

Abstract
Understanding the genetic causes of kidney disease is essential for accurate diagnosis and could lead to improved therapeutic strategies and prognosis. To accurately and promptly identify the genetic background of kidney diseases, we applied a targeted next‐generation sequencing gene panel including 203 genes associated with kidney disease, as well as diseases originating in other organs with mimicking symptoms of kidney disease, to analyze 51 patients with nonspecific nephrogenic symptoms, followed by validation of its efficacy as a diagnostic tool. We simultaneously screened for copy number variants (CNVs) in each patient to obtain a higher diagnostic yield (molecular diagnostic rate: 39.2%). Notably, one patient suspected of having Bartter syndrome presented with chloride‐secreting diarrhea attributable to homozygous SLC26A3 variants. Additionally, in eight patients, NGS confirmed the genetic causes of undefined kidney diseases (8/20, 40%), and initial clinical impression and molecular diagnosis were matched in 11 patients (11/20, 55%). Moreover, we found seven novel pathogenic/likely pathogenic variants in PKD1, PKHD1, COL4A3, and SLC12A1 genes, with a possible pathogenic variant in COL4A3 (c.1229G>A) identified in two unrelated patients. These results suggest that targeted NGS‐panel testing performed with CNV analysis might be advantageous for noninvasive and comprehensive diagnosis of suspected genetic kidney diseases.
Keywords: Kidney disease, Renal disease, NGS panel, Genetic diagnosis, Copy number variant
1. INTRODUCTION
Kidney diseases represent a heterogeneous group of disorders, including monogenic disorders, such as autosomal dominant/recessive polycystic kidney disease (ADPKD/ARPKD), as well as complex genetic diseases, such as steroid resistance nephrotic syndrome (SRNS), Alport syndrome, and congenital anomalies of the kidney and urinary tract (CAKUT). 1 Kidney diseases, especially those with a chronic course, can lead to permanent and irreversible deterioration of renal function. Therefore, accurate and prompt diagnosis is essential for improving outcomes for patients with these diseases; however, this can be difficult due to either nonspecific symptoms and signs or clinically silent symptoms in the early stages of disease. Additionally, this is complicated by several systemic diseases or diseases originating from other organs, that can present symptoms similar to those from confined kidney, including various structural abnormalities, electrolyte imbalances, or metabolic acidosis/alkalosis. For example, renal cysts, which are structural abnormalities in the kidney, can also be identified in various multisystemic diseases, such as tuberous sclerosis complex, oral‐facial‐digital syndrome, and coloboma syndrome, as well as ADPKD or ARPKD. 2 , 3 Therefore, diagnosing the precise underlying causes of kidney diseases with nonspecific symptoms using conventional laboratory and imaging diagnostic tools can be challenging.
An invasive procedure, such as renal biopsy, could often be performed to identify the underlying etiology of this disease group; however, it is limited by the range of conditions that it can successfully confirm and is associated with a risk of complications, including bleeding. 4 , 5 , 6 , 7 Moreover, this diagnostic method often fails to uncover a correct diagnosis in very early or late stages of diseases. 1 , 8
According to several reports, 10% of the population with adult chronic kidney disease (CKD) and almost all pediatric patients who progress to end‐stage renal disease are identified as having inherited kidney disease. 9 , 10 , 11 , 12 Therefore, the importance of genetic testing should not be overlooked during the diagnostic workup of kidney diseases. An accurate genetic diagnosis could provide proper treatment options for the patient in the early phase, leading to prevention of the rapid worsening of the disease and playing a pivotal role in selecting relative kidney donors for transplantation or family planning. However, identifying disease‐causing genes is challenging because of the complexity of the genetic background.
The efforts to improve the capabilities of genetic testing have been developed in recent years, and the revolution of next‐generation sequencing (NGS) has enabled cost‐effective simultaneous sequencing of a broad set of candidate genes. Recently, several studies reported the effectiveness of NGS in identifying various genetic factors in inherited kidney diseases, including glomerular nephropathy, steroid‐resistant nephrotic syndrome, and cystic kidney disease. 2 , 13 , 14 , 15 , 16 , 17 However, most studies are limited by only analyzing well‐known causative genes of inherited kidney disease. Additionally, differential diagnoses of diseases originating in other organs, which could result in symptoms and signs mimicking kidney disease, have not been identified. Moreover, almost all of these studies were performed on patients of mainly European descent; therefore, these results might not represent all ethnic populations.
Here, we applied a targeted NGS panel and simultaneous analysis of copy number variation (CNV) to elucidate the genetic causes of suspected genetic kidney diseases in an Asian population. Additionally, we confirmed the feasibility of this diagnostic method for the differential diagnosis of diseases originating from the kidney or another organ but presenting with overlapping symptoms and signs according to the NGS panel. To validate the diagnostic efficacy of this NGS panel, we tested 51 Korean patients with symptoms of kidney disease and suspected of having inherited kidney disease and referred for evaluation to determine possible genetic causes.
2. MATERIALS AND METHODS
2.1. Patient selection and study design
From January 2018 to August 2019, 51 unrelated, genetically undiagnosed patients were enrolled for targeted NGS testing using a comprehensive kidney disease panel developed in the Department of Clinical Genetics at Severance Children's Hospital (Seoul, Korea). All patients had one or more of the following symptoms/signs: proteinuria, hematuria, electrolyte imbalance, metabolic alkalosis/acidosis, or abnormal kidney structure. We retrospectively reviewed the pedigree information, previous medical history, physical examination findings, and any additional investigative results (e.g., ophthalmologic and otology examinations) in the electronic medical records of each patient. Additionally, we collected the available results based on segregation analyses of family members of each patient. This information was collected under anonymity in a routine diagnostic process, and the study protocol was approved by the Institutional Review Board of the Yonsei University Health System (IRB 4‐2019‐0227). Informed consent for the genetic testing was obtained from each patient or their legal guardians if the patient was aged <19 years.
2.2. Panel design
First, we searched for symptoms and signs resembling those observed in genetic kidney diseases using PubMed, Embase, and MEDLINE. Accordingly, we searched for the following symptoms and signs: proteinuria, hematuria, electrolyte imbalance, metabolic alkalosis/acidosis, and abnormal kidney structure. Based on data from the Human Genome Mutation Database (HGMD), Online Mendelian Inheritance in Man (OMIM) database (http://www.ncbi.nlm.nih.gov/omim), and an extensive literature review using PubMed, we extracted and optimized 203 disease‐causing genes (Table 1). The last search was performed on October 10, 2020.
TABLE 1.
Genes (n = 203) included in the panel of kidney diseases
| Gene | Cytogenic location | Inheritance | Gene accession number | Disease association |
|---|---|---|---|---|
| ACTN4 | 19q13.2 | AD | NM_004924 | Glomerulosclerosis, focal segmental, 1 |
| ADAMTS13 | 9q34.2 | AR | NM_139025 | Thrombotic thrombocytopenic purpura, familial |
| AGTR1 | 3q24 | AR | NM_000685 | Renal tubular dysgenesis |
| AGXT | 2q37.3 | AR | NM_000030 | Hyperoxaluria, primary, type 1 |
| AHI1 | 6q23.3 | AR | NM_017651 | Joubert syndrome 3 |
| ALG8 | 11q14.1 | AR | NM_019109 | Polycystic liver disease 3 with or without kidney cysts |
| ALMS1 | 2p13.1 | AR | NM_015120 | Alström syndrome, retinitis pigmentosa, sensorineural hearing loss |
| ANKS6 | 9q22.33 | AR | NM_173551 | Nephronophthisis 16 |
| AP2S1 | 19q13.32 | AD | NM_001301076 | Hypocalciuric hypercalcemia, type III |
| APRT | 16q24.3 | AR | NM_000485 | Adenine phosphoribosyltransferase deficiency |
| AQP2 | 12q13.12 | AD/AR | NM_000486 | Diabetes insipidus, nephrogenic |
| ARHGDIA | 17q25.3 | AR | NM_001185077 | Nephrotic syndrome, type 8 |
| ARL13B | 3q11.1‐q11.2 | AR | NM_182896 | Joubert syndrome 8 |
| ARNT2 | 15q25.1 | AR | NM_014862 | Webb‐Dattani syndrome |
| ATP6V0A4 | 7q34 | AR | NM_020632 | Renal tubular acidosis, distal, autosomal recessive |
| ATP6V1B1 | 2p13.3 | AR | NM_001692 | Renal tubular acidosis with deafness |
| AVP | 20p13 | AD | NM_000490 | Diabetes insipidus, neurohypophyseal |
| AVPR2 | Xq28 |
XLR |
NM_000054 | Diabetes insipidus, nephrogenic; Nephrogenic syndrome of inappropriate antidiuresis |
| B9D2 | 19q13.2 | AR | NM_030578 | Joubert syndrome 34 |
| BBS10 | 12q21.2 | AR | NM_024685 | Bardet‐Biedl syndrome 10 |
| BBS12 | 4q27 | AR | NM_152618 | Bardet‐Biedl syndrome 12 |
| BBS1 | 11q13.2 | AR/DR | Bardet‐Biedl syndrome 1 | |
| BBS2 | 16q13 | AR | NM_031885 | Bardet‐Biedl syndrome 2 |
| BBS4 | 15q24.1 | AR | NM_033028 | Bardet‐Biedl syndrome 4 |
| BBS9 | 7p14.3 | AR | NM_001033604 | Bardet‐Biedl syndrome 9 |
| BCS1L | 2q35 | AR | NM_004328 | Mitochondrial complex III deficiency, nuclear type 1 |
| BICC1 | 10q21.1 | AD | NM_025044 | Renal dysplasia, cystic, susceptibility to |
| BSND | 1p32.3 |
AR |
NM_057176 | Bartter syndrome, type 4a; Sensorineural deafness with mild renal dysfunction |
| CA2 | 8q21.2 | AR | NM_000067 | Osteopetrosis, autosomal recessive 3, with renal tubular acidosis |
| CA12 | 15q22.2 | AR | NM_001218 | Hyperchlorhidrosis, isolated |
| CASR | 3q13.3‐q21.1 | AD | NM_000388 | Hypocalcemia, autosomal dominant, with Bartter syndrome |
| CC2D2A | 4p15.32 | AR | NM_001080522 | Joubert syndrome 9 |
| CD151 | 11p15.5 | AR | NM_004357 | Nephropathy with pretibial epidermolysis bullosa and deafness |
| CD2AP | 6p12.3 | AD/AR | NM_012120 | Glomerulosclerosis, focal segmental, 3 |
| CEP164 | 11q23.3 | AR | NM_014956 | Nephronophthisis 15 |
| CEP290 | 12q21.32 | AR | NM_025114 | Bardet‐Biedl syndrome 14; Joubert syndrome 5 |
| CEP41 | 7q32.2 | AR | NM_018718 | Joubert syndrome 15 |
| CFH | 1q31.3 | AD/AR | NM_000186 | Hemolytic uremic syndrome, atypical, susceptibility to, 1 |
| CFHR5 | 1q31.3 | AD | NM_030787 | Nephropathy due to CFHR 5 days eficiency |
| CLCN5 | Xp11.23 | XLR | NM_000084 | Dent disease; Nephrolithiasis, type I; Proteinuria, low molecular weight, with hypercalciuric nephrocalcinosis |
| CLCNKB | 1p36.13 |
AR/DR |
NM_000085 |
Bartter syndrome, type 3 Bartter syndrome, type 4b, digenic |
| CLDN10 | 13q32.1 | AR | HELIX syndrome | |
| CLDN16 | 3q28 | AR | NM_006580 | Hypomagnesemia 3, renal |
| CLDN19 | 1p34.2 | AR | NM_148960 | Hypomagnesemia 5, renal, with ocular involvement |
| CNNM2 | 10q24.32 | AD | NM_017649 | Hypomagnesemia 6, renal |
| COL4A1 | 13q34 | AD | NM_001303110 | Angiopathy, hereditary, with nephropathy, aneurysms, and muscle cramps |
| COL4A3 | 2q36.3 | AD/AR | NM_012120 | Alport syndrome |
| COL4A4 | 2q36.3 | AR | NM_000091 | Alport syndrome, familiar hematuria |
| COL4A5 | Xq22.3 | X‐linked | NM_000092 | Alport syndrome |
| COQ2 | 4q21.22‐q21.23 | AR | NM_015697 | Mitochondrial disease, encephalopathy/isolated nephropathy |
| COQ6 | 14q24.3 | AR | NM_182476 | Nephrotic syndrome ± sensorineural deafness |
| CTNS | 17p13.2 | AR | NM_004937 | Cystinosis, nephropathic |
| CUBN | 10p13 | AR | NM_001081 | Imerslund‐Grasbeck syndrome |
| CYP11B2 | 3q24.3 | AR | NM‐000498 | Hypoaldosteronism, congenital, due to CMO I deficiency |
| DGKE | 17q22 | AR | NM_003647 | Nephrotic syndrome, type 7 |
| DGUOK | 2p13.1 | AR | NM_080916 | Mitochondrial DNA depletion syndrome 3 |
| DMP1 | 4q22.1 | AR | NM_001079911 | Hypophosphatemic rickets |
| DHCR7 | 11q13.4 | AR | NM_001360 | Smith‐Lemli‐Opitz syndrome |
| EGF | 10p13 | AR | NM_001178130 | Hypomagnesemia 4, renal |
| EGFR | 7p11.2 | AR | Inflammatory skin and bowel disease, neonatal, 2 | |
| EHHADH | 3q27.2 | AD | NM_001166415 | Fanconi renotubular syndrome 3 |
| EYA1 | 8q13.3 | AD | NM_000503 | Branchiootorenal syndrome 1, with or without cataracts |
| FAM58A | Xq28 | XLD | NM_152274 | STAR syndrome |
| FAN1 | 15q13.3 | AR | NM_014967 | Interstitial nephritis, karyomegalic |
| FGF23 | 12p13.32 | AD | NM_020638 | Hypophosphatemic rickets |
| FN1 | 2q35 | AD | NM_212476 | Glomerulopathy with fibronectin deposits 2 |
| FRAS1 | 4q21.21 | AR | NM_001166133 | Fraser syndrome 1 |
| FREM1 | 9p22.3 | AD/AR | NM_144966 | Bifid nose with or without anorectal and renal anomalies |
| FREM2 | 13q13.3 | AR | NM_207361 | Fraser syndrome 2 |
| FXYD2 | 11q23.3 | AD | NM_021603 | Hypomagnesemia 2, renal |
| GATA3 | 10p14 | AD | NM_001002295 | Hypoparathyroidism, sensorineural deafness, and renal dysplasia |
| GLA | Xq22.1 | XLR | NM_000169 | Fabry disease |
| GLB1 | 3p22.3 | AR | NM_000404 | Mucopolysaccharidosis type IVB (Morquio) |
| GLIS2 | 16p13.3 | AR | NM_032575 | Nephronophthisis 7 |
| GLIS3 | 9p24.2 | AR | NM_152629 | Diabetes mellitus, neonatal |
| GNA11 | 19p13.3 | AD | NM_002067 | Hypocalciuric hypercalcemia, type II |
| HNF1B | 17q12 | AD | NM_000458 | Renal cysts and diabetes syndrome |
| HPRT1 | Xq26.2‐q26.3 | XLR | NM_000194 | HPRT‐related gout, Lesch–Nyhan syndrome |
| HSD11B2 | 16q22.1 | AR | NM_000196 | Apparent mineralocorticoid excess |
| IFT122 | 3q21.3‐q22.1 | AR | NM_018262 | Cranioectodermal dysplasia 1 |
| IFT140 | 16p13.3 | AR | NM_014714 | Short‐rib thoracic dysplasia 9 with or without polydactyly |
| IFT172 | 2p23.3 | AR | NM_015662 | Short‐rib thoracic dysplasia 10 with or without polydactyly |
| INF2 | 14q32.33 | AD | NM_022489 | Glomerulosclerosis, focal segmental, 5 |
| INPP5E | 9q34.3 | AR | NM_019892 | Joubert syndrome 1 |
| INVS | 9q31.1 | AR | NM_014425 | Nephronophthisis 2, infantile |
| IQCB1 | 3q13.33 | AR | NM_014642 | Senior‐Loken syndrome 5 |
| ITGB4 | 17q25.1 | AR | NM_000213 | Epidermolysis bullosa, junctional, with pyloric atresia |
| KAL1 | Xp22.31 | XLR | NM_000216 | Hypogonadotropic hypogonadism 1 with or without anosmia (Kallmann syndrome 1) |
| KANK2 | 19p13.2 | AR | NM_015493 | Nephrotic syndrome, type 16 |
| KCNJ1 | 11q24.3 | AR | NM_000220 | Bartter syndrome, type 2 |
| KCNJ10 | 1q23.2 | AR | NM_002241 | SESAME syndrome |
| KIF7 | 15q26.1 | AR | NM_198525 | Joubert syndrome 12 |
| LAMB2 | 3p21.31 | AR | NM_002292 | Pierson syndrome |
| LCAT | 16q22.1 | AR | NM_000229 | Norum disease |
| LMX1B | 9q33.3 | AD | NM_002316 | Nail patella syndrome; FSGS without extrarenal involvement |
| LRP2 | 2q31.1 | AR | NM_004525 | Donnai‐Barrow syndrome |
| LYZ | 12q15 | AD | NM_000239 | Amyloidosis, renal |
| MAFB | 20q12 | AD | NM_005461 | Multicentric carpotarsal osteolysis syndrome |
| MED28 | 4p15.32 | AR | NM_025205 | nephrotic syndrome |
| MKKS | 20p12.2 | AR | NM_018848 | Bardet‐Biedl syndrome 6 |
| MKS1 | 17q22 | AR | NM_017777 | Bardet‐Biedl syndrome 13, Joubert syndrome 28 |
| MYH9 | 22q12.3 | AD, association | NM_002473 | MYH9‐related disease; Epstein and Fechtner syndromes |
| MMACHC | 1p34.1 | AR | NM_015506 | Methylmalonic aciduria and homocystinuria, cblC type |
| MYO1E | 15q22.2 | AR | NM_004995 | Glomerulosclerosis, focal segmental, 6 |
| NEK1 | 4q33 | AD/AR | NM_001199397 | Short‐rib thoracic dysplasia 6 with or without polydactyly |
| NEK8 | 17q11.2 | AR | NM_178170 | Renal‐hepatic‐pancreatic dysplasia 2 |
| NNT | 5p12 | AR | NM_012343 | Glucocorticoid deficiency 4, with or without mineralocorticoid deficiency |
| NOTCH2 | 1p12 | AD | NM_024408 | Hajdu‐Cheney syndrome |
| NPHP1 | 2q13 | AR | NM_000272 | Joubert syndrome 4, Nephronophthisis 1, juvenile |
| NPHP3 | 3q22.1 | AR | NM_153240 | Nephronophthisis 3 |
| NPHP4 | 1p36.31 | AR | NM_001291593 | Nephronophthisis 4 |
| NPHS1 | 19q13.12 | AR | NM_004646 | Nephrotic syndrome, type 1 |
| NPHS2 | 1q25.2 | AR | NM_014625 | Nephrotic syndrome, type 2 |
| NR0B1 | Xp21.2 | XLR | NM_000475 | Adrenal hypoplasia, congenital |
| NR3C2 | 4q31.23 | AD | NM_000901 | Pseudohypoaldosteronism type I, autosomal dominant |
| NUP214 | 9q34.13 | AR | NM_001318324 | Encephalopathy, acute, infection‐induced, susceptibility to, 9 |
| OCRL | Xq26.1 | XLR | NM_000276 | Dent disease 2, Lowe syndrome |
| OFD1 | Xp22.2 | XLR | NM_003611 | Joubert syndrome 10 |
| PAX2 | 10q24.31 | AD | NM_000278 | Glomerulosclerosis, focal segmental, 7 |
| PCCA | 13q32.3 | AR | NM_000282 | Propionicacidemia |
| PDSS2 | 6q21 | AR | NM_020381 | Leigh syndrome |
| PHEX | Xp22.11 | XLD | NM_000444 | Hypophosphatemic rickets, X‐linked dominant |
| PKD1 | 16p13.3 | AD | NM_000296 | Polycystic kidney disease 1 |
| PKD2 | 4q22.1 | AD | NM_000297 | Polycystic kidney disease 2 |
| PKHD1 | 6p12.3‐p12.2 | AR | NM_138694 | Polycystic kidney disease 4, with or without hepatic disease |
| PLCE1 | 10q23.33 | AR | NM_016341 | Nephrotic syndrome, type 3 |
| PLVAP | 19p13.11 | AR | NM_031310 | Diarrhea 10, protein‐losing enteropathy type |
| POMC | 2p23.3 | AR | NM_001035256 | Obesity, adrenal insufficiency, and red hair due to POMC deficiency |
| PTPRO | 12p12.3 | AR | NM_030667 | Nephrotic syndrome, type 6 |
| REN | 1q32.1 | AR | NM_000537 | Renal tubular dysgenesis |
| RPGRIP1L | 16q12.2 | AR | NM_015272 | Joubert syndrome 7 |
| RRM2B | 8q22.3 | AR | NM_001172477 | Mitochondrial DNA depletion syndrome 8A (encephalomyopathic type with renal tubulopathy) |
| SALL1 | 16q12.1 | AD | NM_002968 | Townes‐Brocks branchiootorenal‐like syndrome |
| SALL4 | 20q13.3 | AD | NM_001318031 | IVIC syndrome |
| SARS2 | 19q13.2 | AR | NM_017827 | Hyperuricemia, pulmonary hypertension, renal failure, and alkalosis |
| SCARB2 | 4q21.1 | AR | NM_005506 | Action myoclonus‐renal failure syndrome ± hearing loss |
| SCNN1A | 12p13.31 | AD | NM_001038 | Liddle syndrome 3, Bronchiectasis with or without elevated sweat chloride 2 |
| SCNN1B | 16p12.2 | AD | NM_000336 | Liddle syndrome 1, Bronchiectasis with or without elevated sweat chloride 1 |
| SCNN1G | 16p12.2 | AD | NM_001039 | Liddle syndrome, Bronchiectasis with or without elevated sweat chloride 3 |
| SDCCAG8 | 1q43‐44 | AR | NM_006642 | Bardet‐Biedl syndrome 16 |
| SIX5 | 19q13.32 | NM_175875 | Branchiootorenal syndrome 2 | |
| SLC12A1 | 15q21.1 | AR | NM_000338 | Bartter syndrome, type 1 |
| SLC12A3 | 16q13 | AR | NM_000339 | Gitelman syndrome |
| SLC22A12 | 11q13.1 | AR | NM_144585 | Hypouricemia, renal |
| SLC26A3 | 7q22.3‐q31.1 | AR | NM_000111 | Diarrhea 1, secretory chloride, congenital |
| SLC2A2 | 3q26.2 | AR | NM_000340 | Fanconi‐Bickel syndrome |
| SLC34A1 | 5q35.3 | AR | NM_003052 | Fanconi renotubular syndrome 2 |
| SLC34A3 | 9q34.3 | AR | NM_080877 | Hypophosphatemic rickets with hypercalciuria |
| SLC3A1 | 2p21 | AD/AR | NM_000341 | Cystinuria |
| SLC4A1 | 17q21.31 | AD/AR | NM_000342 | Renal tubular acidosis, distal |
| SLC4A4 | 4q13.3 | AR | NM_003759 | Renal tubular acidosis, proximal, with ocular abnormalities |
| SLC5A2 | 16p11.2 | AD/AR | NM_003041 | Renal glucosuria |
| SLC6A19 | 5p15.33 | AD | NM_001003841 | Hyperglycinuria |
| SLC6A20 | 3p21.31 | AD | NM_020208 | Hyperglycinuria |
| SLC7A7 | 14q11.2 | AR | NM_001126105 | Lysinuric protein intolerance |
| SLC7A9 | 19q13.11 | AD/AR | NM_001126335 | Cystinuria |
| SLC9A3 | 5p15.33 | AR | Diarrhea 8, secretory sodium, congenital | |
| SLC9A3R1 | 17q25.1 | AD | NM_004252 | Nephrolithiasis/osteoporosis, hypophosphatemic, 2 |
| SMARCAL1 | 2q35 | AR | NM_014140 | Schimke immuno‐osseous dysplasia |
| SOX17 | 8q11.23 | AD | NM_022454 | Vesicoureteral reflux 3 |
| SPINK5 | 5q32 | AR | NM_001127698 | Netherton syndrome |
| SPINT2 | 19q13.2 | AR | NM_001166103 | Diarrhea 3, secretory sodium, congenital, syndromic |
| STAR | 8p11.23 | AR | NM_000349 | Lipoid adrenal hyperplasia |
| TCTN1 | 12q24.11 | AR | NM_024549 | Joubert syndrome 13 |
| TMEM216 | 11q12.2 | AR | NM_016499 | Joubert syndrome 2 |
| TMEM237 | 2q33.1 | AR | NM_152388 | Joubert syndrome 14 |
| TMEM67 | 8q22.1 | AR | NM_153704 | Joubert syndrome 6, Nephronophthisis 11 |
| TRIM32 | 9q33.1 | AR | NM_012210 | Bardet‐Biedl syndrome 11 |
| TRPC6 | 11q22.1 | AD | NM_004621 | Glomerulosclerosis, focal segmental, 2 |
| TTC21B | 2q24.3 | AD/AR | NM_024753 | Nephronophthisis 12 |
| TTC8 | 14q31.3 | AR | NM_144596 | Bardet‐Biedl syndrome 8 |
| UMOD | 16p12.3 | AD | NM_001008389 | Uromodulin‐associated kidney disease |
| UPK3A | 22q13.31 | UD | NM_006953 | Involvement renal dysplasia, possible |
| VIPAS39 | 14q24.3 | AR | NM_022067 | Arthrogryposis, renal dysfunction, and cholestasis 2 |
| VPS33B | 15q26.1 | AR | NM_018668 | Arthrogryposis, renal dysfunction, and cholestasis 1 |
| WDR19 | 4p14 | AR | NM_001317924 | Nephronophthisis 13, Senior‐Loken syndrome 8 |
| WDR35 | 2p24.1 | AR | NM_020779 | Short‐rib thoracic dysplasia 7 with or without polydactyly |
| WNK1 | 12p13.33 | AD | NM_018979 | Pseudohypoaldosteronism, type IIC |
| WNK4 | 17q21.2 | AD | NM_001321299 | Pseudohypoaldosteronism, type IIB |
| WNT4 | 1p36.12 | AD | NM_030761 | Mullerian aplasia and hyperandrogenism |
| WT1 | 11p13 | AD | NM_000378 | Nephrotic syndrome, type 4; Denys–Drash and Frasier syndrome |
| XPNPEP3 | 22q13.2 | AR | NM_022098 | Nephronophthisis‐like nephropathy 1 |
| ZMPSTE24 | 1p34.2 | AR | NM_005857 | Mandibuloacral dysplasia with type B lipodystrophy |
| ZNF423 | 16q12.1 | AD/AR | 604 557 | Joubert syndrome 19; Nephronophthisis 14 |
Abbreviations: AD, autosomal dominant; AR, autosomal recessive; UD, undetermined.
2.3. DNA preparation
We collected 3 ml of peripheral blood in EDTA tubes from each patient and extracted the genomic DNA from leukocytes using a DNeasy blood and tissue kit (Qiagen, Hilden, Germany) according to manufacturer instructions. The quality of isolated DNA was checked using a Nanodrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
2.4. Library preparation, target capture, and DNA sequencing
We constructed a DNA sequencing library using the TruSeq DNA Library Prep Kits protocol according to manufacturer instructions (TruSeq DNA Library Prep Kits,, FC‐121‐2003; Illumina, Carlsbad, CA, USA). Briefly, DNA in each sample was sheared to 250bp sequences, tagged, and then purified according to fragment size with magnetic beads (AMPure XP, Beckman Coulter, IN, USA). We subsequently performed repair, phosphorylation, and adenylation of the 3′ ends and isolated the precaptured amplified 300‐ to 500‐bp fragments. We then performed targeted sequence, up to 15 bp from the exon of the target genes based on hg19, capture according to TruSeq Custom Enrichment Kit protocol (FC‐123‐1096, Illumina). DNA sequencing was performed on a V2 flow cell using MiSeq sequencer (Illumina), generating 150 bp paired‐end reads. Image analysis and base calling were performed using the Illumina pipeline. The yield of each DNA sample averaged 2 GB of raw data with a 150‐fold mean sequencing depth of the targeted regions. 18 , 19 Sequenced reads were mapped to the human reference genome (GRCh37), and sequencing alignment was performed using the Burrows‐Wheeler Aligner software package. 20
2.5. CNV analysis
For CNV detection, we applied ExomeDepth with default parameters. 21 ExomeDepth uses a robust model of the read count data to call CNVs by comparing the test exome data to a matched optimized‐aggregate reference set, which is built with combined exomes from the same batch to maximize the power to handle technical variability between samples. This method was applied to the targeted NGS sequencing data sets to detect pathogenic CNVs, and the identified CNVs were confirmed through multiplex ligation‐dependent probe amplification or real‐time quantitative PCR (RT‐qPCR).
2.6. Sanger sequencing and RT‐qPCR
We performed Sanger sequencing for validation and segregation analysis of variants detected by NGS testing. RT‐qPCR was performed for segregation analysis of CNVs using primers designed according to the oligonucleotide sequences of the variants.
2.7. Interpretation and analysis of the detected variants
We examined the NGS data and prioritized DNA variants according to clinical relevance using the following parameters: (1) sequence quality; (2) allele frequency (according to the Exome Aggregation Consortium [ExAC], dbSNP database, and Korean Reference Genome Database [KRGDB; http://coda.nih.go.kr/]); and (3) presence in HGMD, OMIM, dbSNP, or ClinVar. Real or possible damage of variants was predicted using in silico prediction algorithms, including Polymorphism Phenotyping version 2 (PolyPhen‐2) and Sorting Tolerant from Intolerant (SIFT; https://sift.bii.a-star.edu.sg/). After compressive analysis of all results, we classified the identified variants into a five‐tier system as pathogenic (P), likely pathogenic (LP), variant of unknown significance (VUS), likely benign, or benign according to the American College of Medical Genetics and Genomics (ACMG) guidelines. 22 According to the inheritance pattern of the disease, we considered results positive if one or two P/LP variants in one disease‐related gene was identified according to the inheritance patterns of diseases. If only a VUS or one P/LP variant was detected in a gene with an autosomal recessive (AR) inheritance pattern, we considered the result nondiagnostic. We reported cases without any VUS or P/LP as negative results.
3. RESULTS
3.1. Patient characteristics
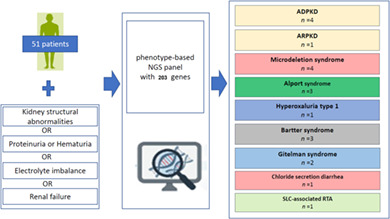
A total of 51 unrelated patients were included in this study (median age: 11.6 years [range: 0–46 years]). Among these, 36 (70.6%) were male, and 15 (29.4%) were female. Five patients (9.8%) had a family history related to kidney disease, and three (5.9%) had undergone renal biopsy due to hematuria before the NGS panel test. The reasons for referral were as follows: structural abnormalities in kidneys detected by computed tomography or sonogram in 16 patients, urinalysis abnormalities in 21 patients, electrolyte imbalances in 12 patients, and renal failure in two patients (Table 2).
TABLE 2.
The clinical causes for using NGS in the renal disease panel test
| Reasons for NGS | Patient number (n) | |
|---|---|---|
| Structural abnormalities in kidney | 16 | |
| Polycystic kidney disease | 13 | |
| Medullary sponge kidney | 1 | |
| Renal agenesis | 1 | |
| Bilateral hydronephrosis | 1 | |
| Urinalysis abnormality | 21 | |
| Proteinuria | 7 | |
| Hematuria | 11 | |
| Proteinuria and hematuria | 3 | |
| Electrolyte imbalance | 12 | |
| Renal failure | 2 | |
| Total | 51 | |
Abbreviation: NGS, next‐generation sequencing.
3.2. Diagnostic yield of NGS
In total, the final diagnostic yield was 39.2% (20/51), which included the diagnostic group when P/LP variants or CNV abnormalities in our NGS panel test matched the symptoms of patients. The final molecular diagnosis confirmed by our targeted NGS panel classified patients into the following diseases: four patients with ADPKD, one patient with ARPKD, three patients with Alport syndrome, one patient with hyperoxaluria type 1, three patients with Bartter syndrome, two patients with Gitelman syndrome, one patient with SLC4A1‐associated renal tubular acidosis, and one patient with congenital chloride secretory diarrhea. Moreover, we identified four patients exhibiting dysmorphic features and global delayed development in addition to several renal cysts in both kidneys with CNV abnormalities on chromosome (1p36 [2 patients] and 17q12 [2 patients]). Nondiagnostic results were showed for 31 patients with VUSs only, as well as for four patients with only one P/LP variant in the disease gene with an AR‐inheritance trait.
The first clinical symptoms of the diagnosed patients were as follows: urinalysis abnormality, including hematuria or proteinuria (4/21; 19.0%), structural abnormalities in the kidney (9/16; 56.3%), and electrolyte imbalances (7/12; 58.3%). The details of the patients confirmed by molecular diagnosis are summarized in Table 3.
TABLE 3.
Clinical and genetic data of patients in whom disease‐causative gene variants were identified
| ID | Gender | Age | Fx | Clinical presentation‐renal | Clinical presentation‐extrarenal | Final diagnosis | Gene | Inheritance | Sequence variant | ACMG class | Zygosity | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patients referred for cystic kidney disease | ||||||||||||
| 01 | M | 3 m | N | Several renal cysts, both kidney |
Sensorineural hearing loss, Rt. Atrial septal defect Umbilical hernia |
1p36.32 microdeletion syndrome | 1p36.32 microdeletion | Hetero | ||||
| 02 | F | 11 y | N | Multiple renal cysts, Metabolic acidosis | Delayed development epilepsy | 1p36 microdeletion syndrome | 1p36 microdeletion | Hetero | ||||
| 03 | F | 2 y | N | Multiple renal cysts, | Delayed development | 17q12 microdeletion syndrome | 17q12 microdeletion | Hetero | ||||
| 04 | M | 6 y | N | Multiple renal cysts, Nephronophthisis | Delayed development | 17q12 microdeletion syndrome | 17q12 microdeletion | Hetero | ||||
| 05 | M | 1 m | N | Multiple renal cysts with variable size Decrease kidney size | Atrial septal defect | ADPKD | PKD1 | AD | c.5303C>A, (p.Thr1768Asn) | 4 | Hetero | |
| ID | Gender | Age | Fx | Clinical presentation‐Renal | Clinical presentation‐Extrarenal | Final diagnosis | Gene | Inheritance | Sequence variant | ACMG class | Zygosity | |
| 06 | M | 18 y | Y | A hemorrhagic component in the multiple renal cysts, both kidney | ADPKD | PKD1 | AD | c.975T>G (p.Tyr325Ter) a | 4 | Hetero | ||
| 07 | F | 50 y | N |
Multiple renal cysts, both kidney Chronic renal failure |
Liver cyst | ADPKD | PKD1 | AD | c.8056C>T (p.Gln2686Ter) | 5 | Hetero | |
| 08 | F | 42 y | N | Multiple renal cysts, both kidney | Liver cyst | ADPKD | PKD1 | AD | c.12060C>A (p.Cys4020Ter) a | 4 | Hetero | |
| 09 | F | 5 d | N | Pulmonary hypoplasia, Polycystic dysplastic kidney | ARPKD | PKHD1 | AR |
c.4879G>T (p.Val1627Phe)(p) a c.11212_11213delAT (p.lle3738SerfsTer19)(m) a |
4 5 |
Compound hetero | ||
| Patients referred for hematuria +/− proteinuria | ||||||||||||
| 10 | F | 7 y | Y | Recurrent HU | Asthma, atopic dermatitis | Alport syndrome | COL4A3 | AD, AR | c.417delG (p.Thr140HisfsTer13) a | 4 | Hetero | |
| 11 | F | 21 y | N |
Recurrent HU GBM irregularity, suggestive of hereditary nephritis |
Sensorihearing loss, both | Alport syndrome | COL4A3 |
AD, AR |
c.1029 + 1G>A a | 4 | Hetero | |
| 12 | M | 4 y | N | Hematuria | Alport syndrome | COL4A4 | AD, AR |
c.2084G>A (p.Gly695Asp)(p) c.1327_1344del (p.Pro444‐Leu449del)(m) |
4 5 |
Compound hetero | ||
| 13 | F | 5 y | N | Hematuria, nephrolithiasis | Short stature | Hyperoxaluria type1 | AGXT | AR | c.331 T>C (p.Arg111Ter) | 5 | Homo | |
| ID | Gender | Age | Fx | Clinical presentation‐Renal | Clinical presentation‐Extrarenal | Final diagnosis | Gene | Inheritance | Sequence variant | ACMG class | Zygosity | |
| Patients referred for electrolyte imbalance | ||||||||||||
| 14 | M | 4 y | N |
polyhydramnios Hx. Hypokalemia |
Bartter syndrome | CLCNKB | AR |
c.371C>T (p.Pro124Leu)(p) Exon 4 del(m) b |
5 | Compound hetero | ||
| 15 | F | 27 y | N |
Hypokalemia Hypochloremia |
Hearing impairment tremor | Bartter syndrome | CLCNKB | AR |
Exon 1–14 del(p) b c.1830G>A (p.Trp610Ter)(m) |
5 | Compound hetero | |
| 16 | M | 15 y | N | Hypokalemia | Hearing impairment tremor | Bartter syndrome | SLC12A1 | AR |
c.888delG (p) a c.1522G>A (p.Ala400Thr) (m) |
5 4 |
Compound hetero | |
| 17 | F | 10 y | N | Hypokalemia Hypomagnesemia | Gitelman syndrome | SLC12A3 | AR |
c.1664C>T (p.Ser555Leu)(p) c. 2186G>A (p.Gly741Arg)(m) |
5 5 |
Compoundhetero | ||
| 18 | M | 23 y | N | Hypokalemia | Dystonia, tremor | Gitelman syndrome | SLC12A3 | AR |
c.1919A>G (p.Asn640Ser)(p) c.1868T>C (p.Leu623Pro)(m) |
5 5 |
Compound hetero | |
| 19 | F | 2 y | Y | Renal tubular acidosis | SLC4A1‐associated renal tubular acidosis | SLC4A1 | AD | c.1765C>T (p.Arg589Cys) | 5 | Hetero | ||
| 20 | M | 11 m | N |
Hypokalemic alkalosis Diffusely bilateral renal enlargement with increased cortical echogenicity |
Colon segmental resection, d/t colon ischemia | Congenital secretory diarrhea, chloride type | SLC26A3 | AR | c.2063‐1G>T (p,m) | 4 | Homo | |
Abbreviations: ACMG, American College of Medical Genetics and Genomics; AD, autosomal dominant; AR, autosomal recessive; Fx, family history; F, female; GBM, glomerular basement membrane; Hetero, heterozygous; HU, hematuria; M, male; m, maternal; PU, proteinuria; p, paternal.
Novel pathogenic/likely pathogenic variant.
Novel exonal deletion.
Among five patients with related familial medical histories, three harbored pathogenic variants associated with their symptoms. Their final diagnosis was ADPKD (one patient), Alport syndrome (one patient), and SLC4A1‐associated distal renal tubular acidosis (one patient). Among the three patients who had undergone renal biopsy due to hematuria before the NGS panel test, one was determined to have one novel LP variant in COL4A3, and two were found to have the same VUS in COL4A3, which was known to be associated with Alport syndrome. These results were consistent with their histological diagnosis.
3.3. Detection of genetic variants and CNVs
Targeted NGS analysis identified 169 variants in 84 genes, with every patient having at least one variant. On average, we detected 2.0 variants, with a maximum of 12 per patient. We detected 27 P/LP variants of 10 genes in 24 patients. Of these variants, 18 (66.7%) had been formerly reported as P/LP, whereas 9 (33.3%) had not yet been reported at the time of our investigation. The mutational types of these 27 P/LP variants were as follows: 11 missense variants, six nonsense variants, three frameshift, three small insertion/deletions, two exonal deletions, and two splicing errors. The most frequently detected P/LP variants were observed in PKD1 (n = 4; 14.8%), CLCNKB (n = 4; 14.8%), and SLC12A3 (n = 4; 14.8%).
Additionally, all patients harbored one or more VUS, with 142 VUSs detected in 70 genes. Among these VUSs, the most frequently involved genes were PKD1 (n = 15; 10.6%), PKHD1 (n = 6; 4.2%), and ALMS1 (n = 6; 4.2%). Additionally, we detected five heterozygous CNVs in five patients, although only two of the CNVs (the 1p36 and 17q12 microdeletions) found in four patients were revealed as pathogenic according to the phenotype of the patients, segregation analysis, and our literature review.
3.4. Novel variants
Among the 20 patients with confirmed disease according to our molecular diagnosis, seven novel variants in seven patients absent from population databases and our in‐house database were identified. Moreover, two novel exonal deletions were identified in CLCNKB for two patients (Patient 14 [exon 4 deletions] and Patient 15 [exon 1–14 deletion]).
Among patients with ADPKD with multiple renal cysts, two novel LP variants of PKD1 were identified, with both were shown to be nonsense variants (Patient 6 [p.Tyr325Ter] and Patient 8 [p.Cys4020Ter]). Further, a 5 day old patient with ARPKD and presenting with polycystic dysplastic kidney disease and hypoplastic lung was found to carry two novel P/LP novel variants in PKHD1. One was a paternally inherited missense LP variant (p.Val1627Phe), whereas the other was a maternally inherited frameshift P variant (p.lle3738SerfsTer19).
In the two patients with Alport syndrome, we detected two different novel P/LP variants of COL4A3. Patient 10, a 7‐year‐old girl, showed recurrent hematuria, and her genetic investigation revealed a paternally inherited P variant (p.Thr140HisfsTer13) in COL4A3. Her father also had a history of nephritis and was diagnosed with Alport syndrome after genetic testing. Additionally, Patient 11 presented with recurrent hematuria and sensory hearing loss and was revealed to have a novel LP variant in COL4A3 that could cause a splicing error (1029 + 1G>A).
Further, we detected a novel P variant (c.888delG) of SLC12A1 in a 15‐year‐old boy with recurrent hypokalemia and symptoms of hearing impairment and tremor. His genetic testing showed a paternally inherited novel variant (c.888delG) combined with a maternally inherited missense P variant (p.Ala400Thr) previously reported in SLC12A1. Accordingly, he was diagnosed with Bartter syndrome based on these results.
3.5. Noteworthy VUSs
Notably, we identified a meaningful VUS in the COL4A3 gene of two unrelated patients (patients 21 and 22). This missense variant was clinically classified as a VUS in a well‐defined disease gene of Alport syndrome (c.1229G>A, p.Gly410Glu) according to ACMG guidelines. Patients 21 and 22 were presenting with proteinuria and hematuria since her 20s and the age of 5. Their renal pathologic findings revealed an irregular thickening of the GBM. Based on the clinical symptoms and biopsy findings, they were clinically suspected of having Alport syndrome. Their NGS panel testing identified the same heterozygous VUS (c.1229G>A, p.Gly410Glu) in COL4A3, with Sanger sequencing confirming the detected variant. The variant was absent from public genome databases (ExAC, 1000 Genomes data, and our in‐house database), and the structure and function of the protein were predicted as likely damaged according to Polyphen2 (PPH2 score: 1.0). The altered residue was revealed to be highly conserved across vertebrate species. Given that this variant was consistently identified in two unrelated patients with highly suspected Alport syndrome from their clinical symptoms and pathological results, along with the results of variant analysis, this suggested a high probability of being reclassified as an LP/P variant. Although further analyses of the genetic consequences based on the family members were recommended to assess the exact pathogenic indication of this VUS, these tests could not be conducted due to lack of agreements by the family members of the patients (Table 4).
TABLE 4.
Clinical and genetic data of patients in whom a noteworthy VUS was identified
| ID | Gender | Age | Fx | Clinical presentation‐Renal | Clinical presentation‐Extrarenal | Final diagnosis | Gene | Inheritance | Sequence variant | ACMG class | Zygosity |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Patients referred for hematuria +/− proteinuria | |||||||||||
| 21 | F | 36 y | N | HU/ PU since 20′ irregular thickening of GBM | Alport syndrome | COL4A3 | AD, AR | c.1229G>A (p.Gly410Glu) | 3 | Hetero | |
| 22 | F | 15 y | N | Consistent HU irregular thickening of GBM | Alport syndrome | COL4A3 | AD, AR | c.1229G>A (p.Gly410Glu) | 3 | Hetero | |
Abbreviations: ACMG, American College of Medical Genetics and Genomics; AR, autosomal recessive; Fx, family history; F, female; GBM, glomerular basement membrane; Hetero, heterozygous; HU, hematuria; M, male; m, maternal; PU, proteinuria; p, paternal; VUS, variant of uncertain significance.
3.6. Correlation between clinically suspected diseases and molecular diagnosis
Of the 20 patients for whom disease was confirmed through molecular diagnosis based on our genetic test, clinically suspected diagnosis and genetic diagnosis were matched in 11 patients (11/20, 55%), including three patients with ADPKD, one patient with ARPKD, two patients with Alport syndrome, three patients with Bartter syndrome, and two patients with Gitelman syndrome. By contrast, the results of our NGS panel reclassified the final diagnosis from initial clinical diagnosis in one patient (5%). This 11‐month‐old boy was initially clinically diagnosed with Bartter syndrome before genetic testing and finally diagnosed with chloride‐secretion diarrhea originating from the intestine and not from the kidneys (Patient 20).
In eight patients, the NGS panel test played an essential role in confirming the genetic cause of their previously undefined kidney disease (8/20, 40%), including one patient with ADPKD with cystic kidney (Patient 5), one patient with Alport syndrome (Patient 12), one patient with hyperoxaluria type 1 (Patient 13), one patient with SLC4A1‐associated renal tubular acidosis (Patient 19) and four patients with CNVs abnormalities (Patients 1, 2, 3 and 4). Detailed results are shown in Figure 1.
FIGURE 1.
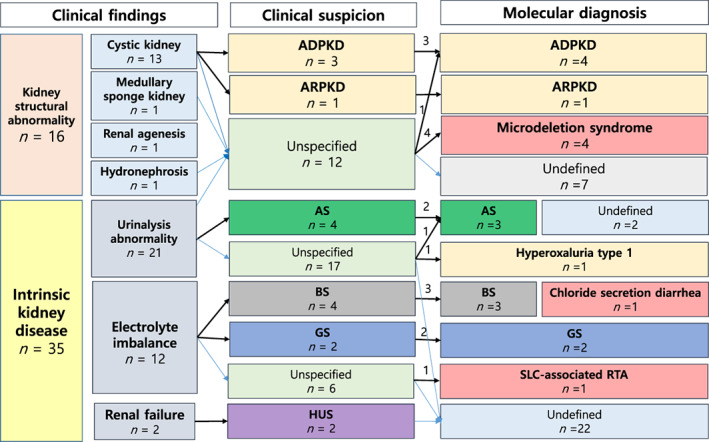
Correlations between clinical suspicion and results of molecular analysis. ADPKD, autosomal dominant polycystic kidney disease; AS, Alport syndrome; BS, Bartter syndrome; GS, Gitelman syndrome; HUS, hemolytic uremic syndrome; TBMD, thin basement membrane disease; SRNS, steroid resistance nephrotic syndrome [Colour figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
In this study, we analyzed the genetic diagnosis of 51 unrelated patients with clinically suspected, inherited kidney disease using a customized NGS panel that included genes related to broad symptoms of kidney disease. Consequently, a total of 39.2% (20/51) of patients were confirmed as having a genetic disease.
Several studies analyzed results using targeted NGS panels to diagnose inherited kidney diseases. Sen et al. reported the results of analysis of an SRNS‐targeted diagnostic gene panel performed for 302 patients, confirming genetic diagnoses in 26.6% of the patients. 23 Another study of 44 patients with typical PKD who underwent targeted NGS testing with 63 related genes revealed 48 related mutation sites in PKD1 and PKD2. 24
It should be noted that in the present study, the NGS panel used did not focus on genes limited to kidney diseases but rather included a broad set of 203 genes related to diseases that might mimic the symptoms of inherited kidney diseases, even though diseases originate from other organs. This approach could facilitate the accurate diagnosis of inherited kidney diseases, as well as the prompt differentiation of genetic diseases with overlapping symptoms, whether these might have originated in the confined kidney or other organs.
One of the advantageous characteristics of this NGS panel was evident based on the notable case of patient 20. This patient visited our hospital for the first time exhibiting lethargy. He had been born at 35.4 weeks of gestation from healthy parents and had two healthy older brothers, with no other notable family history. Fifteen days after birth, he developed abdominal distension suggestive of neonatal necrotizing enterocolitis and received ileostomy surgery. After 2 months, he received another surgery for segmental resection of a 7.9‐cm ischemic ileum lesion and to repair the ileostomy site. On admission, he showed severe hyponatremia and hypokalemic hypochloremic metabolic alkalosis (serum sodium: 128 mmol/L; potassium: 2.5 mmol/L; and chloride: 67 mmol/L; pH 7.652; pCO2: 32.5 mmHg; pO2: 103.0 mmHg; and HCO3: 36.3 mmol/L). Abdominal sonography showed diffuse renal disease with bilateral renal enlargement. Given these results, Bartter syndrome was initially suspected as a diagnosis, and he was referred to our study to precisely identify the genetic etiology of his condition. According to the genetic test, we discovered two homozygous splice‐site pathogenic mutations in SLC26A3 (c.2063‐1G>T), which encodes a transmembrane glycoprotein that exchanges chloride and bicarbonate ions across the cell membrane. His parents were identified as the asymptomatic carriers of c.2063‐1G>T. To confirm the molecular diagnosis, we analyzed the stool of the patient, finding a sodium level of 120 mmol/L. Although there were symptoms that could be mistaken as a disease originating from the kidney, he was finally diagnosed with chloride‐secreting diarrhea, which differed from the first clinical impression. This result of the molecular diagnosis offered the chance of appropriate treatment, focusing salt substitution therapy according to the treatment protocol of chloride‐secreting diarrhea and he has maintained good condition.
Additionally, our findings emphasized the usefulness of CNV analysis. Recent studies report that most CNVs are likely benign, but that some specific variants might be related to genetic diseases, such as neurodevelopmental diseases and various cancers. 25 , 26 , 27 , 28 NGS based CNV detection has a reported sensitivity of up to 92% and specificity of up to 100% for detecting duplications as small as 300 bp and deletions as small as 180 bp in specific genes. 29 , 30 In some inherited kidney diseases, CNVs could also affect susceptibility, and previous studies have highlighted the need for analyzing CNVs in inherited kidney diseases, such as CAKUT. 29 , 31 , 32 In the present study, we detected two known pathogenic CNVs in four patients (Patients 1–4), with both identified as known CNVs that could lead to kidney‐ related symptoms in addition to systemic manifestations. Details of abnormalities in CNVs and patients are described in Table 3.
Regarding the gene variants detected in this study, we found seven novel P/LP variants and two novel exonal deletions among our patient group. Further, there was a noticeable VUS identified in COL4A3, which was found in two unrelated patients. Evidence suggested that this VUS should be reclassified as P/LP, even though it is yet assumed to be a VUS according to ACMG guidelines. Further functional research and segregation analysis of the family of these patients would help define the pathogenicity of this variant.
The present study had a major advantage (ie, using a targeted NGS panel focusing on the phenotype of kidney diseases) that allowed diagnosis of inherited kidney diseases along with differential diagnoses of diseases based on their origin (kidney or other organs), despite their presenting symptoms similar to those of kidney disease. Moreover, we were able to simultaneously obtain relatively high diagnostic performance for CNV analysis with the NGS panel. However, a limitation of the present study is its single‐center research design and inclusion of a small number of patients. Further studies with a larger number of patients might aid verification of these results in the future.
Diagnostic approaches using NGS technology could enable accurate and early detection of genetic diseases and minimize the need for invasive diagnostic procedures, as well as optimize outcomes by broadening therapeutic options. Moreover, presymptomatic testing based on family history could be used to detect the genetic causes of diseases prior to the appearance of overt symptoms, which might allow application of genetic results for prenatal genetic testing and counseling in order to prevent the disease.
This study confirmed the efficacy of NGS with CNV analysis as a diagnostic tool for patients with suspected inherited kidney disease based on their symptoms. The rapid development of NGS technology would enable further clinical applicability of this approach for the diagnosis of inherited kidney disease.
CONFLICT OF INTEREST
The authors declare no potential conflicts of interest.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/cge.13869.
ACKNOWLEDGEMENTS
The authors express their gratitude to the members of the Clinical Genetics lab at Severance Children's Hospital. The authors received no specific funding for this work.
Oh J, Shin JI, Lee K, Lee CH, Ko Y, Lee J‐S. Clinical application of a phenotype‐based NGS panel for differential diagnosis of inherited kidney disease and beyond. Clinical Genetics. 2021;99:236–249. 10.1111/cge.13869
DATA AVAILABILITY STATEMENT
All relevant data are found within the paper and the Supporting information files.
REFERENCES
- 1. Stokman MF, Renkema KY, Giles RH, Schaefer F, Knoers NV, van Eerde AM. The expanding phenotypic spectra of kidney diseases: insights from genetic studies. Nat Rev Nephrol. 2016;12(8):472‐483. [DOI] [PubMed] [Google Scholar]
- 2. Bullich G, Domingo‐Gallego A, Vargas I, et al. A kidney‐disease gene panel allows a comprehensive genetic diagnosis of cystic and glomerular inherited kidney diseases. Kidney Int. 2018;94(2):363‐371. [DOI] [PubMed] [Google Scholar]
- 3. Cramer MT, Guay‐Woodford LM. Cystic kidney disease: a primer. Adv Chronic Kidney Dis. 2015;22(4):297‐305. [DOI] [PubMed] [Google Scholar]
- 4. Prakash J, Singh M, Tripathi K, Rai US. Complications of percutaneous renal biopsy. J Indian Med Assoc. 1994;92(12):395‐396. [PubMed] [Google Scholar]
- 5. González‐Michaca L, Chew‐Wong A, Soltero L, Gamba G, Correa‐Rotter R. Percutaneous kidney biopsy, analysis of 26 years: complication rate and risk factors; comment. Rev Invest Clin. 2000;52(2):125‐131. [PubMed] [Google Scholar]
- 6. Whittier WL, Korbet SM. Renal biopsy: update. Curr Opin Nephrol Hypertens. 2004;13(6):661‐665. [DOI] [PubMed] [Google Scholar]
- 7. Rianthavorn P, Kerr SJ, Chiengthong K. Safety of paediatric percutaneous native kidney biopsy and factors predicting bleeding complications. Nephrology (Carlton). 2014;19(3):143‐148. [DOI] [PubMed] [Google Scholar]
- 8. Renkema KY, Stokman MF, Giles RH, Knoers NV. Next‐generation sequencing for research and diagnostics in kidney disease. Nat Rev Nephrol. 2014;10(8):433‐444. [DOI] [PubMed] [Google Scholar]
- 9. Mehta L, Jim B. Hereditary renal diseases. Semin Nephrol. 2017;37(4):354‐361. [DOI] [PubMed] [Google Scholar]
- 10. Devuyst O, Knoers NV, Remuzzi G, Schaefer F. Rare inherited kidney diseases: challenges, opportunities, and perspectives. Lancet. 2014;383(9931):1844‐1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mallett A, Patel C, Salisbury A, Wang Z, Healy H, Hoy W. The prevalence and epidemiology of genetic renal disease amongst adults with chronic kidney disease in Australia. Orphanet J Rare Dis. 2014;9:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hildebrandt F. Genetics of kidney diseases. Semin Nephrol. 2016;36(6):472‐474. [DOI] [PubMed] [Google Scholar]
- 13. Fallerini C, Dosa L, Tita R, et al. Unbiased next generation sequencing analysis confirms the existence of autosomal dominant Alport syndrome in a relevant fraction of cases. Clin Genet. 2014;86(3):252‐257. [DOI] [PubMed] [Google Scholar]
- 14. Artuso R, Fallerini C, Dosa L, et al. Advances in Alport syndrome diagnosis using next‐generation sequencing. Eur J Hum Genet. 2012;20(1):50‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tan AY, Michaeel A, Liu G, et al. Molecular diagnosis of autosomal dominant polycystic kidney disease using next‐generation sequencing. J Mol Diagn. 2014;16(2):216‐228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Siji A, Karthik KN, Pardeshi VC, Hari PS, Vasudevan A. Targeted gene panel for genetic testing of south Indian children with steroid resistant nephrotic syndrome. BMC Med Genet. 2018;19(1):200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Riedhammer KM, Braunisch MC, Günthner R, et al. Exome sequencing and identification of phenocopies in patients with clinically presumed hereditary nephropathies. Am J Kidney Dis. 2020;76:460‐470. [DOI] [PubMed] [Google Scholar]
- 18. Shang X, Peng Z, Ye Y, et al. Rapid targeted next‐generation sequencing platform for molecular screening and clinical genotyping in subjects with hemoglobinopathies. EBioMedicine. 2017;23:150‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Picelli S, Björklund AK, Reinius B, Sagasser S, Winberg G, Sandberg R. Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res. 2014;24(12):2033‐2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li H, Durbin R. Fast and accurate long‐read alignment with burrows‐wheeler transform. Bioinformatics. 2010;26(5):589‐595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Plagnol V, Curtis J, Epstein M, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics. 2012;28(21):2747‐2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sen ES, Dean P, Yarram‐Smith L, et al. Clinical genetic testing using a custom‐designed steroid‐resistant nephrotic syndrome gene panel: analysis and recommendations. J Med Genet. 2017;54(12):795‐804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang T, Li Q, Shang S, et al. Identifying gene mutations of Chinese patients with polycystic kidney disease through targeted next‐generation sequencing technology. Mol Genet Genomic Med. 2019;7(6):e720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jo HY, Park MH, Woo HM, et al. Application of whole‐exome sequencing for detecting copy number variants in CMT1A/HNPP. Clin Genet. 2016;90(2):177‐181. [DOI] [PubMed] [Google Scholar]
- 26. Lupski JR. Structural variation in the human genome. N Engl J Med. 2007;356(11):1169‐1171. [DOI] [PubMed] [Google Scholar]
- 27. Gilissen C, Hehir‐Kwa JY, Thung DT, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature. 2014;511(7509):344‐347. [DOI] [PubMed] [Google Scholar]
- 28. Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy‐number alteration across human cancers. Nature. 2010;463(7283):899‐905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nagano C, Nozu K, Morisada N, et al. Detection of copy number variations by pair analysis using next‐generation sequencing data in inherited kidney diseases. Clin Exp Nephrol. 2018;22(4):881‐888. [DOI] [PubMed] [Google Scholar]
- 30. Onsongo G, Baughn LB, Bower M, et al. CNV‐RF is a random Forest‐based copy number variation detection method using next‐generation sequencing. J Mol Diagn. 2016;18(6):872‐881. [DOI] [PubMed] [Google Scholar]
- 31. Westland R, Verbitsky M, Vukojevic K, et al. Copy number variation analysis identifies novel CAKUT candidate genes in children with a solitary functioning kidney. Kidney Int. 2015;88(6):1402‐1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sanna‐Cherchi S, Kiryluk K, Burgess KE, et al. Copy‐number disorders are a common cause of congenital kidney malformations. Am J Hum Genet. 2012;91(6):987‐997. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are found within the paper and the Supporting information files.