev. 2013;34:239‐277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cristina E‐V, Alberto F. Management of familial hyperparathyroidism syndromes: MEN1, MEN2, MEN4, HPT‐jaw tumour, familial isolated hyperparathyroidism, FHH, and neonatal severe hyperparathyroidism. Best Pract. Res. Clin. Endocrinol. Metab. 2018;32:861‐875. [DOI] [PubMed] [Google Scholar]
- 47. Haas NB, Nathanson KL. Hereditary renal cancer syndromes. Adv Chronic Kidney Dis. 2014;1:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schmidt LS, Linehan WM. Genetic predisposition to kidney cancer. Semin Oncol. 2016;43:566‐574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bougeard G et al. Revisiting li‐Fraumeni syndrome from TP53 mutation carriers. J. Clin. Oncol. 2015;33:2345‐2352. [DOI] [PubMed] [Google Scholar]
- 50. Cavaillé M et al. Early onset multiple primary tumors in atypical presentation of Cowden syndrome identified by whole‐exome‐sequencing. Front Genet. 2018;9:353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kresak JL, Walsh M. Neurofibromatosis: a review of NF1, NF2, and Schwannomatosis. J Pediatr Genet. 2016;5:98‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Walsh MF et al. Recommendations for childhood cancer screening and surveillance in DNA repair disorders. Clin Cancer Res. 2017;23:e23‐e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Avril M‐F et al. Recommendations for genetic testing and management of individuals genetically at‐risk of cutaneous melanoma. Ann Dermatol Venereol. 2015;142:26‐36. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental File 1: Variants identified in hereditary breast and ovarian cancer syndrome indication
Supplemental File 2: Variants identified in digestive indications
Supplemental File 3: Variants identified in others indications than hereditary breast and ovarian cancer syndrome and digestive indications
Supplemental File 4: Double mutations identified in our study
Data Availability Statement
All variants of interest identified in this study are in supplemental file. Other data are available on request from the authors upon reasonable request.