

Graphical abstract

Keywords: SARS-CoV-2 Spike RBD, hACE2, Blockade mechanism, Structure-activity relationship, Anti-SARS-CoV-2 drugs screening strategy
Highlights
-
•
Drug property and binding energy were negative correlation for compounds.
-
•
8 kinds of common structures with high anti-SARS-CoV-2 activity were selected.
-
•
The structure-activity relationship between drugs and SARS-CoV-2 therapy was built.
-
•
This is the first study on screening strategy of anti-SARS-CoV-2 drugs.
Abstract
Corona virus disease 2019 has spread worldwide, and appropriate drug design and screening activities are required to overcome the associated pandemic. Using computational simulation, blockade mechanism of SARS-CoV-2 spike receptor binding domain (S RBD) and human angiotensin converting enzyme 2 (hACE2) was clarified based on interactions between RBD and hesperidin. Interactions between anti-SARS-CoV-2 drugs and therapy were investigated based on the binding energy and druggability of the compounds, and they exhibited negative correlations; the compounds were classified into eight common types of structures with highest activity. An anti-SARS-CoV-2 drug screening strategy based on blocking S RBD/hACE2 binding was established according to the first key change (interactions between hesperidin and S RBD/hACE2) vs the second key change (interactions between anti-SARS-CoV-2 drugs and RBD/hACE2) trends. Our findings provide valuable information on the mechanism of RBD/hACE2 binding and on the associated screening strategies for anti-SARS-CoV-2 drugs based on blocking mechanisms of pockets.
1. Introduction
Corona virus disease 2019 (COVID-19) is a highly infectious respiratory disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2); it is transmitted through air and body fluids and secretions, with most patients experiencing fever, cough, and dyspnea (Guan et al., 2020, Huang et al., 2020, Phan et al., 2020, Zhu et al., 2020). The COVID-19 pandemic has become a serious threat to human physical and global security (Park et al., 2020, Yuan et al., 2020, Zaki et al., 2012). Since the number of COVID-19-infected individuals worldwide is increasing, it is necessary to investigate the infection mechanism of SARS-CoV-2 in host cells and provide specific drugs for COVID-19 pandemic treatment.
It has been confirmed that SARS-CoV-2 mainly infects human cells by recognizing human angiotensin converting enzyme 2 (hACE2) (Lan et al., 2020, Li et al., 2003, Li et al., 2005, Zhou et al., 2020). According to its infection mechanism in human cells, SARS-CoV-2 spike is divided into S1 and S2 subtypes: S1 subtype binds to hACE2, whereas S2 subtype participates in cell membrane fusion. Full-length hACE2 contains N-terminal PD domain and C-terminal CLD domain, in which PD domain interacts with S1 (Hoffmann et al., 2020, Jin et al., 2020, Li et al., 2003, Prabakaran et al., 2003). The cryo-EM structure of SARS-CoV-2 S RBD-hACE2/hB0AT1 complex showed that in the interaction between two S RBD domains and a heterodimer of hACE2/hB0AT1, S RBD only binds to closed type of hACE2 PD domain in a 1:1 mode (Lv et al., 2020, Song et al., 2018, Yan et al., 2020, Zheng et al., 2006). The loop structure of S1 RBD domain as hACE2 binding area binds to hACE2 PD domain, whereas CLD of hACE2 participates in binding of hB0AT1. Therefore, blockade of binding of SARS-CoV-2 spike receptor binding domain (S RBD) to hACE2 PD, which blocks binding of S RBD to hACE2, can block combination of SARS-CoV-2 and hACE2 (Bao et al., 2020, Burgueño et al., 2020, Fan et al., 2020, Li et al., 2006). Since the mechanism underlying the interaction and blockade of S RBD/hACE2 have been clarified via the interaction type of S RBD/hACE2 and combination of hesperidin (HES) with Spike (Wu et al., 2020), respectively. By blocking the interaction of S RBD/hACE2 to host cells protected from SARS-CoV-2 infection significantly helped mitigate COVID-19. Thereafter, blockers of the interaction between S RBD and hACE2 need to be studied in detail (Haggag et al., 2020, Wang et al., 2020).
Currently, a growing number of drugs are designed based on computational virtual screening and molecular simulation because of low cost, efficiency, and accuracy of these methods (Buonanno et al., 2020, Goyal et al., 2020, Jagtap et al., 2020, Li et al., 2020). Computational simulation technology has largely facilitated drug design and set up quantitative structure-function relationship studies in virtual pharmacological screening and has been widely used (Akhtar et al., 2020, Basu et al., 2020). Computational virtual screening and molecular simulations are based on computational chemistry approaches to obtain lead compounds that are designed and optimized in silico to interact with target receptors (Nguyen et al., 2011, Oliveira et al., 2020). Molecular docking is based on stoichiometric simulation to identify and predict the structure of receptor and ligand and subsequently clarify the mechanism underlying the receptor-ligand interaction (Vivek-Ananth et al., 2020). However, published studies have only shown screening data of SARS-CoV-2, and binding mechanism of compounds to hACE2 binding area and screening strategy for anti-SARS-CoV-2 drugs via hACE2 binding area remain unclear. Therefore, we propose a novel pathway to illustrate structure-activity relationship between anti-SARS-CoV-2 drugs and COVID-19 treatment and established a screening strategy for anti-SARS-CoV-2 drugs using S RBD/hACE2 pockets.
In this study, blockade mechanism of S RBD/hACE2 based on most active compounds was proposed using binding energy in order of Z601-7647/P3 > 2159-4156/P3 > 4491–0722/P3 > HES (as control drug) /RBD > 2595-0160/P2 > P496-0785/P1 > C200-9609/P2 > Y041-7672/P2 > 8020-8119/P1. The binding residues included LEU455, PHE456, TYR489, GLN493 (for P1) or LEU441, ASP442, LYS444, ASN450, TYR451 (for P2) or PHE456, ARG457, LYS458, ASP467, GLU471 (for P3), and virtual screening pockets (P1, P2, and P3), while interaction types included hydrogen-bonds, π-π interaction, hydrophobic interaction, and Van der Waals' (VDW) forces. The key functional groups included —NH2, —OH, phenyl group, nitrogen-containing conjugate ring, —O—, —CO—, —CONH—, and —COO—. There were negative correlations between most active compounds and anti-SARS-CoV-2 therapy, based on binding energy and druggability of compounds for anti-SARS-CoV-2, and compounds were classified into eight types of common structures with the highest activity. The screening strategy of anti-SARS-CoV-2 drugs via blocking of binding between S RBD/hACE2 was established based on the first key change (interactions between HES and S RBD/hACE2) vs the second key change (interactions between anti-SARS-CoV-2 drugs and S RBD/hACE2) trends.
2. Methods
2.1. File processing for virtual receptor screening
The original model of the SARS-CoV-2 S RBD/PD ACE2/hB0AT1 complex (PDB ID 6M17) was successfully downloaded from Protein Data Bank (www.rcsb.org). The computational virtual screening of receptors was performed using Molecular Operating Environment software (MOE 19.01); the RBD model was developed based on the ACE2 binding area of the original structure of the SARS-CoV-2 S RBD/PD ACE2/hB0AT1 complex by the Sequence Editor function in MOE, and models for three pockets (P1, P2, and P3) were prepared using the Site Finder function in MOE. Before screening, P1, P2, P3, and RBD were protonated by adding hydrogen atoms and minimized for energy optimization. Subsequently, the P1, P2, and P3 receptor files were acquired using the apopdb2receptor tool in OpenEye (Release 3.2.0.2), and then the P1, P2, P3, and RBD receptor files were saved as .pdb files.
2.2. Prepared compounds database
The compound database was obtained from a Chemdiv subset (Version 2019), which contains 1,535,478 small molecules with skeleton diversity. To ensure the global conformation of compounds with computational virtual screening processing, on average, each compound generated approximately 50 conformations using the Omega2 tool in OpenEye software. Finally, 1,535,478 small molecules of Chemdiv were converted into 50,679,311 conformations.
2.3. Screening compounds by binding energy
For pocket-based computational virtual screening, ligands were continuously resiliently made to docked with the P1, P2, and P3 receptors, which were represented in binding energy by FRED software on the system workstation of Ubuntu kylin 15.10 (CPU: 40; Memory: 64; SSD: 512G) to identify drug candidates. Compounds of each conformation were scored according to the FRED, and component scores were as true, hitlist size was set at 30,000, the docked molecule file was set as .sdf type, and other parameters set at default values. Through visual inspection, compounds outside the active site, as well as those that fit weakly to the active site, were eliminated.
2.4. Screening compounds based on druggability
Ligands were docked with P1, P2, and P3 receptors, which were represented in druggability by Stardrop (version 6.5.0). Approved screening principle for oral non-central nervous system drugs for identifying possible druggability scores, and the druggability index included logS, logP, molecular weight (MW), number of rotatable bonds, hydrogen bond, topological polar surface area of molecule, the binding affinity to 2C9, the binding affinity to 2D6, the binding affinity to hERG, human intestine absorption, blood brain barrier category, P-glycoprotein, plasma protein binding (>90%).
There are 175 compounds binding to P1 and the druggability score ranged from 0.15 to 0.85. The 175 compounds were cluster analysis by common substructure. Based on the similarity of 175 compounds the MCS cluster set as 0.6, 0.7, 0.8, 0.9, then 175 compounds generated four groups and the number of each group as 29, 33, 33, 21, respectively. The compounds presented as multiple skeleton classifications and lots number of compounds in each group, when MCS cluster set as 0.7. Finally, 175 compounds binding to P1 confirmed 0.7 as the MCS cluster.
There are 610 compounds binding to P2 and the druggability score ranged from 0.04 to 0.87. The 610 compounds were cluster analysis by common substructure. Based on the similarity of 610 compounds the MCS cluster set as 0.3, 0.4, 0.5, 0.6, then 610 compounds generated four groups and the number of each group as 59, 64, 86, 96, respectively. The compounds presented as multiple skeleton classifications and lots number of compounds in each group, when MCS cluster set as 0.5. Finally, 610 compounds binding to P2 confirmed 0.5 as the MCS cluster.
There are 423 compounds binding to P3 and the druggability score ranged from 0.01 to 0.82. The 423 compounds were cluster analysis by common substructure. Based on the similarity of 423 compounds the MCS cluster set as 0.5, 0.6, 0.7, 0.8, then 423 compounds generated four groups and the number of each group as 53, 59, 57, 64, respectively. The compounds presented as multiple skeleton classifications and lots number of compounds in each group, when MCS cluster set as 0.6. Finally, 423 compounds binding to P3 confirmed 0.6 as the MCS cluster.
2.5. Compounds data processing
Firstly, the initial data processing of binding energy and druggability scores of compounds binding to P1, P2, P3 were evaluated using curve graphs, which facilitated the preliminary correlation analysis of binding energy and druggability. Afterward, the initial binding energy data and druggability scores of compounds binding to P1, P2, and P3 were normalized could be expressed as follows: y = (x − xmin)/(xmax − xmin), where x, xmin, and xmax were any arbitrary value, the minimum value, and the maximum value of the initial data of binding energy or druggability score, respectively. The normalized data processing of binding energy and druggability of compounds binding to P1, P2, P3 were estimated using curve graphs, which facilitated the performing of a correlation analysis between binding energy and druggability. Similarly, the normalized druggability data were named η1 and the normalized binding energy data were named η2, which were used to estimate level of druggability and binding energy, respectively. Afterward, the complex level of druggability and binding energy were expressed as the compositive relationship of η1 and η2. The sums of η1 + η2 were used to estimate the ultimate level of druggability and binding energy. What’s more, the high level of η1 or η2 ranged from 0.7 to 1, the middle level of η1 or η2 ranged from 0.4 to 0.7, and the low level of η1 or η2 ranged from 0 to 0.4. The high level of η1 + η2 was ranged from 1.2 to 2, the middle level of η1 + η2 ranged from 0.8 to 1.2, and the low level of η1 + η2 ranged from 0 to 0.8. Therefore, the initial processing of binding energy and druggability score data of compounds binding to P1, P2, and P3 were evaluated using a collective diagram, which yielded the preliminary correlation analysis and the ultimate druggability level and binding energy. Finally, the sum of the normalized binding energy and druggability data for compounds binding to P1, P2, P3 were assessed using a curve graph, combined with the preliminary correlation analysis, and the correlation analysis of binding energy and druggability, which revealed the compounds with the highest activity binding to receptors.
2.6. Molecular docking simulation
The P1, P2, P3, and RBD receptor files were obtained from 2.1. The ligand model for HES was obtained from Chemical Book Web site (http://www.chemicalbook.cn/), and the ligand models for the compounds with the highest activity were prepared using the Molecule Builder function in MOE. Before molecular simulation calculations, receptor P1, P2, P3, and RBD were protonated by adding hydrogen atoms, and small molecule ligands were minimized for energy optimization. Subsequently, the interactions between P1, P2, P3, RBD receptor, and HES or the most active compounds were simulated, and key parameters such as total combination areas, hydrogen-bonds, and VDW for the main interaction sites were obtained.
3. Results
3.1. Pocket structures
The binding of hACE2 binding area loop domain and hACE2 PD was key mechanism of interaction with S RBD/hACE2, and the KD value of S and hACE2 was 15 nM, indicating that it was difficult for small molecules to achieve direct contact with hACE2 binding area (Wrapp et al., 2020, Zhou et al., 2020). Therefore, small molecules could block binding of S and hACE2 by occupying hACE2 binding area loop residues. Accordingly, we selected P1, P2, and P3 as virtual screening regions and interaction domains for small molecules and confirmed RBD as interaction region for HES. All regions above were located near hACE2 binding area loop structure (Fig. 1 a). The positions and residues of P1, P2, and P3 receptors in hACE2 binding area are presented, with P1 located within hACE2 binding area, and P2 and P3 located near hACE2 binding area (Fig. 1b). The P1 receptor was determined from loop structure of hACE2 binding area, and its residues included LYS417, TYR449, TYR453, GLN474, PHE486, ASN487, TYR489, GLN493, TYR495, GLN498, THR500, ASN501, and TYR505 (Fig. 1c). The space structure of P2 receptor was shallow and long; it could accommodate approximately 32 residues, had a druggability score of 1.96, and included 15 highly hydrophobic residues, and space structure of P3 receptor was deep; it could accommodate approximately 25 residues, had a druggability score of 0.76, and included 12 highly hydrophobic residues (Fig. S1). The residues of P2 and P3 receptors included THR345, ARG346, PHE347, ALA348, SER349, TYR351, ALA352, LEU441, ASP442, LYS444, ASN448, ASN450, TYR451, LEU452, ARG509 and ARG454, and PHE456, ARG457, LYS458, SER459, ASP467, SER469, THR470, GLU471, ILE472, TYR473, and PRO491, respectively (Fig. 1d). The space structure of RBD could accommodate approximately 59 residues, including 12 highly hydrophobic residues; its residues included ARG454, PHE456, ARG457, LYS458, ASP467, SER469, THR470, GLU471, ILE472, TYR473, and PRO491 (Fig. S3a).
Fig. 1.

Structure of the SARS-CoV-2 S RBD-hACE2/hB0AT1 complex and the P1, P2, and P3 receptors. (a) Cryo-EM structure of the SARS-CoV-2 S RBD-hACE2/hB0AT1 complex. (b) Positions of P1, P2, and P3 on the hACE2 binding area. (c) Residues details of P1. (d) Residue details of P2 and P3.
3.2. Binding energies of compounds
A large number of compounds were acquired via computational virtual screening (Fig. 2 ), and top 30,000 compounds binding to P1, P2, and P3 were obtained according to primary binding energy, respectively. The relationship between binding energy and logS of top 30,000 compounds binding to P1, P2, and P3 indicated that binding energy was more than −12 kcal/mol and less than −4 kcal/mol. The binding energy of compounds binding to P1 and P2 was mostly between −7 kcal/mol and −4 kcal/mol; compounds with binding energy greater than −7 kcal/mol were considered compounds with high activity and labeled on a blue and pink background. There were 175 higher active compounds binding to P1 as well as 610 higher active compounds binding to P2 (Fig. S2a, Fig. S2e). The binding energy of compounds binding to P3 was mostly between −8.5 kcal/mol and −6 kcal/mol; compounds with binding energy greater than −8.5 kcal/mol were considered to have high activity and labeled on a blue and pink background. There were 423 higher active compounds binding to P3 (Fig. S2i).
Fig. 2.

Computational virtual screening of the P1, P2, and P3 pockets.
3.3. Druggability of the compounds
The 175 compounds with high activity binding to P1 were subjected to diversity analysis in different MCS clusters (Fig. S2b). In total, 33 compounds were selected from 175 compounds with high activity binding to P1 according to binding energy and druggability in MCS cluster of 0.7 (Fig. S2c). The 175 compounds were divided into 33 groups in MCS cluster of 0.7, and 18 groups with higher skeletal diversity were selected from 33 groups. Based on binding energy, druggability, and logS, 33 compounds with more than 18 common skeletons were acquired from 18 groups; here, compounds from group 12 and group 15 with lower druggability scores but higher binding energies were reserved. The binding energy and druggability of 33 compounds binding to P1 were well correlated (Fig. S2d). The 33 compounds binding to P1 with 400 Da conformed to Lipinski’s rule of five. However, 8011-6589 of 33 compounds were confirmed to have low total druggability, and Y042-6589 could be metabolized by CYP 2D6 enzyme.
The 610 compounds with high activity binding to P2 were subjected to diversity analysis in different MCS clusters (Fig. S2f). In total, 85 compounds were selected from 610 compounds with high activity binding to P2 according to binding energy and druggability in MCS cluster of 0.5 (Fig. S2g). The 610 compounds with high activity were divided into 86 groups in MCS cluster of 0.5, and 41 groups with higher skeletal diversity were selected from 86 groups. Based on binding energy, druggability, and logS, 85 compounds with more than 41 common skeletons were acquired from 41 groups; here, compounds of group 0 (8017-3526), group 4 (D585-0103), and group 21 (8012-4236) with lower druggability scores but higher binding energies were reserved. The binding energy and druggability of 85 compounds binding to P2 were well correlated (Fig. S2h). The 85 compounds binding to P2 with 400 Da conformed to Lipinski’s rule of five. However, G786-2290, D306-0541, D691-0064, 8006-6737, D058-0264, Y042-6425, T040-0452, and 8019-9741 could be metabolized by P450 enzyme, and Y020-5878, with a predicted IC50 value lower than 10 μM, was assumed to pose a potent risk to heart safety. Moreover, 0353-0054, 1530-0255, and Y021-0068 could pose a risk of crossing blood-brain barrier.
The 423 compounds with high activity binding to P3 were processed by diversity analysis in different MCS clusters (Fig. S2j). In total, 65 compounds were selected from 423 compounds with high activity binding to P3 according to binding energy and druggability in MCS cluster of 0.6 (Fig. S2k). The 423 compounds with high activity were divided into 59 groups in MCS cluster of 0.6, and 30 groups with higher skeletal diversity were selected from 59 groups. Based on binding energy, druggability, and logS, 65 compounds with more than 30 common skeletons were acquired from 30 groups; here, compounds of group 41 (7462-0039) with lower druggability scores but higher binding energies were reserved. The binding energy and druggability of 65 compounds binding to P3 were well correlated (Fig. S2l). The 65 compounds binding to P3 with 400 Da conformed to Lipinski’s rule of five. However, 7462-0039, D278-0612, Y042-7003, and 8006-9126 of 65 compounds could be metabolized by P450; moreover, 7462-0039 and G582-3109, with predicted IC50 values lower than 10 μM, were assumed to pose a potent risk to heart safety, and E822-0518, G582-2789, C514-1484, and G582-3109 could pose a risk of crossing blood-brain barrier.
3.4. Most active compounds
Based on curve graph, collective diagram, and ultimate level in initial data and normalized data of binding energy and druggability of compounds binding to P1, P2, and P3, preliminary negative correlation analysis of binding energy and druggability was conducted (Fig. S4, Fig. S5, Fig. 3 a, c, e, Table S1). Furthermore, based on curve graph of the sum of normalized data for binding energy and druggability, compounds with highest activity were obtained as follows. From 33 compounds binding to P1, 8 compounds with highest activity were obtained: P1-1, P1-2, P1-3, P1-11, P1-12, P1-17, P1-19, and P1-25. From 85 compounds binding to P2, 17 compounds with highest activity were identified: P2-1, P2-3, P2-6, P2-9, P2-11, P2-17, P2-20, P2-21, P2-26, P2-34, P2-37, P2-39, P2-43, P2-45, P2-46, P2-48, and P2-57. From 65 compounds binding to P3, 17 compounds with highest activity were acquired: P3-1, P3-2, P3-3, P3-5, P3-6, P3-7, P3-8, P3-11, P3-16, P3-17, P3-19, P3-23, P3-24, P3-25, P3-28, P3-36, and P3-44 (Fig. 3b, d, f, Table S1, Table S2).
Fig. 3.

Compounds data processing.
3.5. Molecular mechanisms underlying the receptor binding of the compounds with the highest activity
To better elucidate molecular mechanism underlying binding of eight compounds with highest activity to P1, these eight compounds were divided into two groups by structural similarity: first group consisted of P1-2, P1-11, P1-12, P1-17, and P1-19, and second group consisted of P1-1, P1-3, and P1-25. Next, P1-17 (P496-0785) and P1-1 (8020-8119) were selected for molecular docking simulation with P1. Between P496-0785 and 8020-8119 with P1, there was a reversible binding step through hydrogen bonding as well as hydrophobic interactions involving LEU455, PHE456, TYR489, and GLN493 residues. Accordingly, hydrogen bonds, π-π interactions, hydrophobic interactions, and VDW in main interaction sites were selected as key parameters to evaluate blockade mechanism of compounds with highest activity binding to P1. Molecular simulation provided information on the hydrogen bonds, π-π interactions, hydrophobic interactions, VDW of P496-0785 and 8020-8119 binding to P1. Compared with those of HES and RBD, the hydrogen bonds of P496-0785 and 8020-8119 with P1 were obviously decreased, and hydrophobic interactions of P496-0785 and 8020-8119 were obviously increased (Fig. 4 a, b, Fig. S3d). The —CONH- and —NH2 groups of P496-0785 and 8020-8119 interacted with LEU455, PHE456, and GLN493 through hydrogen bonds and phenyl group, whereas oxygen-containing groups interacted with TYR489 via hydrophobic interactions, including those among pharmacophore features, such as hydrogen bond acceptors (Acc), hydrogen bond donors (Don) & Acc, and aromatic centers (Aro) (Fig. 4i, j, Fig. S6a, Fig. S6b, Fig. S7a, Fig. S7b, Table S3). Therefore, —NH2, —CONH-, nitrogen-containing conjugate ring, and phenyl group were identified as key pharmacophores in interactions between compounds with highest activity and P1. The binding energy of P496-0785 and 8020-8119 to P1 was −9.02 kcal/mol and −8.30 kcal/mol, respectively, with an average of −8.66 kcal/mol (Table S3). The binding energies of P496-0785 and 8020-8119 to P1 were lower than those of HES and RBD, but the difference was not significant.
Fig. 4.

The hydrogen bond and hydrophobic interaction map of the compounds with the highest activity and the interaction between the compounds with the highest activity with P1, P2, or P3. (a ~ h) The VDW map of the compounds with the highest activity; the red area represents a hydrogen bond, the green area represents a hydrophobic interaction. (i ~ j) Interaction between the compounds with the highest activity and P1. (k ~ m) Interaction between the compounds with the highest activity and P2. (n ~ p) Interaction between the compounds with the highest activity and P3. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Similarly, to better understand molecular mechanism underlying binding of 17 compounds with highest activity to P2, these compounds were divided into three groups by structural similarity: first group consisted of P2-6, P2-43, P2-48, and P2-3; second group included P2-9, P2-34, P2-37, P2-39, P2-45, P2-46, and P2-1; and third group comprised P2-11, P2-17, P2-20, P2-21, P2-26, and P2-57. Next, P2-48 (2595-0160), P2-45 (C200-9609), and P2-26 (Y041-7672) were selected for molecular docking simulation with P2. Between 2595-0160, C200-9609, and Y041-7672 with P2, there was a reversible binding step through hydrogen bonding as well as hydrophobic interactions involving LEU441, ASP442, LYS444, ASN450, and TYR451 residues. Accordingly, hydrogen bonds, π-π interactions, hydrophobic interactions, and VDW in main interaction sites were selected as key parameters to evaluate blockade mechanism of compounds with highest activity binding to P2. The molecular simulation provided information on hydrogen bonds, π-π interactions, hydrophobic interactions, and VDW of 2595-0160, C200-9609, and Y041-7672 binding to P2. Compared with those of HES and RBD, hydrogen bonds of 2595-0160, C200-9609, and Y041-7672 with P2 were obviously decreased, and hydrophobic interactions of 2595-0160, C200-9609, and Y041-7672 were obviously increased (Fig. 4c, d, e, Fig. S3d). The —O—, —CO—, —OH, N—, —CONH—, and —NH— groups of 2595-0160, C200-9609, and Y041-7672 interacted with LEU441, ASP442, ASN450, and TYR451 through hydrogen bonds; phenyl group and the nitrogen-containing conjugate ring of C200-9609 and Y041-7672 interacted with LYS444 through hydrophobic interactions, and the —CO— group of 2595-0160 interacted with LYS444 through hydrogen bonds, including those among pharmacophore groups, such as Acc, Don & Acc, Don, Aro, and Hyd|Aro (Fig. 4k, l, m, Fig. S6c, Fig. S6d, Fig. S6e, Fig. S7c, Fig. S7d, Fig. S7e, Table S3). Therefore, —O—, —CO—, —OH, N—, —CONH—, —NH—, phenyl group, and nitrogen-containing conjugate ring were identified as key pharmacophores in interactions between compounds with highest activity and P2. The binding energy of 2595-0160, C200-9609, and Y041-7672 to P2 was −9.27 kcal/mol, −8.96 kcal/mol, and −8.68 kcal/mol, respectively, with an average of −8.97 kcal/mol (Table S3). The binding energies of 2595-0160, C200-9609, and Y041-7672 to P2 were lower than those of HES and RBD, but the difference was not significant.
Equally, to better understand molecular mechanism underlying binding of 17 compounds with highest activity to P3, these compounds were divided into 3 groups by structural similarity: first group consisted of P3-1, P3-2, P3-5, P3-7, P3-24, P3-25, P3-28, and P3-44; second group included P3-3, P3-6, P3-8, P3-11, P3-16, P3-17, P3-19, and P3-23; and third group comprised P3-36. Next, P3-44 (Z601-7647), P3-16 (4491-0722), and P3-36 (2159-4156) were selected for molecular docking simulation with P3. Between Z601-7647, 4491-0722, and 2159-4156 with P3, there was a reversible binding step through hydrogen bonding as well as hydrophobic interactions involving PHE456, ARG457, LYS458, ASP467, and GLU471 residues. Accordingly, hydrogen bonds, π-π interactions, hydrophobic interactions, and VDW in main interaction sites were selected as key parameters for evaluating blockade mechanism of compounds with highest activity binding to P3. Molecular simulation provided information on hydrogen bonds, π-π interactions, hydrophobic interactions, and VDW of Z601-7647, 4491-0722, and 2159-4156 binding to P3. Compared with those of HES and RBD, hydrogen bonds of Z601-7647, 4491-0722, and 2159-4156 with P3 were obviously decreased, and hydrophobic interactions of Z601-7647, 4491-0722, and 2159-4156 were obviously increased (Fig. 4f, g, h, Fig. S3d). The —NH—, —CONH—, —CO—, and N— of Z601-7647, 4491-0722, and 2159-4156 interacted with PHE456, LYS458, ASP467, and GLU471 through hydrogen bonds; moreover, phenyl group and nitrogen-containing conjugate ring interacted with PHE456, ARG457, and LYS458 via hydrophobic interactions, including those among pharmacophore groups, such as Acc, Don, and Aro (Fig. 4n, o, p, Fig. S6f, Fig. S6g, Fig. S6h, Fig. S7f, Fig. S7g, Fig. S7h, Table S3). Therefore, —NH—, —CONH—, —CO—, N—, phenyl group and nitrogen-containing conjugate ring were identified as key pharmacophores in interactions between compounds with highest activity and P3. The binding energy of Z601-7647, 4491-0722, and 2159-4156 to P3 was −11.20 kcal/mol, −10.32 kcal/mol, and −10.61 kcal/mol, respectively, with an average of −10.71 kcal/mol (Table S3). Compared with those of HES and RBD, binding energies of Z601-7647, 4491-0722, and 2159-4156 to P3 were obviously increased.
Between HES and RBD, there was a reversible binding step through hydrogen bonding involving PHE456, ARG457, LYS458, ASP467, and GLU471 residues. Accordingly, hydrogen bonds and VDW in main interaction sites were selected as key parameters to evaluate blockade mechanism of HES binding to RBD. Molecular simulation provided information on hydrogen bonds and VDW of HES binding to RBD. Compared with those of compounds with highest activity for P1, P2, or P3 receptors, hydrogen bonds of HES with RBD were obviously increased (Fig. S3b, Fig. S3d). The —O—, —OH, and —CO— of HES interacted with PHE456, ARG457, LYS458, ASP467, and GLU471 through hydrogen bonds (Fig. S3e, Fig. S3f, Fig. S3g, Fig. S3h, Table S3). Therefore, —O—, —OH, and —CO— were identified as key pharmacophores in interactions between HES and RBD, and phenyl group and oxygen-containing group were considered significant for combination of HES and RBD by basic structure of HES. The binding energy of HES and RBD was −9.36 kcal/mol (Table S3), showing greater interaction. The order of binding energy of HES with RBD and compounds with highest activity with P1, P2, or P3 was as follows: compounds with highest activity with P3 (−10.71 kcal/mol) > HES with RBD (−9.36 kcal/mol) > compounds with highest activity with P2 (−8.97 kcal/mol) > compounds with highest activity with P1 (−8.66 kcal/mol). The sequence for total score of hydrogen-bonds with highest active compounds and Spike is as follow: 2595-0160 (397.3%) > HES (210.2%) > Y041-7672 (163.1%) > C200-9609 (150.6%) > 2159-4156 (137.0%) > 8020-8119 (136.6%) > P496-0785 (90.4%) > 4491-0722 (77.6%) > Z601-7647 (54.2%) (Fig. S8, Table S3). The active group —CO— of 2595-0160, having a higher total score than HES, interacted with LYS444 (basic amino acid), the —O— group interacted with ASP442 (acidic amino acid), the —CO— and —O— groups interacted with LEU441, ASN450, TYR451 (neutral amino acid). The active groups —O—, —OH, and —CO— in HES interacted with ARG457, LYS458 (basic amino acid), the —OH group interacted with GLU471 (acidic amino acid), the —OH group interacted with PHE456 (neutral amino acid). Similarly, key active groups of other highest active compounds interacting with Spike protein included —O—, —CO—, —OH, phenyl group, and nitrogen-containing conjugate ring.
3.6. Common structures of compounds with the highest activity
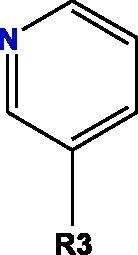
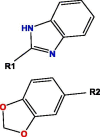
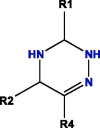
Based on blockade mechanism and key pharmacophores, we obtained common structures of most active compounds binding to P1, P2, and P3 (Table 1 , Table S4). Accordingly, benzene and nitrogen-containing conjugate rings were key groups constituting common structures of most active compounds. The common structures with compounds with highest activity binding to P1 were two types, including P1a and P1b, and could displace each other. Furthermore, substituent group R1 of P1a were —Cl or —CH3, R2 was —CH3, and R3 was —CH3. Conversely, substituent group R1 of P1b was nitrogen-containing conjugate ring (Table S4), and R2 were —CH2CH2NH2, —CH2COOH, or —CH3. The common structures of compounds with highest activity binding to P2 were three types, including P2a, P2b, and P2c, and could substitute each other. Furthermore, substituent R1 groups of P2a were —H, —NH2, phenyl group, methylbenzene, fluorobenzene, anisole, R2 were —OH, —SH, or —SCH2CONH2, R3 were —H, —OH, —NH2, or —CN or specific structure in Table S4, and R4 were —COOCH3, —NHCH3, or —SCH3. The substituent R1 groups of P2b were —H, —CH2OH, or specific structure in Table S4, and R2 groups were —SCH2CONHCH2CH2CH3, —SCH2CONHCH2CH2CH2CH3, —SCH2CONHCH2CH2CH(CH3)2, —H, or specific structure in Table S4. In addition, substituent R3 groups were —OH or —NH2, R1 substituent groups of P2c were —NH2, —NHCH3, or O, R2 substituent groups were —CH2Cl, —Cl, or O, and R3 and R4 were specific structure presented in Table S4.
Table 1.
Common structures of the most active compounds.
| Common structures | ||
|---|---|---|
| P1a |  |
 |
| P1b | 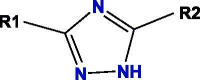 |
 |
| P2a | 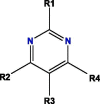 |
 |
| P2b | 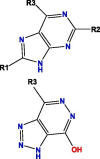 |
 |
| P2c | 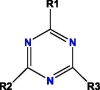 |
 |
| P3a | 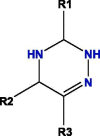 |
|
| P3b | 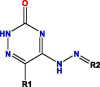 |
|
| P3c |  |
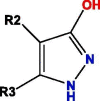 |
Notes: R1, R2, R3, R4 and R5 represent different functional groups in Table S4.
The common structures of compounds with highest activity binding to P3 were three types, including P3a, P3b, and P3c, and P3c could be displaced by two other types. Furthermore, substituent group R1 of P3a was O, substituent R2 was O, and substituent R3 was specific structure displayed in Table S4, substituent group R1 of P3b was —H or —CH3, and R2 was specific structure in Table S4. The substituent group R1 groups of P3c were —OCH3 or —H, and R2 groups were —H, —Cl, or —CH3, or specific structure in Table S4, while R3 groups were —H or —CH3, and R4 groups were —CONHCH2CH2COOH, —CONHCH2CH2CH2COOH, —CONHCH2CH2NH2, —CONHCH2CH2CH2N(CH3)2, —CH3, or specific structure in Table S4. The R5 groups were -H or specific structure shown in Table S4.
4. Discussions
Before elucidating the blockade mechanism of SARS-CoV-2 S RBD and hACE2, the binding mechanism should be described as the interaction between the loop domain of ACE2 binding area and the PD structure of ACE2 based on the polar residues of LYS417, TYR453, GLN474, PHE486, GLN498, THR500, and ASN50; the key interactions were via hydrogen bonding and hydrophobic interactions (Zhou et al., 2020). The results based on the interactions between HES and RBD indicated that the key sites were PHE456, ARG457, LYS458, ASP467, and GLU471, and disrupted contact between S and hACE2, and the significant interactions were in the form of hydrogen-bonds and VDW, while —O—, —OH, —CO—, phenyl group and oxygen-containing group were the key pharmacophores, with a binding energy of −9.36 kcal/mol. The interactions between the most active compounds and P1 indicated that the key sites at which contact between S and hACE2 was disrupted were LEU455, PHE456, TYR489, and GLN493, and the significant interactions were in the form of hydrogen-bonds, π-π interaction, hydrophobic interactions, and VDW, and —NH2, —CONH—, nitrogen-containing conjugate ring, and phenyl group were the key pharmacophores, with a binding energy range of −10 ~ −7 kcal/mol. The interactions between the most active compounds and P2 indicated that the key sites at which contact between S and hACE2 was disrupted were LEU441, ASP442, LYS444, ASN450, and TYR451, and the significant interactions were in the form of hydrogen-bonds, π-π interaction, hydrophobic interaction, and VDW, and —O—, —CO—, —OH, N—, —CONH—, —NH—, phenyl group, and nitrogen-containing conjugate ring were the key pharmacophores, with a binding energy range of −10 ~ −7 kcal/mol. Interaction between the most active compounds and P3 indicated that the key sites at which contact between S and hACE2 was disrupted were PHE456, ARG457, LYS458, ASP467, and GLU471, and the significant interactions were in the form of hydrogen-bonds, π-π interaction, hydrophobic interaction, and VDW; and —NH—, —CONH—, —CO—, N—, phenyl group, and nitrogen-containing conjugate ring were the key pharmacophores, with a binding energy range of −12 ~ −8 kcal/mol. Accordingly, the blockade mechanism of S and ACE2 was the binding of the most active compounds with ACE2 by LEU455, PHE456, TYR489, GLN493 or LEU441, ASP442, LYS444, ASN450, TYR451 or PHE456, ARG457, LYS458, ASP467, and GLU471 residues, and the significant interactions were in the form of hydrogen-bonds, π-π interaction, hydrophobic interaction, and VDW. —NH2, —CONH—, nitrogen-containing conjugate ring, phenyl group or —O—, —CO—, —OH, N—, —CONH—, —NH—, phenyl group, nitrogen-containing conjugate ring or —NH—, —CONH—, —CO—, N—, phenyl group, nitrogen-containing conjugate ring, or —O—, —OH, —CO—, phenyl group, and oxygen-containing group were identified as the key pharmacophores, and the range of binding energy was −12 ~ −7 kcal/mol.
In summary, we present the blockade mechanism of S and ACE2 as binding between the most active compounds and ACE2-binding area, with binding energy in the order of Z601-7647/P3 > 2159-4156/P3 > 4491-0722/P3 > HES/RBD > 2595-0160/P2 > P496-0785/P1 > C200-9609/P2 > Y041-7672/P2 > 8020-8119/P1. The average binding energy for the most active compounds, P1, P2, and P3, were −8.66 kcal/mol, −8.97 kcal/mol, −10.71 kcal/mol, respectively, and the binding residues were LEU455, PHE456, TYR489, and GLN493 (for P1), LEU441, ASP442, LYS444, ASN450, and TYR451 (for P2), and PHE456, ARG457, LYS458, ASP467, and GLU471 (for P3), respectively. The significant interactions were in the form of hydrogen-bonds, π-π interaction, hydrophobic interaction, and VDW, and the hydrophilic groups (—NH2, —OH), the conjugate rings (phenyl group, nitrogen-containing conjugate ring), and oxygen-containing groups (—O—, —CO—, —CONH—, —COO—) were the key functional groups.
Based on the structures of small molecules and the interactions between small molecules and P1, P2, P3, or RBD, all the compounds exhibited the basic and important structure of the phenyl group, and HES and RBD was contained an oxygen-containing group, and the compounds that interacted the most with P1, P2, or P3 had a nitrogen-containing conjugate ring. Compared to HES and RBD, the most active compounds interacting with P1, P2, and P3 were had a —CONH— group. Compared to the compounds exhibiting the greatest activity with P1, HES, and RBD, the most active compounds with P2 or P3 included a —CO— group. In addition, compared to the compounds with the greatest activity with P1 and P3, HES and RBD and the most active compounds with P2 had —O— and —OH. Compared to HES and RBD and the most active compounds with P1, the highest active compounds with P2 or P3 included —NH— and N— groups. Compared to HES and RBD and the highest activity with P2 or P3, the compounds with the highest activity with P1 included —NH2. The order of binding energy was Z601-7647/P3 > 2159-4156/P3 > 4491-0722/P3 > HES/RBD > 2595-0160/P2 > P496-0785/P1 > C200-9609/P2 > Y041-7672/P2 > 8020-8119/P1; meanwhile the order of average binding energy was P3 > P2 > P1.
The correlation between the most active compounds and anti-SARS-CoV-2 therapy was investigated in the present study. The binding energy and druggability of compounds for anti-SARS-CoV-2 were negatively correlated, and the structures of compounds were consistent with the eight types of common structures with the highest activity, and the range of binding energy among the compounds with the greatest activity with ACE2 binding area was −12–−7 kcal/mol, and the binding residues were LEU455, PHE456, TYR489, GLN493, LEU441, ASP442, LYS444, ASN450, TYR451, PHE456, ARG457, LYS458, ASP467, and GLU471, and the significant interactions were in the form of hydrogen-bonds, π-π interaction, hydrophobic interaction, and VDW, and —NH2, —OH, —O—, —CO—, —CONH—, —COO—, phenyl group, nitrogen-containing conjugate ring were the key functional groups. The common structures of the most active compounds binding to P1, P2 and P3 are shown in Table 1.
Rather than their interaction mechanism, changes in the blocking mechanism of SARS-CoV-2 S RBD and hACE2 were identified as the first key change, where compounds interacted with ACE2 binding areas via LEU455, PHE456, TYR489, GLN493 or LEU441, ASP442, LYS444, ASN450, TYR451 or PHE456, ARG457, LYS458, ASP467, and GLU471 residues. Furthermore, significant interactions included hydrogen-bonds, π-π interactions, hydrophobic interactions, and VDW forces. The —NH2, —CONH—, nitrogen-containing conjugate ring, and phenyl group or —O—, —CO—, —OH, N—, —CONH—, —NH—, phenyl group, and nitrogen-containing conjugate ring, or —NH—, —CONH—, —CO—, N—, phenyl group, and nitrogen-containing conjugate ring or —O—, —OH, —CO—, phenyl group, and oxygen-containing group were the key pharmacophores with a binding energy ranging −12 ~ −7 kcal/mol.
Based on the binding and block mechanisms of SARS-CoV-2 S RBD and hACE2, and the structure-activity relationships, we have put forward a screening strategy for antiviral compounds to block the binding of SARS-CoV-2/hACE2 and anti-SARS-CoV-2 therapy, since numerous compounds were acquired by computational virtual screening. The mechanisms of interaction between new compound and ACE2 binding area were determined based molecular docking, and the second key change was obtained by comparing the mechanism of interaction to the blockade mechanism. The final critical procedure was exploring the first and second key change trends. If the change trends were consistent, new compound was recognized as compound with the highest activity; alternatively, the new compound was removed (Fig. 5 ). The eight most active compounds binding to P1, P2, and P3 were explored as binding energy and residues were consistent with the first key change, and the sum of η1 + η2 was>1.2, which falls within the high range. The screening strategy details for the most active compounds were consistent with blockade mechanism of SARS-CoV-2 S RBD/hACE2, and the structure-activity relationships of the most active compounds and anti-SARS-CoV-2 therapy.
Fig. 5.

Screening strategy for anti-SARS-CoV-2 drugs.
5. Conclusions
Based on computer virtual screening and molecular docking simulation, the blockade mechanism of S and ACE2, as the compounds with the highest activity with ACE2 binding area, was proposed while taking into account of the order of binding energy, which was Z601-7647/P3 > 2159-4156/P3 > 4491-0722/P3 > HES/RBD > 2595-0160/P2 > P496-0785/P1 > C200-9609/P2 > Y041-7672/P2 > 8020-8119/P1, and the order of average binding energy, which was P3 > P2 > P1. The interaction between anti-SARS-CoV-2 drugs and SARS-CoV-2 S RBD/hACE2 pockets involved LEU455, PHE456, TYR489, GLN493, LEU441, ASP442, LYS444, ASN450, TYR451, PHE456, ARG457, LYS458, ASP467, and GLU471 binding residues. Furthermore, the key interactions among the highest active compounds and SARS-CoV-2 Spike included hydrogen-bonds, π-π interactions, hydrophobic interactions, and VDW forces, and the key functional groups were nitrogen-containing groups (—NH2, —OH), oxygen-containing groups (—O—, —CO—, —CONH—, and —COO—), and conjugate rings (phenyl group, nitrogen-containing conjugate rings). The correlation between the compounds with the highest activity and anti-SARS-CoV-2 therapy, based on binding energy and druggability of compounds for anti-SARS-CoV-2, was negative. Furthermore, the compounds exhibited structures that were consistent eight common types of structures with the highest activity. The screening strategy of anti-SARS-CoV-2 drugs via blocking binding between SARS-CoV-2 S RBD and hACE2 was established based on the first key change (interactions between HES and SARS-CoV-2 S RBD/hACE2) vs the second key change (interactions between anti-SARS-CoV-2 drugs and SARS-CoV-2 S RBD/hACE2). In the present work, the results indicated that the novel screening strategy could be applied in screening of anti-coronavirus drugs in general based on the inhibition of binding between coronaviruses and hosts’ receptors.
6. Author statement
This work clarified the A novel screening strategy of anti-SARS-CoV-2 drugs via blocking interaction between Spike RBD and ACE2.
In this work, the aims are shown as following:
-
(a)
Using computational simulation, blockade mechanism of SARS-CoV-2 spike receptor binding domain (S RBD) and human angiotensin converting enzyme 2 (hACE2) was clarified based on interactions between RBD and hesperidin.
-
(b)
Interactions between anti-SARS-CoV-2 drugs and therapy were investigated based on the binding energy and druggability of the compounds, and they exhibited negative correlations; the compounds were classified into eight common types of structures with highest activity.
-
(c)
An anti-SARS-CoV-2 drug screening strategy based on blocking S RBD/hACE2 binding was established according to the first key change (interactions between hesperidin and S RBD/hACE2) vs the second key change (interactions between anti-SARS-CoV-2 drugs and RBD/hACE2) trends.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgements
Funding.
This work was supported by the Special Fund for Military Medical Science [grant numbers AWS17J009] and the National Defense Science and Technology Research Project [19-163-12-ZD-019-002-01].
Author contributions
The authors would like to thank X.N.W., C.X.Y, Y.A.W., and Y.L. designed the research; X.N.W., C.X.Y, Y.Y.S., X.S., T.Z, Q.W., S.W., J.Y, W.J.Y., F.Y.L., M.M.Z, Y.A.W., and Y.L. performed the computational virtual screening and molecular simulation; X.N.W., C.X.Y, Q.W., Y.A.W., and Y.L. analysed the data; X.N.W., C.X.Y, T.Z., Q.W., Y.A.W., and Y.L. contributed to the discussion and manuscript; X.N.W., C.X.Y, Y.A.W., and Y.L. wrote the paper.
Handling Editor: Frederic Coulon
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.envint.2020.106361.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- Akhtar S., Benter I.F., Danjuma M.I., Doi S.A.R., Syed S.S., Habib A.M. Pharmacotherapy in COVID-19 patients: A review of ACE2-raising drugs and their clinical safety. J. Drug. Target. 2020;28(7–8):683–699. doi: 10.1080/1061186X.2020.1797754. [DOI] [PubMed] [Google Scholar]
- Bao L.L., Deng W., Huang B.Y., Gao H., Liu J.N., Ren L.L., Wei Q., Yu P., Xu Y.F., Qi F.F., Qu Y.J., Li F.D., Lv Q., Wang W.L., Xue J., Gong S.R., Liu M.Y., Wang G.P., Wang S.Y., Song Z.Q., Zhao L.N., Liu P.P., Zhao L., Ye F., Wang H.J., Zhou W.M., Zhu N., Zhen W., Yu H.S., Zhang X.J., Guo L., Chen L., Wang C.H., Wang Y., Wang X.M., Xiao Y., Sun Q.M., Liu H.Q., Zhu F.L., Ma C.X., Yan L.M., Yang M.L., Han J., Xu W.B., Tan W.J., Peng X.Z., Jin Q., Wu G.Z., Qin C. The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature. 2020;583(7818):830–833. doi: 10.1038/s41586-020-2312-y. [DOI] [PubMed] [Google Scholar]
- Basu A., Sarkar A., Maulik U. Molecular docking study of potential phytochemicals and their effects on the complex of SARS-CoV2 spike protein and human ACE2. Sci. Rep-UK. 2020;10(1):17699. doi: 10.1038/s41598-020-74715-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buonanno G., Morawska L., Stabile L. Quantitative assessment of the risk of airborne transmission of SARS-CoV-2 infection: prospective and retrospective applications. Environ. Int. 2020;145 doi: 10.1016/j.envint.2020.106112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgueño J.F., Reich A., Hazime H., Quintero M.A., Fernandez I., Fritsch J., Santander A.M., Brito N., Damas O.M., Deshpande A., Kerman D.H., Zhang L.Y., Gao Z., Ban Y.G., Wang L.L., Pignac-Kobinger J., Abreu M.T. Expression of SARS-CoV-2 entry molecules ACE2 and TMPRSS2 in the gut of patients With IBD. Inflamm. Bowel. Dis. 2020;26(6):797–808. doi: 10.1093/ibd/izaa085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X., Cao D., Kong L., Zhang X. Cryo-EM analysis of the post-fusion structure of the SARS-CoV spike glycoprotein. Nat. Commun. 2020;11(1):3618. doi: 10.1038/s41467-020-17371-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal R.K., Majeed J., Tonk R., Dhobi M., Apparsundaram S. Current targets and drug candidates for prevention and treatment of SARS-CoV-2 (COVID-19) infection. Rev. Cardiov. Asc. Med. 2020;21(3):365–384. doi: 10.31083/j.rcm.2020.03.118. [DOI] [PubMed] [Google Scholar]
- Guan W.J., Ni Z.Y., Hu Y., Liang W.H., Ou C.Q., He J.X., Liu L., Shan H., Lei C.L., Hui D.S.C., Du B., Li L.J., Zeng G., Yuen K.Y., Chen R.C., Tang C.L., Wang T., Chen P.Y., Xiang J., Li S.Y., Wang J.L., Liang Z.J., Peng Y.X., Wei L., Liu Y., Hu Y.H., Peng P., Wang J.M., Liu J.Y., Chen Z., Li G., Zheng Z.J., Qiu S.Q., Luo J., Ye C.J., Zhu S.Y., Zhong N.S. Clinical characteristics of 2019 novel coronavirus infection in China. N. Engl. J. Med. 2020;382(18):1708–1720. doi: 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggag Y.A., El-Ashmawy N.E., Okasha K.M. Is Hesperidin essential for prophylaxis and treatment of COVID-19 infection? Med. Hypotheses. 2020;144 doi: 10.1016/j.mehy.2020.109957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann, M., Hannah, K.W., Nadine, K., Marcel, M., Christian, D., Stefan, P., 2020. The novel coronavirus 2019 (2019-nCoV) uses the SARS-coronavirus receptor ACE2 and the cellular protease TMPRSS2 for entry into target cells. BioRxiv.
- Huang C.L., Wang Y.M., Li X.W., Ren L.L., Zhao J.P., Hu Y., Zhang L., Fan G.H., Xu J.Y., Gu X.Y., Cheng Z.S., Yu T., Xia J.A., Wei Y., Wu W.J., Xie X.L., Yin W., Li H., Liu M., Xiao Y., Gao H., Guo L., Xie J.G., Wang G.F., Jiang R.M., Gao Z.C., Jin Q., Wang J., Cao B. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagtap, D., Kumar, S., Mahale, S., Patel, V., 2020. Modelling and docking of Indian SARS-CoV-2 spike protein 1 with ACE2: implications for co-morbidity and therapeutic intervention. ArXiv.
- Jin Z.M., Du X.Y., Xu Y.C., Deng Y.Q., Liu M.Q., Zhao Y., Zhang B., Li X.F., Zhang L.K., Peng C., Duan Y.K., Yu J., Wang L., Yang K.L., Liu F.J., Jiang R.D., Yang X.L., You T., Liu X.C., Yang X.N., Fang Bai F., Liu H., Liu X., Guddat L.W., Xu W.Q., Xiao G.F., Qin C.F., Shi Z.L., Jiang H.L., Rao Z.H., Yang H.T. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582(7811):289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Lan J., Ge J.W., Yu J.F., Shan S.S., Zhou H., Fan S.L., Zhang Q., Shi X.L., Wang Q.S., Zhang L.Q., Wang X.Q. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581(7807):215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- Li D., Sangion A., Li L. Evaluating consumer exposure to disinfecting chemicals against coronavirus disease 2019 (COVID-19) and associated health risks. Environ. Int. 2020;145 doi: 10.1016/j.envint.2020.106108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.H., Choe H., Farzan M. Insights from the association of SARS-CoV S-protein with its receptor, ACE2. Adv. Exp. Med. Biol. 2006;581:209–218. doi: 10.1007/978-0-387-33012-9_36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.H., Moore M.J., Vasilieva N., Sui J.H., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C., Choe H., Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.H., Zhang C.S., Sui J.H., Kuhn J.H., Moore M.J., Luo S.W., Wong S.K., Huang I.C., Xu K.M., Vasilieva N., Murakami A., He Y.Q., Marasco W.A., Guan Y., Choe H., Farzan M. Receptor and viral determinants of SARS coronavirus adaptation to human ACE2. Embo. J. 2005;24(8):1634–1643. doi: 10.1038/sj.emboj.7600640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv H.B., Wu N.C., Owen T.Y.T., Meng Y., Perera R.A.P.M., Leung W.S., So R.T.Y., Chan J.M.C., Yip G.K., Chik T.S.H., Wang Y.Q., Choi C.Y.C., Lin Y.H., Ng W.W., Zhao J.C., Poon L.L.M., Peiris J.S.M., Wilson I.A., Mok C.K.P. Cross-reactive Antibody Response between SARS-CoV-2 and SARS-CoV Infections. Cell. Rep. 2020;31(9) doi: 10.1016/j.celrep.2020.107725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T., Ryu H.J., Lee S.H., Hwang S.W., Breton V., Rhee J., Kim D. Virtual screening identification of novel severe acute respiratory syndrome 3C-like protease inhibitors and in vitro confirmation. Bioorg. Med. Chem. Lett. 2011;21(10):3088–3091. doi: 10.1016/j.bmcl.2011.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira O.D., Rocha G.B., Paluch A.S., Costa L.T. Repurposing approved drugs as inhibitors of SARS-CoV-2 S-protein from molecular modeling and virtual screening. J. Biomol. Struct. Dyn. 2020:1–10. doi: 10.1080/07391102.2020.1772885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H.H., Kim H.N., Kim H., Yoo Y., Lee W. Acetylated K676 TGFBIp as a severity diagnostic blood biomarker for SARS-CoV-2 pneumonia. Sci. Adv. 2020;6(31):eabc1564. doi: 10.1126/sciadv.abc1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan L.T., Nguyen T.V., Luong Q.C., Nguyen T.V., Nguyen H.T., Le H.Q., Nguyen T.T., Cao T.M., Pham Q.D. Importation and human-to-human transmission of a novel coronavirus in Vietnam. N. Engl. J. Med. 2020;382(9):872–874. doi: 10.1056/NEJMc2001272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabakaran P., Xiao X., Dimitrov D.S. A model of the ACE2 structure and function as a SARS-CoV receptor. Biochem. Bioph. Res. Co. 2003;314(1):235–241. doi: 10.1016/j.bbrc.2003.12.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W.F., Gui M., Wang X.Q., Xiang Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. Plos. Pathog. 2018;14(8) doi: 10.1371/journal.ppat.1007236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivek-Ananth R.P., Rana A., Rajan N., Biswal H.S., Samal A. In silico identification of potential natural product inhibitors of human proteases key to SARS-CoV-2 infection. Molecules. 2020;25(17):3822. doi: 10.3390/molecules25173822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.H., Zhang Y.F., Wu L.L., Niu S., Song C.L., Zhang Z.Y., Lu G.W., Qiao C.P., Hu Y., Yuen K.Y., Wang Q.S., Zhou H., Yan J.H., Qi J.X. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell. 2020;181(4):894–904. doi: 10.1016/j.cell.2020.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrapp D., Wang N., Corbett K., Goldsmith J., Hsieh C.L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367(6483):1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.R., Liu Y., Yang Y.Y., Zhang P., Zhong W., Wang Y.L., Wang Q.Q., Xu Y., Li M.X., Li X.Z., Zheng M.Z., Chen L.X., Li H. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta. Pharm. Sin. B. 2020;10(5):766–788. doi: 10.1016/j.apsb.2020.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R.H., Zhang Y.Y., Li Y.N., Xia L., Guo Y.Y., Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367(6485):1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S.F., Chu H., Huang J.J., Zhao X.Y., Ye Z.W., Lai P.M., Wen L., Cai J.P., Mo Y.F., Cao J.L., Liang R.H., Poon V.K.M., Sze K.H., Zhou J., To K.K.W., Chen Z.W., Chen H.L., Jin D.Y., Chan J.F.W., Yuen K.Y. Viruses harness YxxØ motif to interact with host AP2M1 for replication: A vulnerable broad-spectrum antiviral target. Sci. Adv. 2020;6(35):eaba7910. doi: 10.1126/sciadv.aba7910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki A.M., van Boheemen S., Bestebroer T.M., Osterhaus A.D., Fouchier R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012;367(19):1814–1820. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]
- Zheng Q., Deng Y.Q., Liu J., Hoek L.V.D., Ben B., Lu M. Core structure of S2 from the human coronavirus NL63 spike glycoprotein. Biochemistry-US. 2006;45(51):15205–15215. doi: 10.1021/bi061686w. [DOI] [PubMed] [Google Scholar]
- Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L., Chen H.D., Chen J., Luo Y., Guo H., Jiang R.D., Liu M.Q., Chen Y., Shen X.R., Wang X., Zheng X.S., Zhao K., Chen Q.J., Deng F., Liu L.L., Yan B., Zhan F.X., Wang Y.Y., Xiao G.F., Shi Z.L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N., Zhang D.Y., Wang W.L., Li X.W., Yang B., Song J.D., Zhao X., Huang B.Y., Shi W.F., Lu R.J., Niu P.H., Zhan F.X., Ma X.J., Wang D.Y., Xu W.B., Wu G.Z., Gao G.F., Tan W.J. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020;382(8):727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.