

Abstract
The genetic causes of multiple congenital anomalies are incompletely understood. Here, we report novel heterozygous predicted loss-of-function (LoF) and predicted damaging missense variants in the WW domain binding protein 11 (WBP11) gene in seven unrelated families with a variety of overlapping congenital malformations, including cardiac, vertebral, tracheo-esophageal, renal and limb defects. WBP11 encodes a component of the spliceosome with the ability to activate pre-messenger RNA splicing. We generated a Wbp11 null allele in mouse using CRISPR-Cas9 targeting. Wbp11 homozygous null embryos die prior to E8.5, indicating that Wbp11 is essential for development. Fewer Wbp11 heterozygous null mice are found than expected due to embryonic and postnatal death. Importantly, Wbp11 heterozygous null mice are small and exhibit defects in axial skeleton, kidneys and esophagus, similar to the affected individuals, supporting the role of WBP11 haploinsufficiency in the development of congenital malformations in humans. LoF WBP11 variants should be considered as a possible cause of VACTERL association as well as isolated Klippel-Feil syndrome, renal agenesis or esophageal atresia.
Introduction
Congenital malformations affect 3–6% of live human births and are associated with higher rates of fetal demise (1). Malformations occur in isolation or in combination, aggravating the burden of disease on families and society. In 80% of cases, the underlying etiology is unknown (2). A genetic diagnosis enables surveillance for other complications, recurrence prevention and targeted treatment.
Historically, pathogenic variants have been identified by studying families or groups of individuals who display similar phenotypic features that then typify a syndrome. Multiple congenital anomalies have been associated with pathogenic variants in pleiotropic genes involved in cellular processes such as pre-messenger RNA (mRNA) splicing (3), DNA stability (4,5), gene expression (6,7) or nicotinamide adenine dinucleotide (NAD) synthesis (8,9). In the absence of a discernible syndromic diagnosis and if three or more of vertebral, anal, cardiac, tracheo-esophageal (TE), renal or limb anomalies occur in an individual, then a diagnosis of VACTERL association may be made (10). VACTERL association is epitomized by its diversity of clinical features and causative factors, most of which are still unknown. While cases of VACTERL association lack a syndromic diagnosis, genetic factors contribute to its causation (11–13).
Over the years, massively parallel sequencing has introduced new approaches to gene discovery, including the development of global communities sharing information about novel variants in uncharacterized genes, enabling the identification of additional cases with variants in those genes and characterization of associated phenotypic features.
Here, we present individuals from seven unrelated families, with anomalies in one or more organs, each found to have a predicted loss-of-function (LoF), truncating or predicted damaging missense variant in the WW domain binding protein 11 (WBP11, MIM 618083) gene. Variants in WBP11 have not previously been associated with human disease. WBP11 (also known as NpwBP, SIPP1, SNP70 and PPP1R165) is a recognized component of the splicing machinery and colocalizes with SC35 in nuclear speckles (14), the site of splicing assembly, storage and regulation (15). WBP11 copurifies with complex B as part of the PRP19 complex of the spliceosome (16,17), a dynamic ribonucleoprotein (RNP) complex responsible for the removal of non-coding segments, exon ligation (coding segments) and regulation of differential/alternative splicing of precursor mRNA (18). We targeted the Wbp11 locus in mouse, confirming a link between WBP11 LoF and congenital disease. Our findings indicate that WBP11 haploinsufficiency causes a pleiotropic malformation syndrome affecting the cardiac, skeletal, gastrointestinal and renal systems.
Results
Predicted damaging WBP11 variants are the top candidates for disrupting embryogenesis in patients with related developmental anomalies
Through exome sequencing by several groups investigating patient cohorts with vertebral malformations, syndromic esophageal atresia (EA), renal hypodysplasia or multiple congenital anomalies (see Supplemental Note: Case Reports), novel predicted damaging variants in the WBP11 gene were identified independently as the variants most likely to cause disease. Phenotypic and mutational comparisons were subsequently facilitated via GeneMatcher (19).
Novel variants predicted to truncate WBP11 were identified in two unrelated families (Families 1 and 2, Table 1) through exome sequencing of an extended vertebral malformation cohort. Patient 3 had clinical trio-exome sequencing to determine the cause of his multiple congenital anomalies. The predicted truncating variant in Family 4 was identified as the part of an ongoing effort to determine the causes of syndromic forms of EA. Patients 5 and 6 were from a cohort of patients with renal hypodysplasia. Patient 7 and her unaffected mother underwent exome sequencing to investigate her congenital anomalies, short stature and microcephaly. In total, 13 affected individuals from seven unrelated families had a WBP11 variant considered as the top candidate in each case. Each of these variants is absent from the population databases. Of these, five WBP11 variants are predicted to be truncating, four of which are expected to result in LoF alleles as transcripts including them will be subject to exon junction complex (EJC)-promoted nonsense-mediated decay (20). LoF variants in WBP11 are underrepresented in the gnomAD database [probability of being LoF-intolerant (pLI), score = 1, with 1 LoF variant observed and 32 expected] (21), which is consistent with the hypothesis that heterozygous LoF WBP11 alleles are pathogenic. Transcripts including the c.1559dup p.(Gly521TrpfsTer28) variant (Patient 3) should escape the EJC-promoted nonsense-mediated decay as the variant is in the final exon (Fig. 1A), and therefore are likely to express a WBP11 protein lacking 121 amino acids from the C-terminus. Finally, Patient 6 harbored a novel missense variant c.169A>G p.(Met57Val), which was predicted to be damaging by the combined annotation dependent depletion (CADD) and Polyphen-2 metrics (Supplementary Material, Table S1). A methionine at this position in WBP11 orthologues is conserved in mammals, amphibians and fish.
Table 1.
Heterozygous predicted damaging WBP11 variants identified in patients with overlapping organ defects
| Patient | 1A | 1B | 2 | 3 | 4 | 4A | 4B | 4C | 4D | 5 | 6 | 7 | 7A |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WBP11 variant NM_016312.2 | c.280C > T p.(Arg94Ter) | c.280C > T p.(Arg94Ter) | c.484C > T p.(Gln162Ter) | c.1559dup p.(Gly521TrpfsTer28) | c.688C > T p.(Arg230Ter) | c.688C > T p.(Arg230Ter) | c.688C > T p.(Arg230Ter) | c.688C > T p.(Arg230Ter) | c.688C > T p.(Arg230Ter) | c.612del p.(Gly205ValfsTer19) | c.169A > G p.(Met57Val) | c.612del p.(Gly205ValfsTer19) | c.612del p.(Gly205ValfsTer19) |
| Inheritance | Maternal | De novo | De novo | U | Maternal | U | U | Presumed paternal | |||||
| Gender | M | M | F | M | F | M | M | F | F | M | F | F | F |
| Growth | Birth at 40 WG, W: 2.67 kg (−2 SD). Short stature | Birth at 42 WG, W: 3.69 kg (−0.2 SD) | W, H: U | History of short stature. Normal at 12 years 10 months, W: 35.1 kg (−1.4 SD), H: 143.1 cm (−1.7 SD) | H: 157 cm (−0.7 SD), HC: 52.5 cm (−2 SD). | TOP at 27 WG, W: ~−1 SD, H, HC: 0 SD | At 11 years, W: 0 SD, H: +1.5 SD, HC: −2 SD | Fetal death at 16 WG, HC: 0 SD | Birth at 28 WG, W: −1.6 SD, H: −1 SD, HC: −1.5 SD. At 2 years, W: −2.5 SD, H: −1.5 SD, HC: −2.5 SD. At 8 years, W: −1.5 SD, H: normal, HC: −2 SD | U | W, H, HC: < − 2 SD. At 23 months, H: 73 cm (−4.9 SD) | Birth at 39 WG, W: 2.498 kg (−2 SD). At age 11 years 9 months, W: 26 kg (−2.6 SD), H: 129.8 cm (−2.6 SD), HC: 50 cm (−2.4 SD) | U |
| Cardiac, 8/13 | - | PS, resolved by age 7 years | VSD | TAPVR, VSD, vascular ring, abnormal subclavian artery morphology | U | Hypoplastic left ventricle. Small VSD. Single atrium with anomalous pulmonary venous drainage. BAV. Interrupted aortic arch. | - | Muscular VSD | Muscular VSD in infancy, resolved by age 1 year | U | ASD | PFO, VSD, PDA that resolved spontaneously | U |
| Gastrointestinal, 7/13 | - | - | - | TEF, EA. Duodenal atresia | U | TEF, EA | - | TEF, EA | TOF, EA. Persistent feeding difficulties | U | TEF, EA | TEF, EA | Duodenal atresia |
| Vertebral, 6/13 | C3-C4, C6-C7 block vertebrae; C5 spina bifida occulta; bilateral omovertebral bones from C5; mild spinal canal stenosis at C4-C5 and C5-C6 | T12 butterfly vertebra, congenital scoliosis | C6-C7 fused cervical vertebrae. T1, T4 butterfly vertebrae. Left T2 fused thoracic vertebra. Abnormality of the right upper ribs | C1-C3, C4-C6 fused cervical vertebrae. T8 butterfly vertebra | U | U | U | U | Hypoplasia of first costal arch | U | U | Abnormal L5 vertebra | U |
| Other musculoskeletal, 5/13 | Sprengel deformity suggested by scapular elevation | Mild pectus excavatum | - | - | U | - | Sprengel deformity. Reduced mobility of left shoulder | U | Sprengel deformity—right thoraco-scapular aplasia | U | U | - | Sprengel deformity |
| Limb, 5/13 | Clinodactyly of the fifth finger | - | - | Unilateral abnormality of radial rayand left thumb morphology | Tapered fingers. Hypoplasia of the right thumb with absent phalangeal crease | Deviation of toes, hypoplasia of the toenails | Bilateral hypoplasia of the thumbs, camptodactyly of fifth fingers, unilateral camptodactyly of third and fourth fingers | - | - | U | U | - | U |
| Renal, 5/13 | Right enlarged kidney (mild) | - | - | - | U | - | U | Right renal agenesis; left small, pelvic kidney | Relatively small kidneys | Renal agenesis | Renal agenesis | - | U |
| Cerebral and/or neurodevelopmental, 4/13 | - | Suspected ADHD | Microcephaly. DD | Mild ID | - | NR | - | NR | - | U | - | Agenesis of the corpus callosum, ventriculomegaly on head ultrasound at birth. Global DD, learning difficulties, ADHD | U |
| Craniofacial, 7/13 | Facial asymmetry. Preauricular pit. Low posterior hairline; slightly webbed neck | Broad forehead; deeply set eyes | Choanal atresia | Submucous cleft palate. Velopharyngeal insufficiency. No facial dysmorphism | - | Low-set, dysplastic right ear. Retrognathia. Wide nose | Right preauricular skin tags, dysplastic and anteverted ears | - | Plagiocephaly. Asymmetric crying face | U | - | - | U |
| Other | Mega cisterna magna, normal variant. Decreased testicular size, resolved. History of delayed puberty. Café au lait spot on back | Hypopigmented macule on left arm | - | Cryptorchidism. Stable conductive hearing impairment. Low CD19 and CD8 cells, hypogammaglobulinemia; no recurrent infections | - | Hypertrichosis | - | - | - | - | Right inguinal hernia | U | |
Total number of patients with defects indicated under each organ system, over total number of affected patients. F, female; M, male. +; present; −, no abnormality detected; NR, not relevant; U, unknown; W, weight; H, height/length; HC, head circumference; cm, centimeters; kg, kilograms; SD, standard deviation; C, cervical; T, thoracic; L, lumbar; ASD, atrial septal defect; BAV, bicuspid aortic valve; PDA, patent ductus arteriosus, PFO: patent foramen ovale; PS, pulmonic stenosis; TAPVR, total anomalous pulmonary venous return; VSD, ventricular septal defect; TEF, tracheo-esophageal fistula; EA, esophageal atresia; ADHD, attention deficit hyperactivity disorder; DD, developmental delay; ID, intellectual disability; TOP, termination of pregnancy; WG, weeks of gestation.
Figure 1.
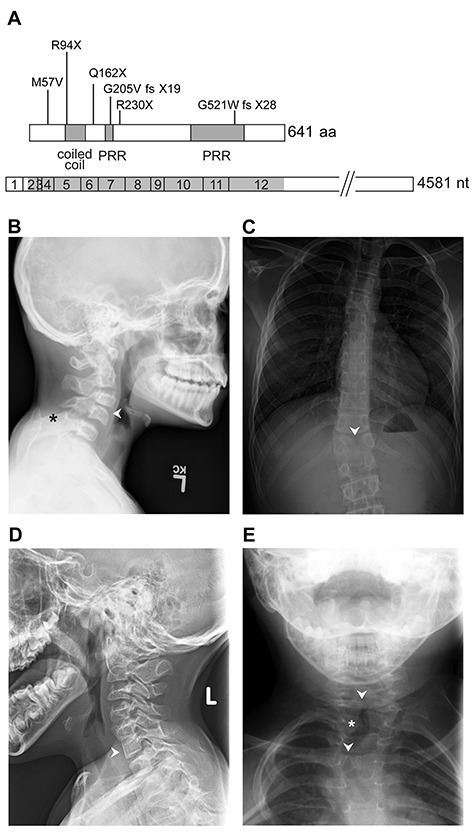
WBP11 variants and vertebral segmentation defects in selected individuals. (A) Schematic representation of the WBP11 open reading frame and cDNA (bottom) with variants and protein structure (top). PRR, proline-rich region. (B) Patient 1A has C3-C4 (arrowhead), C6-C7 fusion/block vertebrae (not shown) and bilateral omovertebral bones from C5 (asterisk). (C) Patient 1B has T12 butterfly vertebrae (arrowhead) and congenital scoliosis. Patient 2 has C6-C7 fusion (D, arrowhead) and T2 left hemifusion (E, asterisk) and T1, T4 butterfly vertebrae (E, arrowheads).
The inheritance of WBP11 variants from the apparently unaffected mother in Family 1 indicates that heterozygous LoF alleles are a partially penetrant cause of disease (Supplementary Material, Fig. S1). Patients 1A and 1B had different phenotypic features and the affected members of Family 4 had even more variable phenotypic expression, ranging from minor dysmorphology in the mother to major malformations resulting in fetal demise. Despite harboring the same frameshift WBP11 variant, affected individuals in Families 5 and 7 exhibited a variety of clinical presentations. Patient 5 only had renal agenesis reported, whereas Patient 7 had normal renal imaging but instead had cerebral, cardiac, TE and vertebral malformations. Again demonstrating variable disease expression even within the same family, her sister Patient 7A had duodenal atresia and Sprengel deformity. Two other sisters in Family 7 were also found to be heterozygous for the WBP11 variant (Supplementary Material, Fig. S1), and they are awaiting investigations for malformations. The WBP11 variant did not segregate with the unaffected mother in Family 7, and paternal DNA was not available. Inheritance in Family 7 might be presumed paternal and indeed might explain the report of short stature and short neck in the father and his brother. Parental DNA was not available in Families 5 and 6 to determine variant inheritance. Patients 2 and 3 had de novo WBP11 variants.
Following identification of predicted damaging WBP11 variants in these families, each group independently screened our respective research cohorts for similar variants in WBP11. Among our cohorts of patients with vertebral malformation, syndromic EA, renal hypodysplasia or multiple congenital anomalies, one additional WBP11 variant was detected in a patient with Klippel-Feil syndrome. The WBP11 insertion variant (c.191-2_191-1insAA) in this patient is in the splice acceptor site but does not modify the splice acceptor dinucleotide and is not predicted to alter WBP11 splicing according to SpliceAI (22), Spliceogen (23) and maxEntScan (24); it is found in one individual in the gnomAD database. This patient also has a nonsense variant in the KIAA1217 gene [NM_019590:c.4219A>T p.(Lys1407Ter)], which is considered the more likely genetic cause (25).
Patients with predicted LoF, truncating or damaging missense WBP11 variants had overlapping phenotypes involving the heart, vertebrae, trachea, esophagus, kidney and limbs (Table 1). Short stature and/or decreased body weight were reported in five cases. Eight individuals from six families had congenital heart disease. Ventricular septal defect (VSD) was the most common, occurring as an isolated finding in three cases and as part of a complex heart defect in Patients 3, 4A and 7. Patient 1B had self-resolved pulmonary stenosis and Patient 6 had an atrial septal defect (ASD). All the patients who have had vertebral imaging had abnormal vertebral morphology. These were identified in five families, presenting as fused cervical vertebrae and/or thoracic butterfly vertebrae in four individuals and as a hypoplastic first costal arch in Patient 4D (Fig. 1B–E). Patient 1A also had bilateral omovertebral bones and scapular elevation, with a short neck and low posterior hairline associated with his cervical vertebrae fusion. Patient 1B had congenital scoliosis, while Patient 2 had associated abnormality of the ribs. Patient 7 had abnormal L5 vertebra (undefined), while her sister, Patient 7A, had Sprengel deformity. Additionally, Patients 4B and 4D had Sprengel deformity (Supplementary Material, Fig. S3). EA with tracheo-esophageal fistula (TEF) was found in Families 3, 4, 6 and 7, affecting six individuals in total. Patients 3 and 7A had duodenal atresia. Abnormal renal morphology was found in five cases from four families, the most severe being renal agenesis in Patients 4C, 5 and 6. Limb abnormalities were identified in five cases from three families. Notably, Patient 3 had hypoplasia of the left thumb (Supplementary Material, Fig. S2), and Family 4 had three members with a range of anomalies, including tapered fingers, camptodactyly and hypoplasia of the thumb (Supplementary Material, Fig. S3). Patient 1A had clinodactyly of the fifth finger. Dysmorphic facial features were not constant but include facial asymmetry, reported in Patients 1A and 4D. Features reported in single cases include choanal atresia, submucous cleft palate with velopharyngeal insufficiency and conductive hearing impairment, broad forehead with deeply set eyes, hypertrichosis, low-set dysplastic ear with retrognathia, preauricular pit, preauricular skin tags, cryptorchidism and inguinal hernia. In Family 4, head circumferences (HCs) measured from early childhood were at least two standard deviations (SDs) below the mean, including that of the mother; but HC measured before term gestation ranged between 1.5 and 0 SD from the mean. Growth parameters of Patient 6 were all more than 2 SD below the mean. Patient 7 had microcephaly with agenesis of the corpus callosum and short stature. Patient 2 had both microcephaly and chromosome 1q21.1 deletion in common with her mother, who had no other anomalies. This 374 kb deletion does not include disease-associated genes and does not overlap with the recognized 1.35 megabase microdeletion in chromosome 1q21.1 (MIM 612474) (26) (see Supplemental Note: Case Reports). Moreover, Patient 2 was delayed in achieving her developmental milestones, Patient 3 had mild intellectual disability (ID), Patient 1B had suspected attention deficit hyperactivity disorder (ADHD) and Patient 7 had global developmental delay (DD) and ADHD. Limited information regarding extra-renal manifestations was available for Patients 5 and 6. Ophthalmological features were not reported in any of the patients. There was no documented history of antenatal teratogen exposure. Patients 1A, 3, 4D, 5, 6 and 7 had a normal chromosome microarray, and Patients 3 and 4D also had normal chromosome breakage studies.
Given that vertebral defects were common in patients identified with WBP11 variants, we investigated if a cohort of vertebral malformation patients harbored an enrichment in WBP11 variants compared with controls. Rare variant enrichment analysis (27) was carried out, testing for enrichment in the number of WBP11 variants in the exome data of 58 vertebral malformation patients compared with 194 control exomes that passed quality control. Variation in WBP11 was significantly enriched in the vertebral malformation cohort compared with controls, either when considering all coding variants [except synonymous single nucleotide variant (SNVs)] and splicing variants [Table 2, Set1, P = 0.01911] or when only predicted damaging variants were included [Table 2, Set2, P = 0.02032].
Table 2.
Enrichment of WBP11 variants in vertebral malformation cases
| Variant set | P-value |
|---|---|
| Set 1 | 0.01911 |
| Set 2 | 0.02032 |
P-values for tests of WBP11 variant enrichment in vertebral malformation cases (58 samples) over controls (194 samples). SNP-seq (Sequence) Kernel Association Test-Optimized (SKAT-O) was used for enrichment testing. Variants selected for gene enrichment had ExAC MAF < 0.01 (1%). Variant types selected were—Set 1: stop gain, frameshift (insertion/deletion), non-synonymous single nucleotide variant (SNV) and splicing variants; Set 2: stop gain, frameshift (insertion/deletion) and variants that exceeded PolyPhen-2 HVAR score >=0.446 and combined annotation dependent depletion (CADD) Phred score > 15.
Wbp11 heterozygous null mice model variable penetrance and expressivity observed in patients
To link the WBP11 predicted LoF variants identified in the affected individuals with the constellation of their organ defects, we generated a Wbp11 null allele in mice using CRISPR-Cas9. Since targeting C57BL/6J zygotes was unsuccessful, exon 5 was targeted on an FVB/N × C57BL/6J F1 hybrid background generating a single founder carrying an 8 bp deletion, causing premature termination of the protein six amino acids downstream (Supplementary Material, Fig. S4A). This site is 8 bp away from the equivalent position of variant c.280C>T p.(Arg94Ter) identified in Family 1.
In some families with putative heterozygous LoF variants, there is incomplete penetrance and variable expressivity. The same variability is seen in the mouse model. Mice carrying the Wbp11 null allele were not found in the expected Mendelian ratio in progeny from Wbp11+/+ × Wbp11+/− matings, with only a third of live mice being heterozygous null post-weaning (Fig. 2A, P = 0.0001). Wbp11+/− mice were also underrepresented at postnatal day 12 (P12) and at E17.5, indicating that a proportion of Wbp11+/− mice died during gestation and shortly after birth (Fig. 2A, E17.5 P = 0.0263, P12 P = 0.0412). The percentage of Wbp11+/− mice that died increased with subsequent backcrosses onto C57BL/6J or FVB/N strains (Supplementary Material, Fig. S4C–F), presumably because heterosis (hybrid vigor) of the FVB/N × C57BL/6J hybrid founder is lost through successive backcrosses to inbred strains. Significantly, more male Wbp11+/− mice died or needed to be culled on either background compared with Wbp11+/− females (Fig. 2B, FVB/N P = 0.0063, C57BL/6J P = 0.0049). Because the partial lethality of the Wbp11 null allele worsened as the mice were backcrossed to inbred FVB/N and C57BL/6J strains, subsequent skeletal and internal soft tissue analysis was restricted to the second FVB/N backcross and comparisons made to wildtype littermates.
Figure 2.
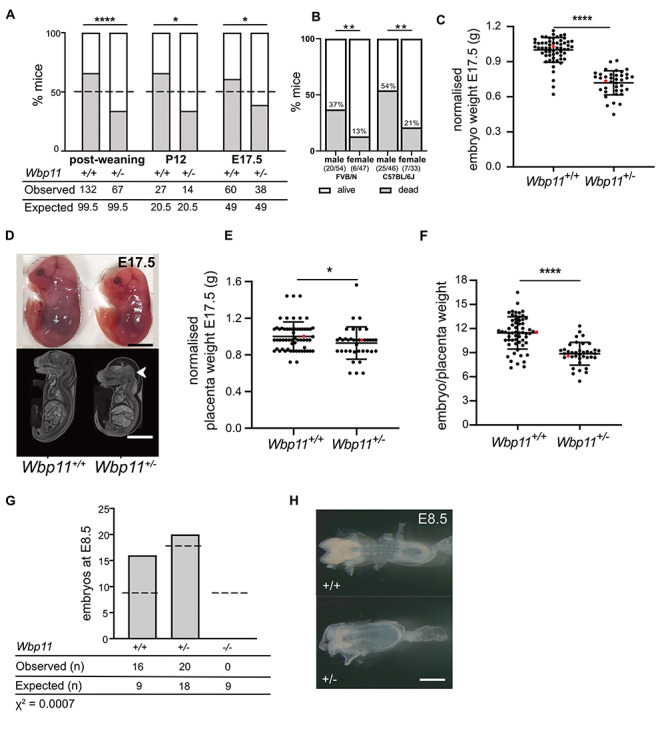
Wbp11−/− and Wbp11+/− embryos and mice do not survive in expected numbers. (A) Number of Wbp11+/− and Wbp11+/+ mice observed compared with the expected Mendelian ratio (dotted line) at E17.5 (backcross 2, *P = 0.0263), P12 (backcross 2, *P = 0.0412) and post-weaning (backcrosses 1 and 2, ****P = 0.0001). Deviation from expected Mendelian ratios for the Wbp11+/− × Wbp11+/+ breeding scheme was calculated by chi-squared test. Dead mice that could not be genotyped were omitted. (B) Percentage of male and female Wbp11+/− mice that died postnatally. On the left, FVB/N background backcrosses 1–4, **P = 0.0063. On the right, C57BL/6J background crosses 1–4, **P = 0.0049. Fisher’s exact test was used to determine significance. (C) Comparison of Wbp11+/+ and Wbp11+/− embryo weights at E17.5 normalized to average wildtype body weight (n = 98, ****P < 0.0001). (D) Photomicrographs and contrast-enhanced micro-computed tomography (micro-CT) scans of E17.5 embryos illustrating the smaller size of Wbp11+/− embryos and the presence of edema (arrowhead) compared with wildtype embryos from the same litter. (E) Placenta weight of E17.5 embryos (FVB/N) after dissection, normalized to average wildtype placenta weight (n = 98, *P = 0.0239). (F) Wbp11+/− and Wbp11+/+ E17.5 embryo weights normalized to the weights of their placentas (****P < 0.0001). (G) Embryo genotypes observed at E8.5 in litters from Wbp11+/− × Wbp11+/− matings (backcrosses 2 and 3). Deviation from expected Mendelian ratios (dotted lines) was calculated by chi-squared test (P = 0.0007). (H) Photomicrographs of Wbp11+/− and wildtype E8.5 embryos. Error bars represent SD. (C, E and F) Unpaired t-test was used to determine significance, and red dots indicate the embryos in (D). Scale bars: 5 mm (D), 500 μm (H).
E17.5 Wbp11+/− embryos weighed 30% less on average than their wildtype littermates (Fig. 2C, P < 0.0001) and exhibited the appropriate developmental landmarks (wrinkled skin, disappearance of umbilical hernia and parallel fingers and toes), indicating that their reduced weight was not due to delayed development (Fig. 2D). Even though placentas of Wbp11+/− embryos weighed 10% less than those of Wbp11+/+ embryos (Fig. 2E, P = 0.0239), the ratio of fetal weight to placental weight was 22% lower in Wbp11+/− embryos compared with wildtype littermates, suggesting either an inherent growth defect in the embryo or that Wbp11 heterozygosity impaired placental efficiency (Fig. 2F, P < 0.0001).
Given that a proportion of Wbp11+/− mice died postnatally, we hypothesized that embryos homozygous null for Wbp11 may not be viable. To test this, we carried out Wbp11+/− × Wbp11+/− matings and harvested embryos at E8.5. Wbp11+/− embryos at this stage had rounded head folds and it was difficult to discern their somites, indicative of a slight delay in development (~half a day) compared with wildtype littermates (Fig. 2H). Wbp11 homozygous null embryos were not found at E8.5 (Fig. 2G, P = 0.0007), indicating that Wbp11 is an essential gene for development.
Wbp11 heterozygous mutant mice display several axial skeletal defects
Of the families reported here, most had skeletal abnormalities including cervical vertebral fusions and rib abnormalities. To determine if the same was true in mice, we assessed the skeletons of Wbp11+/− mice at P12 by micro-computed tomography (micro-CT). All seven Wbp11+/− pups analyzed had cervical vertebral fusions which were reminiscent of those seen in many of the affected individuals (Fig. 3; Supplementary Material, Table S2; P < 0.0001). Other cervical defects seen in Wbp11+/− mice include absent vertebral elements, asymmetry of the midline, hemi- and butterfly vertebrae, cervical ribs, hypoplastic transverse processes and dyssymphysis. Rib defects (small or absent 13th rib) and sternum malformations (fusions between the 3rd and 4th sternebrae) were also common (Supplementary Material, Table S2, P < 0.0001). The majority of Wbp11+/− pups had five lumbar vertebrae rather than the usual six (Supplementary Material, Table S2, P < 0.0001). Fewer defects were found in the thoracic region with two Wbp11+/− pups having fused vertebrae (Fig. 3D) and another having hypoplastic transverse processes in two thoracic vertebrae. No defects were observed in sacral and caudal regions. Thus, WBP11 haploinsufficiency causes axial skeletal defects in humans and mouse, most commonly in the cervical vertebrae.
Figure 3.
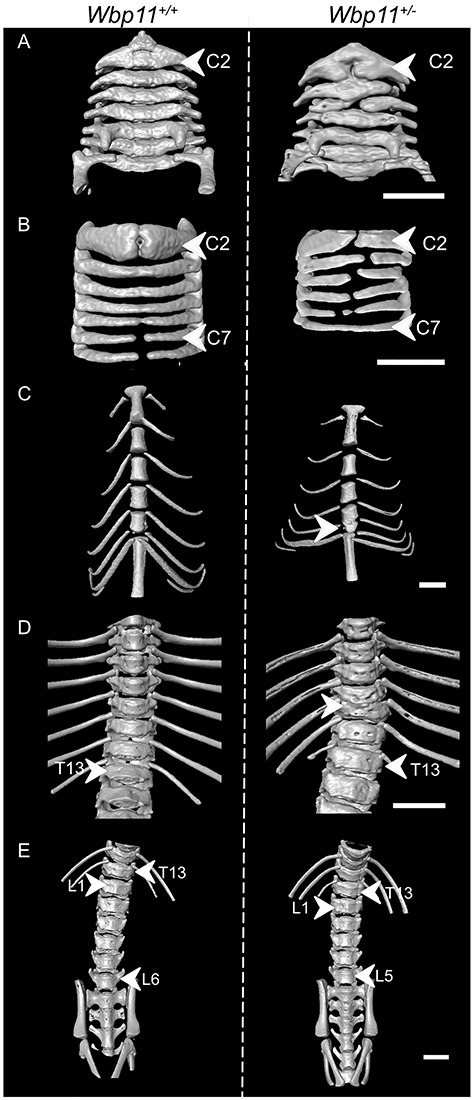
Wbp11+/− mice exhibit axial skeletal defects. Surface views of micro-computed tomography (micro-CT) reconstructions illustrating skeletal defects in Wbp11+/− (right) compared with wildtype P12 pups (left). Pups included in this data are from the second backcross. (A) Anterior and (B) posterior halves of the cervical vertebrae of a Wbp11+/− (right) and a wildtype P12 pup (left). The atlas (C1) has been removed to allow the visualization of defects in the axis (C2). In this Wbp11+/− pup, the axis and C4 are missing vertebral bodies, the anterior arches of the axis are fused to C3, the right anterior arch of C4 is fused to C3 and C5 (A) and the midline of the neural arches (B) is asymmetrical. (C) Anterior portion of the thoracic cage showing fusion of sternebrae 3 and 4, common in Wbp11+/− mice. In this example, a true rib is missing from the right side (arrowhead) of the mouse. (D) Anterior view of the thorax of a Wbp11+/− mouse with a vertebral fusion (T10-T11 fusion). (E) Anterior view of the lumbar vertebrae. Scale bars: 2 mm.
Wbp11 heterozygosity affects multiple organ systems
To detect defects in the organs and other tissues, we stained E17.5 embryos with Lugol’s reagent and performed contrast-enhanced micro-CT, which captures soft tissue anatomy (28). All 10 Wbp11+/− embryos examined had edema in the head (Supplementary Material, Table S3, P < 0.0001) with associated reduced brain volume (Figs 2D and 4A). Volume measurements of the brain confirmed that Wbp11+/− embryos had on average a 24% smaller brain than wildtype littermates (Fig. 4B, P < 0.0001). To evaluate the extent of the reduction, we measured brain anteroposterior (AP) length, cortical width and cortical thickness from 3D micro-CT reconstructions (Fig. 4C) (29,30). All three measurements were significantly lower in the Wbp11+/− embryos compared with the wildtype littermates (Fig. 4D–F; Fig. 4D, P = 0.004, Fig. 4E and F, P < 0.0001), indicating that brain size is reduced but otherwise appeared normal in Wbp11 heterozygotes.
Figure 4.
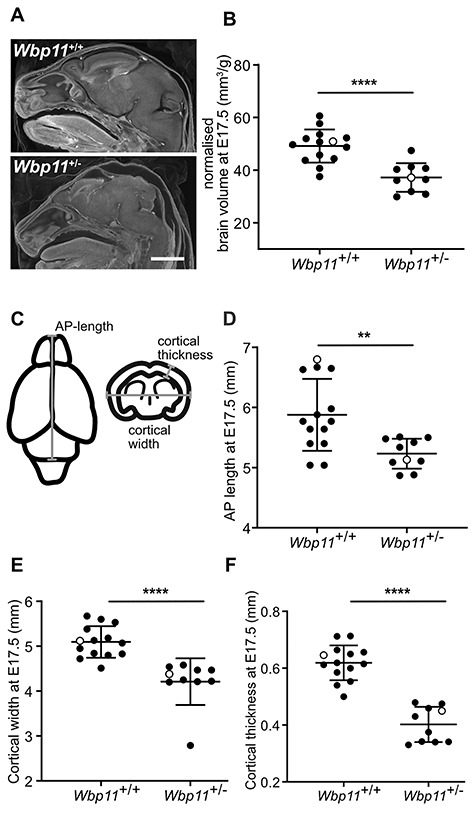
Brains of Wbp11+/− embryos are smaller than Wbp11+/+ littermates. (A) Head of Wbp11+/+ (top) and Wbp11+/− (bottom) E17.5 embryos in sagittal section of contrast-enhanced micro-CT scans. (B) Quantification of the total brain volume normalized to each embryo’s weight (P < 0.0001). (C) Schematic diagram of measurements taken from the brains (E17.5). Anteroposterior (AP) length is the maximum length in the AP axis of the brain. Cortical width is the maximum cross-brain distance at the level of lateral ventricle and third ventricle, while cortical thickness is measured at the level of the third ventricle. (D–F) Comparison of Wbp11+/+ to Wbp11+/− measurements of the brain including AP length (D, **P = 0.004), cortical width (E, ****P < 0.0001) and cortical thickness (F, ****P < 0.0001). Embryos included in this data are from the second backcross. Unpaired t-test was used to determine statistical significance. Error bars represent standard deviation (SD). Open dots indicate the embryos in (A). Scale bar: 2 mm (A).
Wbp11+/− mouse embryos also had kidney and lung defects at E17.5. In 4 out of 10 Wbp11+/− embryos, the kidneys appeared smaller and abnormally elongated (Fig. 5A; Supplementary Material, Table S3; P = 0.019). Volume measurements confirmed that both left and right kidneys were 30% smaller in Wbp11+/− embryos compared with wildtype littermates (Fig. 5A; left kidney, P = 0.0024; right kidney, P = 0.0002). Although smaller than wildtype, kidneys from the Wbp11+/− embryos were structurally normal (Fig. 5B). The lungs were also significantly smaller in Wbp11+/− embryos compared with wildtype littermates (Fig. 5C, P = 0.0034). Unlike affected individuals, heart defects, TEF or EA were not observed in the Wbp11+/− embryos. However, a small blind sac attached to the esophagus was found near the stomach in 2 of the 10 Wbp11+/− embryos examined (Supplementary Material, Fig. S5, Table S3). No obvious craniofacial or stomach defects were found when analyzing E17.5 embryos. However, internal analysis of the head revealed that 3 out of 10 Wbp11+/− embryos had unilateral choanal atresia (Supplementary Material, Fig. S5, Table S3), an anomaly also found in Patient 2. Even though there were no obvious defects in the liver, volume analysis revealed that Wbp11+/− embryos had significantly smaller livers compared with wildtype littermates (Fig. 5D, P = 0.0001). We also examined Wbp11+/− embryos from later FVB/N backcrosses (backcross 3), which exhibit higher rates of postnatal death, at E15.5 in an attempt to detect defects responsible for embryo death by E17.5. Like E17.5 Wbp11+/− embryos from an earlier backcross, E15.5 embryos had edema (Supplementary Material, Table S4, P = 0.01), choanal atresia (Supplementary Material, Table S4, P = 0.01) and a blind sac arising from the esophagus (Supplementary Material, Table S4, P = 0.049), but heart defects and more severe esophageal defects were not found (Supplementary Material, Fig. S6, Table S4). Three out of 8 Wbp11+/− embryos at E15.5 had severe cleft palate, a defect not observed in E17.5 Wbp11+/− embryos, suggesting that the palate fuses by E17.5 in such embryos or that they die before E17.5 (Supplementary Material, Table S4, P = 0.049).
Figure 5.
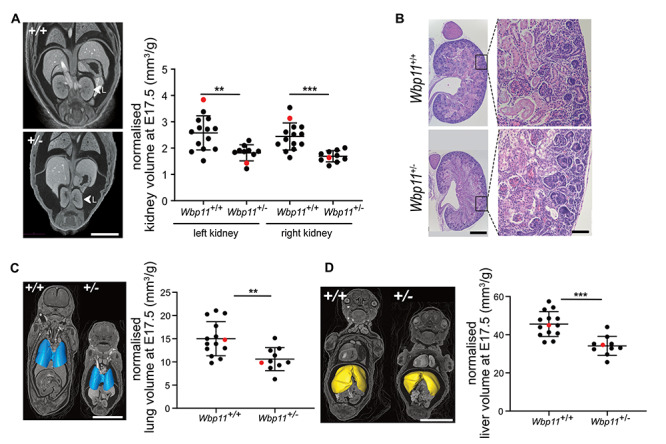
Kidney defects are present in E17.5 Wbp11+/− embryos. (A) Anterior view of micro-computed tomography (micro-CT) reconstructions comparing kidney morphology between Wbp11+/+ and Wbp11+/− littermates at E17.5. Left kidney is indicated. Graph shows comparison of volume measurements of left and right kidneys in Wbp11+/+ and Wbp11+/− littermates at E17.5 (left kidney, **P = 0.0024; right kidney, ***P = 0.0002). Red dots indicate the embryos on the left. (B) Histological sections (sagittal) stained with hematoxylin and eosin of an E17.5 kidney from Wbp11+/+ (top two panels) and Wbp11+/− embryos (bottom two panels). Whole kidney micrographs (left) and high magnifications (right) are shown. (C) 3D surface view of the lungs combined with micro-CT reconstruction slice (anterior view). Volume measurements of the lungs from Wbp11+/+ and Wbp11+/− littermates at E17.5 (**P = 0.0034). Red dots indicate embryos on the left. (D) 3D surface view of the liver combined with micro-CT reconstruction slice (anterior view). Volume measurements of the livers from Wbp11+/+ and Wbp11+/− littermates at E17.5 (***P = 0.0001). Red dots indicate embryos on the left. Embryos included in this data are from the second backcross. Kidney, lung and liver volumes were normalized against the embryo weight. Unpaired t-test was performed to determine statistical significance. Scale bars: 2 mm (A), whole kidney = 500 μm and high magnification = 100 μm (B), 5 mm (C, D). Error bars represent standard deviation (SD).
Given the presence of tissue-specific defects in Wbp11+/− embryos, we sought to establish the expression pattern of WBP11 during development. A polyclonal anti-WBP11 antibody specifically detected transfected FLAG-WBP11 as well as endogenous WBP11 at the expected size of 90 kDa in mouse E9.5 embryo lysates (Fig. 6A) (14). Anti-WBP11 reactivity was largely nuclear in E9.5 mouse embryos, where it was found in the presomitic mesoderm and somites, the progenitors of the vertebrae and all other tissues at this stage, although expression was higher in the surface ectoderm and in fetal blood cells (Fig. 6B). WBP11 was also ubiquitously expressed at Stages E10.5, E11.5 and E12.5 (Fig. 6C–K, not shown). Thus, WBP11 protein expression is not restricted to the precursors of the tissues affected by Wbp11 heterozygosity.
Figure 6.
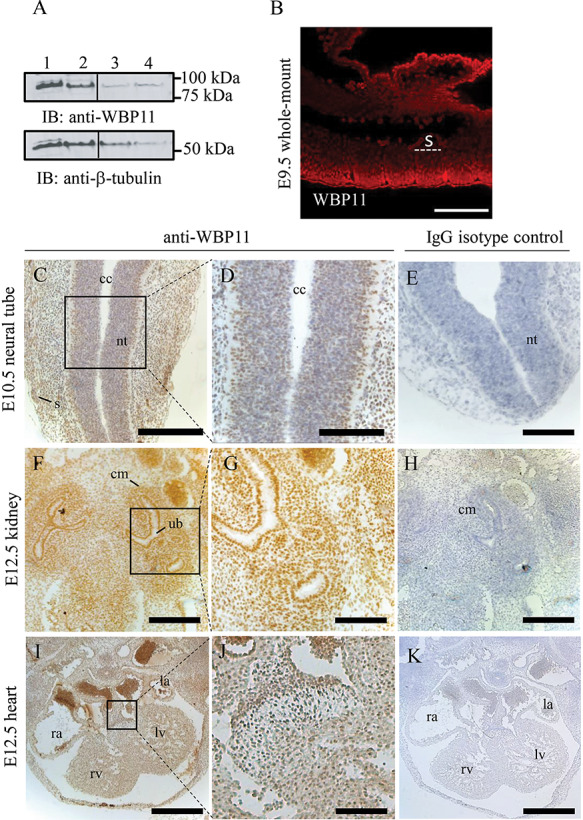
WBP11 is ubiquitously expressed in mouse embryos. (A) Immunoblot showing WBP11 (lane 1) and WBP11-FLAG (lane 2) overexpression in C2C12 mouse cell line and endogenous WBP11 expression in C2C12 (lane 3) and E9.5 embryo lysate (lane 4). Beta-tubulin was used as a loading control. (B) Whole-mount immunofluorescence staining of an E9.5 mouse embryo with anti-WBP11 antibody. Anti-WBP11 reactivity was detected with donkey anti-rabbit RRX. (C–K) Transverse sections of paraffin embedded mouse embryos at different stages labeled with anti-WBP11 antibody (left two columns) or IgG isotype control (right column) followed by detection with anti-rabbit HRP and DAB staining. (C–E) Sections through the neural tube at E10.5. (F–H) Sections through the kidney at E12.5. (I–K) Sections through the heart at E12.5. All paraffin sections were counterstained with hematoxylin, dorsal is to the top and ventral to the bottom. s, somite; nt, neural tube; cc, central canal; cm, condensed mesenchyme; ub, uretric bud; rv, right ventricle; lv, left ventricle; ra, right atrium; la, left atrium. Scale bars: 500 μm (I, K), 300 μm (C, E, F, H), 100 μm (B, D, G, J).
Discussion
WBP11 is an established component of the spliceosome that activates pre-mRNA splicing (14,31–33). Here, we report novel predicted LoF and missense variants in the WBP11 gene in at least 13 individuals from 7 families exhibiting an overlapping range of phenotypes, including vertebral, cardiac, esophageal and renal defects. We also report that Wbp11+/− mouse embryos exhibit anomalies that overlap with those found in the affected individuals.
Transcripts from truncating WBP11 alleles found in the affected individuals, with the exception of c.1559dup p.(Gly521TrpfsTer28), are likely to undergo nonsense-mediated decay and therefore express little or no truncated protein. Consistent with this, Wbp11 expression in Wbp11+/− mice was approximately half (0.53×) of that of wildtype littermates (Supplementary Material, Fig. S4B, P = 0.008). Even if the truncated WBP11 proteins were to be expressed from the alleles found in patients, they are expected to lack the ability to activate splicing since they are shorter than those previously shown to lack splicing activity (33). Thus, the variants described in this study that are predicted to truncate WBP11 are likely to create LoF alleles.
Several additional lines of evidence support the hypothesis that predicted LoF WBP11 variants cause congenital disease. Firstly, we confirm an association between WBP11 variation and congenital disease by demonstrating that predicted damaging variants in WBP11 are significantly enriched in a cohort of patients with vertebral malformations when compared with controls. Secondly, mice heterozygous for a Wbp11 null allele exhibit cervical vertebral, renal and esophageal defects like patients carrying heterozygous predicted LoF WBP11 variants. Finally, WBP11 is among those genes that are intolerant to LoF variation, a group that includes almost all known human haploinsufficient disease genes (34). Thus, the phenotype described in the affected patients is likely caused by the haploinsufficiency of WBP11. Taken together, our findings indicate that LoF variants in WBP11 are a cause of congenital anomalies in humans and in mice.
The American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology (AMP) guidelines (35,36) do not apply to initial reports of novel gene-disease associations. However, once the link between the WBP11 LoF variants and congenital anomalies is recognized, we would be able to interpret the variants reported here using these guidelines. With the exception of c.169A>G p.(Met57Val), the novel predicted LoF variants would then receive a pathogenic or likely pathogenic ACMG–AMP classification.
While WBP11 has not previously been associated with disease, mutation of its binding partner PQBP1 (MIM 300463) causes Renpenning syndrome (MIM 309500) (37). Clinical features in affected patients include ID, microcephaly, short stature, small testes and less commonly, dysmorphic facial features, cleft palate and congenital heart defects (37,38). Like WBP11, PQBP1 is a splicing factor (39) but unlike WBP11, it has roles in innate immunity (40) and neurodegeneration (41). Notably, the c.194A>G p.(Y65C) variant in PQBP1, a cause of Renpenning syndrome (42), disrupts WBP11 binding to PQBP1 and reduces the pre-mRNA splicing in patient lymphoblasts (43). Thus, there may be common aspects to the etiology of both diseases that includes disruption of splicing.
WBP11 and PQBP1 have important roles during early development in Xenopus. Morpholino knockdown of wbp11 in Xenopus embryos causes AP axis truncation and small or absent heads and tails, phenotypes similar to pqbp1 knockdown (44). While we do not have complete growth data for our cohort, growth restriction has been reported in some of the affected individuals. WBP11 knockdown caused a reduced expression of mesoderm markers wnt8, fgf4 and cdx4 but not the pan-mesoderm marker, brachyury (44). Our findings demonstrate a developmental role for Wbp11 in mammals, with partial embryonic and postnatal lethality, axial skeletal, renal and esophageal defects and reduced brain and organ volumes evident in heterozygous null mouse embryos and early embryonic lethality (prior to E8.5) of homozygous null embryos. Abnormal mesoderm specification in Wbp11 heterozygous null embryos may be responsible for defects in the axial skeleton and kidneys.
Phenotypes reported here for WBP11 overlap those associated with variants in other spliceosomal genes: choanal atresia and cardiac malformations are features of Burn-McKeown syndrome (MIM 608572) caused by the variants in the splicing factor TXNL4A (MIM 611595) (45); renal, cardiac and vertebral anomalies are found in some patients with the cerebrocostomandibular syndrome (MIM 117650) caused by SNRPB (MIM 182282) mutation (46,47); cardiac, renal and skeletal anomalies are reported in patients with the Verheij syndrome (MIM 615583) due to pathogenic variants in PUF60 (MIM 604819) (3,48) and vertebral and cardiac malformations are features of the Au-Kline syndrome (MIM 616580) due to pathogenic variants in HNRNPK (MIM 600712) (49). A homozygous likely pathogenic variant in CDK9 (MIM 603251), an important player in alternative RNA splicing (50), has been reported in families sharing similar features of cardiac, renal and skeletal anomalies as well as microcephaly, coloboma and cataracts (51). Interestingly, WBP11 interacts with EFTUD2 (MIM 603892) (52), variants in which cause mandibulofacial dysostosis with EA and renal anomalies (MIM 610536) (53,54). Perhaps most similar to the defects reported here for WBP11 heterozygosity are those caused by truncating variants in the spliceosome component SON (MIM 182465), which cause ZTTK syndrome (MIM 617140) characterized by ID and congenital anomalies affecting the brain, heart, kidney, vertebrae and ribs (55,56). Unlike other diseases caused by mutation of splicing genes (57), patients with predicted damaging variants in WBP11 do not have overt eye anomalies, although they have not all undergone ophthalmological assessment, and ID is not common. Thus, disease caused by disruption of WBP11 has overlapping but distinct features compared with known diseases of this class.
There is considerable variation in the penetrance and expressivity of defects found within families and between families with distinct WBP11 variants. Incomplete penetrance in families with congenital vertebral segmentation defects and congenital heart disease is a recognized phenomenon (58–61). In Family 1, the variant was inherited from the apparently unaffected mother, while the inheritance in Families 5 and 6 is unknown. In Family 7, two of the sisters with the WBP11 variant are yet to demonstrate a phenotype, pending organ imaging. Different phenotypic features are present in the affected individuals from Families 1, 4 and 7. There was limited assessment of extra-renal phenotypes for Patients 5 and 6 as they were ascertained from nephrology services, and it is possible that these patients have additional malformations in common with the rest of the cohort. Given their diverse phenotypes, the patients in our study would not have been considered collectively to have a unifying genetic cause if it was not for the collaborative investigation established via the GeneMatcher (19). Nonetheless, most families shared a combination of vertebral, cardiac, gastrointestinal and/or renal malformations. Consistent with predicted damaging WBP11 variants causing these phenotypes, such variants are significantly enriched in our cohort of patients with vertebral malformation.
The combination of malformations detected in the affected individuals overlap those described in VACTERL association (MIM 192350), with at least five of the patients affected with features in three component organs (10,62). LoF WBP11 variants should be considered when seeking a genetic etiology for patients who fall under the broad spectrum of VACTERL association, even in those who have less than three organs affected. Indeed, family studies have identified component organ abnormalities in first-degree relatives of individuals with VACTERL association, and vertebral malformations are the most prevalent (11,63). The variable phenotypic expression within the affected families is consistent with this observation. WBP11 should also be considered in cases that include the Klippel-Feil syndrome, renal agenesis or EA. Identification of the disease-causing variant enables reverse phenotyping, which may uncover other abnormalities similar to those reported here.
Vertebral and renal anomalies are common between WBP11 patients and Wbp11 heterozygous null mice, confirming Wbp11 mutation as a cause of congenital defects and consistent with our hypothesis that LoF variants in WBP11 lead to congenital disease in humans. EA with TEF was not observed, although minor esophageal defects are present in the Wbp11 heterozygous null mice. Likewise, congenital heart defects observed in eight of the cases were not found in our mouse model. While we noted some loss of Wbp11+/− embryos prior to E17.5 that could be caused by heart abnormalities, we found no evidence of heart defects earlier in development at E15.5. We found that WBP11 expression was not restricted to affected tissues and organs or their progenitors and instead was ubiquitous at the stages of mouse development examined. Abnormalities unique to the mouse model include edema, hypoplastic ectopic thymii, small lungs and liver. A narrow and elongated snout was observed in some Wbp11 heterozygous null mice (not shown), although this was not evident at E17.5. We also noted that some Wbp11 heterozygous null mice ran in circles, suggestive of vestibular dysfunction (not shown).
It is not uncommon for the phenotypes observed in knockout mouse lines to diverge from the human disease that they are intended to model. A study by the International Mouse Phenotyping Consortium (IMPC) showed that only 40% of IMPC mouse lines orthologous to rare disease-gene associations had one or more phenotypes in common with the human disease (64). Phenotypes of mice carrying null alleles do not always correlate well when comparing among multiple genetic backgrounds (65), providing one possible explanation for the observed divergence between the reported phenotypes in mouse and those of the patients. Our own data indicate that, like the affected individuals, Wbp11 heterozygous mice exhibit variable penetrance and expressivity. Postnatal survival of Wbp11 heterozygous mice also differed between the two genetic backgrounds we examined. Given the variability observed among individual mice and backgrounds, it is not surprising that there are some differences in organs/tissues affected in Wbp11 heterozygous mice and in patients with predicted LoF heterozygous variants in WBP11.
Wbp11 heterozygous null mouse embryos have reduced brain volume compared with wildtype littermates but no obvious structural brain anomalies, suggestive of a primary microcephaly (66). It is interesting to note that PQBP1 variants cause primary microcephaly in patients without structural disruption to the brain (67). In patients with WBP11 variants, HC was reduced in several individuals: affected individuals in Family 4 had reduced HC postnatally, and Patients 6 and 7 had growth parameters under the third centile. Microcephaly in Patient 2 and her otherwise unaffected mother may be due to their shared 1q21.1 microdeletion, but the de novo WBP11 variant may have also been contributory. Further work will be required to establish a link between WBP11 mutation and microcephaly.
WBP11 was listed among 3435 candidate developmental lethal genes in mouse that were not yet linked to human disease (68). Here, we report that heterozygous predicted LoF WBP11 variants cause an overlapping constellation of congenital malformations in humans, and in mice, loss of Wbp11 does indeed cause early embryonic lethality. Moreover, fetal and postnatal death of heterozygous null mice is a feature of our Wbp11 mouse model. In this regard, an instance of fetal demise at 16 weeks was recorded in Family 4 in our cohort, raising the possibility that WBP11 heterozygosity also causes partial lethality during development and neonatally in humans. In general, partial lethality in the heterozygous state is not common in mice. Of the 2252 mouse knock-out lines reported thus far that show prenatal lethality, only 203 exhibit lethality in the heterozygous state. This is perhaps due to the difficulties targeting and breeding such lines. A null allele in the gene for WBP11’s binding partner PQBP1 reportedly cannot be generated (44), suggesting that it has an essential cellular function. Wbp11 is also considered essential for cell viability (68), and our attempts to target the Wbp11 locus with CRISPR-Cas9 failed on the C57BL6/J background with targeting on a F1 hybrid background necessary to generate the allele. Consistent with this, on the inbred C57BL6/J background, half of the WBP11 heterozygous null mice die or are culled due to hydrocephalus.
In sum, we report predicted LoF variants in the splicing factor gene WBP11 cause a phenotype characterized by vertebral, cardiac, TEF, renal and limb malformations. Follow-up studies will be required to further define the function of WBP11 and its role in the pathogenesis of the developmental anomalies reported here. Recognition of WBP11 as an essential developmental gene will enable the identification of pathogenic variants in more patients with single or multiple congenital anomalies, improving our ability to provide more accurate recurrence risks and guide surveillance for possible complications in affected families. Conversely, identification of pathogenic WBP11 variants in more patients will extend the phenotypic spectrum related to WBP11 in humans, potentially establishing its association with microcephaly and other organ anomalies.
Materials and Methods
DNA samples, exome sequencing and variant analysis
Families 1–4 and 7 were ascertained through Medical Genetics services, and Families 5 and 6 through Nephrology services. Families 1 and 2 were included in a cohort of 76 probands with segmentation defects of the vertebrae that underwent family-based exome sequencing and analysis. Family 3 underwent clinical trio-exome sequencing. Family 4 was part of a cohort of families with syndromic EA. Families 5 and 6 were enrolled in a case–control study that compared 525 patients with renal hypodysplasia recruited from nephrology clinics and 6585 controls. Family 7 was part of a cohort of 200 trio-exomes, where each proband fulfilled one of the following criteria for inclusion: (a) severe global DD/ID—developmental quotient/intelligence quotient (DQ/IQ) <54; (b) mild–moderate global DD/ID—DQ/IQ <70 and major anomaly or seizures or (c) two congenital major anomalies. Investigations were performed after obtaining informed consent in accordance with the ethics standards of the responsible institutional review boards. Exome sequencing and variant analysis for each family are described in the Supplement.
Rare variant enrichment analysis
Exome data from 76 probands with vertebral malformations and 200 controls (Osteoporosis cases provided by Anglo-Australasian Osteoporosis Genetics Consortium) were considered for rare variant enrichment analysis. Application of Principal Component Analysis and Sample quality control metrics were applied as described in (69), reducing vertebral case samples to 58 and control samples to 194. Rare variants (minor allele frequency (MAF) <0.01) were considered disruptive (nonsense, frameshift and essential splice-site variants) or missense variants rated as predicted damaging by two prediction algorithms (PolyPhen-2 HVAR and CADD Phred). Enrichment analyses were performed on these variants sets using SNP-set (Sequence) Kernel Association Test-Optimized (SKAT-O) test (27) to determine the association of variant burden.
Generation of Wbp11 null mice and genotyping
CRISPR targeting of the Wbp11 locus was carried out by the MEGA team at Australian BioResources (Moss Vale, NSW, Australia). Single guide RNA (AAGGCGTAGAATGCGTTCAA) was designed against exon 5 of Wbp11, with the aim of creating a frameshift mutation to disrupt protein function. Electroporation of CRISPR guide RNA/Cas9 into C57BL/6J/FVB/N F1 zygotes generated a single founder carrying an allele with an 8 bp deletion, which was successfully passed on to the next generation. This deletion causes a reading frameshift and termination of the protein six amino acids downstream (Supplementary Material, Fig. S4A). The genotyping strategy utilizes the fact that the 8 bp deletion removes a XmnI restriction site to distinguish the wildtype and null Wbp11 alleles following PCR amplification of exon 5 (forward primer: TGGGTCACTTCTGGGAACGA; reverse primer: TCACGCTCTTACTCATGTTTACCA).
Breeding and timed matings
This research was performed following the guidelines, and with the approval, of the Garvan Institute of Medical Research/St. Vincent’s Animal Experimentation Ethics Committee, research approvals 15/27 and 18/27. Since C57BL/6J mice are predisposed to developing hydrocephalus (70), the founder and subsequent male heterozygous Wbp11 mutant offspring were backcrossed to FVB/N females to maintain the colony. Mice were examined for vaginal plugs in the morning and the presence of a plug was taken as embryonic day 0.5 (E0.5). Pregnant females were sacrificed at E8.5, E15.5 or E17.5, and uteri were examined for evidence of resorptions before embryos were dissected. E17.5 embryos and their placentas were weighed and inspected for any external morphological defects. P12 pups were sacrificed by carbon dioxide inhalation. FVB/N mice were purchased from Australian BioResources.
Mendelian ratios for all stages (E8.5, E15.5, E17.5, P12 and post-weaning) were analyzed by chi-squared test. For statistical analysis of weights, embryo and placenta weights (E17.5) were normalized against the average weights of wildtype littermates. As embryo and placenta weight data did not follow a normal distribution, a Mann–Whitney (non-parametric) t-test was performed to determine the statistical significance using Prism 8 (GraphPad). To compare mortality between heterozygous null and wildtype littermates in both strains (FVB/N and C57BL/6J) and all backcrosses, a two-tailed Fisher’s exact test was used. To determine whether significantly more heterozygous null males die compared with heterozygous null females, a two-tailed Fisher’s exact test was used.
Western blotting
1.5 × 105 C2C12 cells were seeded on a 60 mm dish. After 24 h, cells were transfected with 5 μg of either mouse Wbp11 (untagged) or FLAG tagged mouse Wbp11. Twenty-four hours post-transfection, cells were scraped and lyzed in 50 μL WCE lysis buffer [20 mM HEPES pH 7.8, 420 mM NaCl, 0.5% NP40, 25% glycerol, 0.2 mM EDTA, 1.5 mM MgCl2, 1 mM PMSF, cOmplete protease inhibitor cocktail (Sigma)] and protein concentration was determined by bicinchononic acid (BCA) assay. To determine the endogenous expression levels of WBP11 in mammalian cells, a confluent 60 mm dish of C2C12 cells was lyzed in 50 μL WCE. To determine endogenous WBP11 protein expression in embryos, proteins were extracted from an E9.5 wildtype embryo. The embryo was lyzed in 30 μL of RIPA buffer [20 mM Tris–HCl pH 7.5, 150 mM NaCl, 2 mM EDTA, 1% NP40, 1% Deoxycholate, 0.1% SDS, 1 mM PMSF, cOmplete protease inhibitor cocktail (Sigma)]. The supernatant was taken after a 15-min spin at maximum speed (4°C) and stored at −80°C. Protein concentration was determined using BCA assay (50 μg of total protein was run on a 4–12% Bis-Tris gel). WBP11 expression was determined by western blotting with WBP11 antibody (1:1000 PA5–31241; Pierce). β-Tubulin (1:5000; Sigma) was used as loading control.
Whole-mount immunofluorescence, histology and immunohistochemistry
Whole-mount immunofluorescence on E9.5 wildtype embryos was performed as described in (71) except that permeabilization was achieved by a 2-h incubation in methanol:dimethyl sulfoxide (DMSO):30% hydrogen peroxide (4:1:1) at room temperature and that embryos were blocked overnight in 10% donkey serum (Sigma D9663) dissolved in phosphate-buffered saline (PBS) containing 0.1% Triton X-100. Primary antibody for WBP11 (1:100) or Isotype control IgG and anti-rabbit RRX secondary antibody (1:100) was used. The embryos were cleared in BA:BB (benzyl alcohol: benzyl benzoate, Merck B6630) before imaging on an AxioObserver Z1 inverted microscope equipped with 710 scan head (Zeiss) using appropriate excitation/emission settings.
For paraffin sectioning, embryos (E10.5, E11.5 and E12.5) and kidneys of E17.5 embryos were fixed in 4% paraformaldehyde (PFA) overnight at 4°C, with paraffin embedded and sectioned in the transverse plane (for whole-mount embryos) and sagittal plane (for E17.5 kidneys) at 7 μm.
Immunohistochemistry was performed to determine the protein expression of WBP11 and Isotype IgG in mouse embryos. The paraffin-embedded tissue sections were deparaffinized and hydrated through xylene and graded ethanol series. Subsequently, the sections were boiled in an antigen unmasking solution (10 mM Tris Base, 1 mM EDTA, 0.05% Tween20, pH 9.0) and blocked for 1 h in 10% donkey serum. For detection of WBP11 and Isotype IgG, a horseradish peroxidase (HRP) donkey anti-rabbit secondary reagent and 3, 3′-diaminobenzidine (DAB) was used. After an hematoxylin staining, sections were mounted with Gelvatol mounting media.
Histological sections of E17.5 kidneys were deparaffinized, stained with hematoxylin (Sigma HHS16) and Eosin (Sigma Y E4382) and were mounted in Depex before imaging on a Brightfield microscope (Leica MC170 HD).
Tomography
For assessment of the skeleton, FVB/N mouse pups were sacrificed at P12 and fixed in 4% PFA for 3 days before scanning. micro-CT imaging of the entire skeletons was performed using the Skyscan 1272 scanner (Bruker) operated at 40 kV (source voltage), 238 μA (source current) and an Al 0.25 mm filter. The magnification was set at 21.5 μm pixel size and images were taken every 0.8° through 360° using 1216 ms exposure, generating 1497 projections in 30 min per field of view. Four fields of view were needed to capture the entire skeleton.
To assess soft tissues, E17.5 and E15.5 FVB/N embryos were dissected in cold 10 mM PBS and fixed in cold 4% PFA and 1% glutaraldehyde. Each embryo and their placenta were individually weighed and assessed for external morphological abnormalities. After 3 days, the embryos were immersed in hydrogel (4% acrylamide, 0.05% bis-acrylamide, 0.25% VA044 initiator and 0.05% saponin made up in PBS) (72) and were incubated for an additional 3 days at 4°C with rocking. The hydrogel was polymerized at 37°C for 3 h using an X-Clarity hydrogel embedding station (Logos) and subsequently removed to retrieve the stabilized embryo. Samples were immersed in 100% Lugol’s solution (Sigma-Aldrich) for 5 days in the dark at room temperature and were scanned using 75 kV source voltage, 238 μA source current and an Al 0.5 mm + Cu 0.038 mm filter. Magnification was set to 2.23 μm pixel resolution, and images were taken every 0.3° through 180° with an exposure of 2700 ms, generating 1923 projections in 1.5 h for three fields of view. Acquired projections were reconstructed using NRecon software (Bruker) with smoothing, ring artifact and post-alignment corrections.
Analysis of micro-CT data, volume measurements and 3D rendering
To assess defects systematically, primary anatomical information was recorded for each embryo using CTvox (Bruker v3.3.0) with the aid of a checklist of 27 points covering cerebral, craniofacial, cardiac, gastrointestinal and limb defects for soft tissue analysis and of 40 points for skeletal defects covering the skull, cervical, thoracic, lumbar and sacral regions. Morphological defects in the affected organs were then quantified using Amira (Thermo Fisher). Rendering was performed in the segmentation editor of Amira by defining a global threshold to select only the skeleton. A surface view of this selection was then rendered. Reconstructed micro-CT datasets were also processed in the segmentation editor of Amira to obtain the 3D volume measurements of the brain, kidneys, lungs and livers. To segment the organ of interest from the rest of the body, the brush tool was used to accurately select the appropriate pixels. Contours of the organ were selected manually using the brush tool every 10–20 slices, interpolated and then adjusted manually where necessary. Volume measurements of the segmented organs were extracted from the material statistics table and were normalized against the embryo weight. Statistical differences between genotypes were established with an unpaired t-test, provided the dataset followed a normal distribution. If that was not the case, a non-parametric Mann–Whitney test was used.
Supplementary Material
Acknowledgements
The authors acknowledge the technical assistance of Melissa Rapadas, advice from Cedric Le Caignec and Vivette D’Agatti, and Matthew Brown for establishing the exome sequencing pipeline (for Families 1 and 2). The authors also acknowledge Robert Brink and David Zahra of the Mouse Engineering at Garvan/Australian Bioresources (MEGA) service for their help in the design and generation of Wbp11 CRISPR targeted allele. The authors gratefully acknowledge the assistance of the Marshfield Clinic, the Pediatrics Department of the University of Wisconsin–Madison, the Scoliosis Research Society and the Malika Ray, Asok K. Ray, M.D., FRCS/(Edin) Initiative for Child Health Research. The authors are grateful to the families for participating in this research.
Conflict of Interest statement. R.D.S. has equity interest in and has received consulting fees from Acer Therapeutics and Censa Pharmaceuticals. He has received grant funding from Alexion, travel support from Pfizer and consulting fees from Raptor, Biomarin, Alexion, E-Scape Bio, Health Advances, Precision for Value and Best Doctors. He is an employee of PreventionGenetics. R.D.S. has patents awarded but not licensed (no revenue) in the area of newborn screening for sterol and bile acid disorders.
Funding
This work was supported by the National Health and Medical Research Council (NHMRC) (project grant ID1044543 to S.L.D, D.B.S. and E.L.D, and fellowships ID1135886 and ID1042002 to S.L.D.); the Office of Health and Medical Research New South Wales Government and the Victor Chang Cardiac Research Institute Innovation Centre, funded by the New South Wales Government; the Agence Nationale de la Recherche [CranioRespiro project and ‘Investissements d’avenir’ program (ANR-10-IAHU-01)] and Merck Sharp and Dohme ameliorer la vie ensemble par l'innovation et la recherche (Devo-Decode project) and the National Institute of Health, National Institute of Diabetes and Digestive and Kidney Diseases (grant R01DK080099). Research reported in this publication was also supported in part by the Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health under Award Number R03HD099516 to P.F.G and by the Baylor-Hopkins Center for Mendelian Genomics (NHGRI UM1 HG006542).
Accession Numbers
The WBP11 variants described in this report have been submitted to ClinVar (SUB6661060).
Web Resources
OMIM, http://www.omim.org/.
gnomAD, https://gnomad.broadinstitute.org/.
References
- 1. Christianson A., Howson C.P. and Modell B. (2006) March of dimes, global report on birth defects March of Dimes Birth Defects Foundation, White Plains, USA.
- 2. Feldkamp M.L., Carey J.C., Byrne J.L.B., Krikov S. and Botto L.D. (2017) Etiology and clinical presentation of birth defects: population based study. BMJ, 357, j2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Low K.J., Ansari M., Jamra R.A., Clarke A., El Chehadeh S., FitzPatrick D.R., Greenslade M., Henderson A., Hurst J., Keller K. et al. (2017) PUF60 variants cause a syndrome of ID, short stature, microcephaly, coloboma, craniofacial, cardiac, renal and spinal features. Eur. J. Hum. Genet., 25, 552–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vetro A., Iascone M., Limongelli I., Ameziane N., Gana S., Della Mina E., Giussani U., Ciccone R., Forlino A., Pezzoli L. et al. (2015) Loss-of-function FANCL mutations associate with severe Fanconi anemia overlapping the VACTERL association. Hum. Mutat., 36, 562–568. [DOI] [PubMed] [Google Scholar]
- 5. Savage S.A., Ballew B.J., Giri N., Laboratory N.D.C.G.R., Chandrasekharappa S.C., Ameziane N., Winter J., Alter B.P. and Group, N.D.C.S.W (2016) Novel FANCI mutations in Fanconi anemia with VACTERL association. Am. J. Med. Genet. A, 170A, 386–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zweier C., Albrecht B., Mitulla B., Behrens R., Beese M., Gillessen-Kaesbach G., Rott H.D. and Rauch A. (2002) ‘Mowat-Wilson’ syndrome with and without Hirschsprung disease is a distinct, recognizable multiple congenital anomalies-mental retardation syndrome caused by mutations in the zinc finger homeo box 1B gene. Am. J. Med. Genet., 108, 177–181. [PubMed] [Google Scholar]
- 7. Kiefer S.M., Ohlemiller K.K., Yang J., McDill B.W., Kohlhase J. and Rauchman M. (2003) Expression of a truncated Sall1 transcriptional repressor is responsible for Townes-Brocks syndrome birth defects. Hum. Mol. Genet., 12, 2221–2227. [DOI] [PubMed] [Google Scholar]
- 8. Shi H., Enriquez A., Rapadas M., Martin E., Wang R., Moreau J., Lim C.K., Szot J.O., Ip E., Hughes J.N. et al. (2017) NAD deficiency, congenital malformations, and niacin supplementation. N. Engl. J. Med., 377, 544–552. [DOI] [PubMed] [Google Scholar]
- 9. Szot J.O., Campagnolo C., Cao Y., Iyer K.R., Cuny H., Drysdale T., Flores-Daboub J.A., Bi W., Westerfield L., Liu P. et al. (2020) Bi-allelic mutations in NADSYN1 cause multiple organ defects and expand the genotypic spectrum of congenital NAD deficiency disorders. Am. J. Hum. Genet., 106, 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Husain M., Dutra-Clarke M., Lemieux B., Wencel M., Solomon B.D. and Kimonis V. (2018) Phenotypic diversity of patients diagnosed with VACTERL association. Am. J. Med. Genet. A, 176, 1830–1837. [DOI] [PubMed] [Google Scholar]
- 11. Solomon B.D., Pineda-Alvarez D.E., Raam M.S. and Cummings D.A. (2010) Evidence for inheritance in patients with VACTERL association. Hum. Genet., 127, 731–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Solomon B.D. (2018) The etiology of VACTERL association: current knowledge and hypotheses. Am. J. Med. Genet. C Semin. Med. Genet., 178, 440–446. [DOI] [PubMed] [Google Scholar]
- 13. Bartels E., Jenetzky E., Solomon B.D., Ludwig M., Schmiedeke E., Grasshoff-Derr S., Schmidt D., Marzheuser S., Hosie S., Weih S. et al. (2012) Inheritance of the VATER/VACTERL association. Pediatr. Surg. Int., 28, 681–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Craggs G., Finan P.M., Lawson D., Wingfield J., Perera T., Gadher S., Totty N.F. and Kellie S. (2001) A nuclear SH3 domain-binding protein that colocalizes with mRNA splicing factors and intermediate filament-containing perinuclear networks. J. Biol. Chem., 276, 30552–30560. [DOI] [PubMed] [Google Scholar]
- 15. Galganski L., Urbanek M.O. and Krzyzosiak W.J. (2017) Nuclear speckles: molecular organization, biological function and role in disease. Nucleic Acids Res., 45, 10350–10368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Deckert J., Hartmuth K., Boehringer D., Behzadnia N., Will C.L., Kastner B., Stark H., Urlaub H. and Lührmann R. (2006) Protein composition and electron microscopy structure of affinity-purified human spliceosomal B complexes isolated under physiological conditions. Mol. Cell. Biol., 26, 5528–5543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Agafonov D.E., Deckert J., Wolf E., Odenwalder P., Bessonov S., Will C.L., Urlaub H. and Luhrmann R. (2011) Semiquantitative proteomic analysis of the human spliceosome via a novel two-dimensional gel electrophoresis method. Mol. Cell. Biol., 31, 2667–2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fredericks A.M., Cygan K.J., Brown B.A. and Fairbrother W.G. (2015) RNA-binding proteins: splicing factors and disease. Biomol. Ther., 5, 893–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sobreira N., Schiettecatte F., Valle D. and Hamosh A. (2015) GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat., 36, 928–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kurosaki T. and Maquat L.E. (2016) Nonsense-mediated mRNA decay in humans at a glance. J. Cell Sci., 129, 461–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alfoldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P. et al. (2020) The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581, 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jaganathan K., Panagiotopoulou S.K., McRae J.F., Darbandi S.F., Knowles D., Li Y.I., Kosmicki J.A., Arbelaez J., Cui W., Schwartz G.B. et al. (2019) Predicting splicing from primary sequence with deep learning. Cell, 176, 535–548.e524. [DOI] [PubMed] [Google Scholar]
- 23. Monger S., Troup M., Ip E., Dunwoodie S.L. and Giannoulatou E. (2019) Spliceogen: an integrative, scalable tool for the discovery of splice-altering variants. Bioinformatics, 35, 4405–4407. [DOI] [PubMed] [Google Scholar]
- 24. Yeo G. and Burge C.B. (2004) Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol., 11, 377–394. [DOI] [PubMed] [Google Scholar]
- 25. Al Dhaheri N., Wu N., Zhao S., Wu Z., Blank R.D., Zhang J., Raggio C., Halanski M., Shen J., Noonan K. et al. (2020) KIAA1217: a novel candidate gene associated with isolated and syndromic vertebral malformations. Am. J. Med. Genet. A, 182, 1664–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bernier R., Steinman K.J., Reilly B., Wallace A.S., Sherr E.H., Pojman N., Mefford H.C., Gerdts J., Earl R., Hanson E. et al. (2016) Clinical phenotype of the recurrent 1q21.1 copy-number variant. Genet. Med., 18, 341–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee S., Emond M.J., Bamshad M.J., Barnes K.C., Rieder M.J., Nickerson D.A., Team N.G.E.S.P.-E.L.P., Christiani D.C., Wurfel M.M. and Lin X. (2012) Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am. J. Hum. Genet., 91, 224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hsu C.W., Wong L., Rasmussen T.L., Kalaga S., McElwee M.L., Keith L.C., Bohat R., Seavitt J.R., Beaudet A.L. and Dickinson M.E. (2016) Three-dimensional microCT imaging of mouse development from early post-implantation to early postnatal stages. Dev. Biol., 419, 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang D.W., Xie Z.Y., Stephenson D., Morton D., Hicks C.D., Brown T.M., Sriram R., O'Neill S., Raunig D. and Bocan T. (2011) Volumetric MRI and MRS provide sensitive measures of Alzheimer’s disease neuropathology in inducible tau transgenic mice (rTg4510). NeuroImage, 54, 2652–2658. [DOI] [PubMed] [Google Scholar]
- 30. Harrison S.J., Nishinakamura R., Jones K.R. and Monaghan A.P. (2012) Sall1 regulates cortical neurogenesis and laminar fate specification in mice: implications for neural abnormalities in Townes-brocks syndrome. Dis. Model. Mech., 5, 351–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bedford M.T., Reed R. and Leder P. (1998) WW domain-mediated interactions reveal a spliceosome-associated protein that binds a third class of proline-rich motif: the proline glycine and methionine-rich motif. Proc. Natl. Acad. Sci. U. S. A., 95, 10602–10607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Llorian M., Beullens M., Andrés I., Ortiz J.-M. and Bollen M. (2004) SIPP1, a novel pre-mRNA splicing factor and interactor of protein phosphatase-1. Biochem. J., 378, 229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Llorian M., Beullens M., Lesage B., Nicolaescu E., Beke L., Landuyt W., Ortiz J.-M. and Bollen M. (2005) Nucleocytoplasmic shuttling of the splicing factor SIPP1. J. Biol. Chem., 280, 38862–38869. [DOI] [PubMed] [Google Scholar]
- 34. Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O'Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B. et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Abou Tayoun A.N., Pesaran T., DiStefano M.T., Oza A., Rehm H.L., Biesecker L.G., Harrison S.M. and ClinGen Sequence Variant Interpretation Working, G (2018) Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat., 39, 1517–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E. et al. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med., 17, 405–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stevenson R.E., Bennett C.W., Abidi F., Kleefstra T., Porteous M., Simensen R.J., Lubs H.A., Hamel B.C.J. and Schwartz C.E. (2005) Renpenning syndrome comes into focus. Am. J. Med. Genet. A, 134A, 415–421. [DOI] [PubMed] [Google Scholar]
- 38. Germanaud D., Rossi M., Bussy G., Gérard D., Hertz-Pannier L., Blanchet P., Dollfus H., Giuliano F., Bennouna-Greene V., Sarda P. et al. (2010) The Renpenning syndrome spectrum: new clinical insights supported by 13 new PQBP1-mutated males. Clin. Genet., 79, 225–235. [DOI] [PubMed] [Google Scholar]
- 39. Wang Q., Moore M.J., Adelmant G., Marto J.A. and Silver P.A. (2013) PQBP1, a factor linked to intellectual disability, affects alternative splicing associated with neurite outgrowth. Genes Dev., 27, 615–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yoh S.M., Schneider M., Seifried J., Soonthornvacharin S., Akleh R.E., Olivieri K.C., De Jesus P.D., Ruan C., Castro E., Ruiz P.A. et al. (2015) PQBP1 is a proximal sensor of the cGAS-dependent innate response to HIV-1. Cell, 161, 1293–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Okazawa H. (2018) PQBP1, an intrinsically disordered/denatured protein at the crossroad of intellectual disability and neurodegenerative diseases. Neurochem. Int., 119, 17–25. [DOI] [PubMed] [Google Scholar]
- 42. Lubs H., Abidi F.E., Echeverri R., Holloway L., Meindl A., Stevenson R.E. and Schwartz C.E. (2006) Golabi-Ito-Hall syndrome results from a missense mutation in the WW domain of the PQBP1 gene. J. Med. Genet., 43, e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tapia V.E., Nicolaescu E., McDonald C.B., Musi V., Oka T., Inayoshi Y., Satteson A.C., Mazack V., Humbert J., Gaffney C.J. et al. (2010) Y65C missense mutation in the WW domain of the Golabi-Ito-Hall syndrome protein PQBP1 affects its binding activity and deregulates pre-mRNA splicing. J. Biol. Chem., 285, 19391–19401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Iwasaki Y. and Thomsen G.H. (2014) The splicing factor PQBP1 regulates mesodermal and neural development through FGF signaling. Development, 141, 3740–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wieczorek D., Newman W.G., Wieland T., Berulava T., Kaffe M., Falkenstein D., Beetz C., Graf E., Schwarzmayr T., Douzgou S. et al. (2014) Compound heterozygosity of low-frequency promoter deletions and rare loss-of-function mutations in TXNL4A causes Burn-McKeown syndrome. Am. J. Hum. Genet., 95, 698–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tooley M., Lynch D., Bernier F., Parboosingh J., Bhoj E., Zackai E., Calder A., Itasaki N., Wakeling E., Scott R. et al. (2016) Cerebro-costo-mandibular syndrome: clinical, radiological, and genetic findings. Am. J. Med. Genet. A, 170A, 1115–1126. [DOI] [PubMed] [Google Scholar]
- 47. Lynch D.C., Revil T., Schwartzentruber J., Bhoj E.J., Innes A.M., Lamont R.E., Lemire E.G., Chodirker B.N., Taylor J.P., Zackai E.H. et al. (2014) Disrupted auto-regulation of the spliceosomal gene SNRPB causes cerebro-costo-mandibular syndrome. Nat. Commun., 5, 4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. El Chehadeh S., Kerstjens-Frederikse W.S., Thevenon J., Kuentz P., Bruel A.-L., Thauvin-Robinet C., Bensignor C., Dollfus H., Laugel V., Rivière J.-B. et al. (2017) Dominant variants in the splicing factor PUF60 cause a recognizable syndrome with intellectual disability, heart defects and short stature. Eur. J. Hum. Genet., 25, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Au P.Y.B., You J., Caluseriu O., Schwartzentruber J., Majewski J., Bernier F.P., Ferguson M., Care for Rare Canada, C, Valle D., Parboosingh J.S. et al. (2015) GeneMatcher aids in the identification of a new malformation syndrome with intellectual disability, unique facial dysmorphisms, and skeletal and connective tissue abnormalities caused by de novo variants in HNRNPK. Hum. Mutat., 36, 1009–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yang J., Zhao Y., Kalita M., Li X., Jamaluddin M., Tian B., Edeh C.B., Wiktorowicz J.E., Kudlicki A. and Brasier A.R. (2015) Systematic determination of human cyclin dependent kinase (CDK)-9 interactome identifies novel functions in RNA splicing mediated by the DEAD box (DDX)-5/17 RNA helicases. Mol. Cell. Proteomics, 14, 2701–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shaheen R., Patel N., Shamseldin H., Alzahrani F., Al-Yamany R., Almoisheer A., Ewida N., Anazi S., Alnemer M., Elsheikh M. et al. (2016) Accelerating matchmaking of novel dysmorphology syndromes through clinical and genomic characterization of a large cohort. Genet. Med., 18, 686–695. [DOI] [PubMed] [Google Scholar]
- 52. Malinová A., Cvačková Z., Matějů D., Hořejší Z., Abéza C., Vandermoere F., Bertrand E., Staněk D. and Verheggen C. (2017) Assembly of the U5 snRNP component PRPF8 is controlled by the HSP90/R2TP chaperones. J. Cell Biol., 216, 1579–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lines M.A., Huang L., Schwartzentruber J., Douglas S.L., Lynch D.C., Beaulieu C., Guion-Almeida M.L., Zechi-Ceide R.M., Gener B., Gillessen Kaesbach G. et al. (2012) Haploinsufficiency of a spliceosomal GTPase encoded by EFTUD2 causes mandibulofacial dysostosis with microcephaly. Am. J. Hum. Genet., 90, 369–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gordon C.T., Petit F., Oufadem M., Decaestecker C., Jourdain A.-S., Andrieux J., Malan V., Alessandri J.-L., Baujat G., Baumann C. et al. (2012) EFTUD2 haploinsufficiency leads to syndromic oesophageal atresia. J. Med. Genet., 49, 737–746. [DOI] [PubMed] [Google Scholar]
- 55. Kim J.-H., Shinde D.N., Reijnders M.R.F., Hauser N.S., Belmonte R.L., Wilson G.R., Bosch D.G.M., Bubulya P.A., Shashi V., Petrovski S. et al. (2016) De novo mutations in SON disrupt RNA splicing of genes essential for brain development and metabolism, causing an intellectual-disability syndrome. Am. J. Hum. Genet., 99, 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tokita M.J., Braxton A.A., Shao Y., Lewis A.M., Vincent M., Küry S., Besnard T., Isidor B., Latypova X., Bézieau S. et al. (2016) De novo truncating variants in SON cause intellectual disability, congenital malformations, and failure to thrive. Am. J. Hum. Genet., 99, 720–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Xu M., Xie Y.A., Abouzeid H., Gordon C.T., Fiorentino A., Sun Z., Lehman A., Osman I.S., Dharmat R., Riveiro-Alvarez R. et al. (2017) Mutations in the spliceosome component CWC27 cause retinal degeneration with or without additional developmental anomalies. Am. J. Hum. Genet., 100, 592–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ghebranious N., Blank R.D., Raggio C.L., Staubli J., McPherson E., Ivacic L., Rasmussen K., Jacobsen F.S., Faciszewski T., Burmester J.K. et al. (2008) A missense T (Brachyury) mutation contributes to vertebral malformations. J. Bone Miner. Res., 23, 1576–1583. [DOI] [PubMed] [Google Scholar]
- 59. Wu N., Ming X., Xiao J., Wu Z., Chen X., Shinawi M., Shen Y., Yu G., Liu J., Xie H. et al. (2015) TBX6 null variants and a common hypomorphic allele in congenital scoliosis. N. Engl. J. Med., 372, 341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Edwards J.J. and Gelb B.D. (2016) Genetics of congenital heart disease. Curr. Opin. Cardiol., 31, 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sparrow D.B., Chapman G., Smith A.J., Mattar M.Z., Major J.A., O'Reilly V.C., Saga Y., Zackai E.H., Dormans J.P., Alman B.A. et al. (2012) A mechanism for gene-environment interaction in the etiology of congenital scoliosis. Cell, 149, 295–306. [DOI] [PubMed] [Google Scholar]
- 62. Van de Putte R., De Walle H.E., Van Hooijdonk K.J., De Blaauw I., Marcelis C.L., van Heijst A., Giltay J.C., Renkema K.Y., Broens P.M. and Brosens E.J.B.D.R. (2020) Maternal risk associated with the VACTERL association: a case–control study. Birth Defects Res., 112:1495–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hilger A., Schramm C., Draaken M., Mughal S.S., Dworschak G., Bartels E., Hoffmann P., Nothen M.M., Reutter H. and Ludwig M. (2012) Familial occurrence of the VATER/VACTERL association. Pediatr. Surg. Int., 28, 725–729. [DOI] [PubMed] [Google Scholar]
- 64. Meehan T.F., Conte N., West D.B., Jacobsen J.O., Mason J., Warren J., Chen C.K., Tudose I., Relac M., Matthews P. et al. (2017) Disease model discovery from 3,328 gene knockouts by The International Mouse Phenotyping Consortium. Nat. Genet., 49, 1231–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sittig L.J., Carbonetto P., Engel K.A., Krauss K.S., Barrios-Camacho C.M. and Palmer A.A. (2016) Genetic background limits generalizability of genotype-phenotype relationships. Neuron, 91, 1253–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Passemard S., Kaindl A.M. and Verloes A. (2013) Microcephaly. Handb Clin Neurol., ; 129–141. [DOI] [PubMed] [Google Scholar]
- 67. Ito H., Shiwaku H., Yoshida C., Homma H., Luo H., Chen X., Fujita K., Musante L., Fischer U., Frints S.G.M. et al. (2015) In utero gene therapy rescues microcephaly caused by Pqbp1-hypofunction in neural stem progenitor cells. Mol. Psychiatry, 20, 459–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dawes R., Lek M. and Cooper S.T. (2019) Gene discovery informatics toolkit defines candidate genes for unexplained infertility and prenatal or infantile mortality. NPJ Genom. Med., 4, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chapman G., Moreau J.L.M., Ip E., Szot J.O., Iyer K.R., Shi H., Yam M.X., O'Reilly V.C., Enriquez A., Greasby J.A. et al. (2020) Functional genomics and gene-environment interaction highlight the complexity of congenital heart disease caused by Notch pathway variants. Hum. Mol. Genet., 29, 566–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Brayton C. (2013) Spontaneous diseases in commonly used mouse strains/stocks In The Mouse in Biomedical Research, Vol. 2 Academic press A volume in American College of Laboratory Animal Medicine. [Google Scholar]
- 71. Geffers I., Serth K., Chapman G., Jaekel R., Schuster-Gossler K., Cordes R., Sparrow D.B., Kremmer E., Dunwoodie S.L., Klein T. et al. (2007) Divergent functions and distinct localization of the Notch ligands DLL1 and DLL3 in vivo. J. Cell Biol., 178, 465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wong M.D., Spring S. and Henkelman R.M. (2013) Structural stabilization of tissue for embryo phenotyping using micro-CT with iodine staining. PLoS One, 8, e84321. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.