

Abstract
Functional pairing between cellular glycoconjugates and tissue lectins like galectins has wide (patho)physiological significance. Their study is facilitated by nonhydrolysable derivatives of the natural O‐glycans, such as S‐ and Se‐glycosides. The latter enable extensive analyses by specific 77Se NMR spectroscopy, but still remain underexplored. By using the example of selenodigalactoside (SeDG) and the human galectin‐1 and ‐3, we have evaluated diverse 77Se NMR detection methods and propose selective 1H,77Se heteronuclear Hartmann–Hahn transfer for efficient use in competitive NMR screening against a selenoglycoside spy ligand. By fluorescence anisotropy, circular dichroism, and isothermal titration calorimetry (ITC), we show that the affinity and thermodynamics of SeDG binding by galectins are similar to thiodigalactoside (TDG) and N‐acetyllactosamine (LacNAc), confirming that Se substitution has no major impact. ITC data in D2O versus H2O are similar for TDG and LacNAc binding by both galectins, but a solvent effect, indicating solvent rearrangement at the binding site, is hinted at for SeDG and clearly observed for LacNAc dimers with extended chain length.
Keywords: 77Se NMR spectroscopy, calorimetry, circular dichroism, galectins, selenoglycosides
Selenoglycosides offer attractive benefits over natural O‐glycans for studying molecular recognition processes, such as access to specific 77Se NMR spectroscopy. We evaluate NMR methods for 1H,77Se correlation, including the new selective HeHaHa experiment. We show that the thermodynamics of selenodigalactoside binding to human galectins remains unaltered, but a clear solvent effect is observed if the glycan chain is extended.
Introduction
Formation of the glycosidic bond is the molecular means to build messages from carbohydrates, the letters of the third alphabet of life. The generated glycan “words”, as part of cellular glycoconjugates, are increasingly shown to encode wide, (patho)physiologically relevant information. [1] This emerging role as biochemical multipurpose signals has intensified efforts in carbohydrate chemistry to synthesise both naturally occurring oligosaccharides and derivatives with special modifications and properties. For instance, sulfur substitution of the oxygen in the glycosidic linkage between saccharide moieties leads to thioglycosides [2] that resist hydrolysis and show increased flexibility to access and populate secondary conformations more readily. [3] Bioactivity tests confirm that thioglycosides maintain the capacity of natural O‐glycosides to interact with lectins, the “readers” of glycan‐encoded information, [4] and thiodigalactoside (TDG) has emerged as the most potent inhibitor of carbohydrate‐dependent haemagglutination in the pioneering study detecting galectins in vertebrates. [5] This family of tissue lectins, which primarily recognise β‐galactose moieties, has become a research focus for its role in cellular homeostasis and various diseases through functional pairing with glycoconjugate receptors. Nonhydrolysable derivatives like thioglycosides have become important probes for their study, [6] where human galectins 1 and 3 (hGal‐1, hGal‐3) showed higher affinity for TDG than for their canonical ligand lactose (Lac). [7] Evidently, the increased van der Waals radius (1.8 Å for S vs. 1.5 Å for O), C−X bond length (1.8 Å for S vs. 1.4 Å for O), and C(−X−)C distance (2.9 Å for S vs. 2.4 Å for O) as well as decreased C−X−C angle (95° for S vs. 115° for O) do not impair thioglycoside recognition and, therefore, allow their wide use as hydrolysis resistant lectin inhibitors.
The homologous selenoglycosides (Se‐glycosides) offer similar hydrolytic stability and bioactivity to thioglycosides. Thus, the conformational space and dynamics of Se‐glycosides of the histo‐blood group ABH system were found to be rather similar to corresponding thioglycosides, whereas mono‐ and bivalent Se‐glycosides, especially Se‐digalactoside (SeDG), strongly inhibit canonical Lac binding by human galectins. [8] In contrast to sulfur and oxygen, the selenium atom furthermore enables two powerful analytical techniques for high‐resolution studies. First, selenium facilitates the phasing of X‐ray diffraction maps by single‐wavelength anomalous diffraction (SAD), which was exploited to solve the structure of lectins with bound β‐methyl Se‐glycosides. [9] Second, the stable spin‐ isotope 77Se enables heteronuclear NMR spectroscopy, which was used to monitor Se‐glycoside recognition by plant lectins (ConA, PSA, WGA) through direct 77Se detection, [10] and SeDG binding to human hGal‐1 and hGal‐3 through the far more sensitive indirect 77Se detection. [11]
With this work, we further pave the way for a broad use of Se‐glycosides to exploit the great merits of 77Se NMR in glycan recognition studies. As a test system, we again use SeDG, hGal‐1, and hGal‐3. In the first part, we present a quantitative comparison to substantiate the superiority of indirect versus direct 77Se NMR detection and to assess the losses accruing in the former. These mainly derive from evolution of homonuclear 1H,1H′ coupling during the required long 1H,77Se magnetisation transfer steps. We therefore introduce the 1H,77Se heteronuclear Hartmann–Hahn (HeHaHa) transfer experiment, where a simple adjustment suppresses competing 1H,1H’ homonuclear (HoHaHa) transfer and restricts 1H,77Se HeHaHa transfer to the anomeric H1 protons in Se‐glycosides. Compared to the benchmark CPMG‐HSQMBC, [11] this selective H1,77Se HeHaHa experiment yields spectra with maximal simplicity, phase purity, and signal intensity, and should prove most beneficial in competitive screening applications with a Se‐glycoside as spy ligand. These assays identify other ligands binding to the same site, without false positives, and directly report on their affinity relative to the spy ligand. To prepare for such applications, the second part of our study presents a thorough thermodynamic characterisation of SeDG binding to hGal‐1 and hGal‐3, in comparison with the related β‐galactosides LacNAc, TDG, and nonbinding diselenodigalactoside (DSeDG). We thereby validate fluorescence anisotropy (FA) and circular dichroism (CD) as complementary techniques to isothermal calorimetry (ITC) for studying galectin interactions. We then use ITC to measure the enthalpic and entropic contributions to binding, and also derive the thermodynamic contributions to the solvent isotope effect (SIE) by comparing the results obtained in H2O versus D2O. The SIE, which reveals the influence of solvent reorganisation on the thermodynamics of binding, had previously been measured only for two leguminous lectins. [12] We now extend this approach to human galectins, where we also compare the impact on binding affinity and thermodynamics caused by S/Se substitution in the glycosidic bond of the digalactoside or by glycan chain extension in two LacNAc dimers.
Results and Discussion
1H NMR spectroscopy
The binary SeDG + SeDGlc mixture provides a rare example of fully separable 1H NMR spectra (Figure 1) that nevertheless illustrate the widespread 1H signal overlap and complex fine structure (from extensive homonuclear n J HH coupling) typical for carbohydrates. A comparison of this basic 1D 1H NMR spectrum in the absence and presence of hGal‐3 (Figure 1, top) readily indicates selective binding of SeDG by strong attenuation and broadening of all signals. The [1H→]1H saturation transfer difference (STD) spectrum confirms reversible binding of SeDG, where H2 and H4‐H6 show the strongest signals, hence, closest contact with galectin protons (Figure 1, bottom). The anomeric H1, which barely approaches the protein, [13] shows the smallest STD effect despite strongest signal attenuation (−75 %) upon hGal‐3 binding, underscoring that signal attenuation is a very sensitive indicator of molecular interactions even on nuclei outside the binding interface, but therefore unsuitable for mapping the binding epitope.
Figure 1.
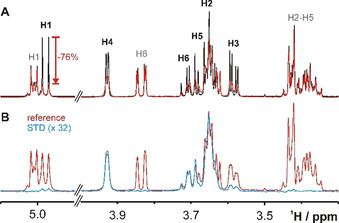
1H NMR spectra of SeDG (4 mm, assignments in black) and SeDGlc (4 mm, assignments in grey) in the absence (black) and presence (red) of hGal‐3 (0.125 mm). A) 1H Spectrum. Strong signal broadening and attenuation indicates binding only for SeDG, where the maximal attenuation (−76 %) is observed for H1. B) [1H→]1H STD spectrum in the presence of hGal‐3. The difference 1H spectrum (blue, 32‐fold upscaled) reveals maximal STD effects of ≥3 % for SeDG protons H2, H4, H5, and H6 while STD effects on H3 and especially on H1 are much weaker. No STD effects are observed for SeDGlc. Residual hGal‐3 1H signals were suppressed by a 100 ms T1ρ filter.
77Se NMR spectroscopy
Contrary to oxygen and sulfur, selenium has a naturally occurring spin‐ isotope, 77Se, that is detectable by heteronuclear high‐resolution NMR. 77Se NMR offers decisive advantages over conventional 1H NMR such as absence of background signals, specificity for selenium‐containing compounds, and enormous spectral simplification from a much wider dispersion of signals without fine structure (due to a lack of homonuclear 77Se coupling partners). Selenoglycosides open the door to exploit these unique benefits of 77Se NMR in glycan‐(ga)lectin recognition studies. Yet, the cost is a significant sensitivity loss relative to 1H NMR due to the inherently lower polarisation and isotopic abundance of 77Se as well as possibly higher relaxation rates, lower detection sensitivity by the hardware, etc. In the following, we deconvolute and quantify these loss factors, and summarise routes to greatly alleviate them.
The 77Se direct‐detection NMR spectrum in the absence and presence of hGal‐3 confirms selective binding of SeDG via similar signal attenuation and broadening as in the 1H spectrum, but with prohibitively low sensitivity (Figure 2 A). Yet, if the molecule shows sizable heteronuclear n J H,Se couplings, important sensitivity gains up to (γ H/γ Se)≈5.2 and further (γ H/γ Se)1⋅5≈12 could be realised by initial 1H→77Se polarisation transfer and indirect 77Se detection on 1H, respectively. [11] Thus, the combination of both enhancement schemes in 1H↔77Se out‐and‐back transfer experiments can yield up to 60‐fold higher sensitivity, as suggested by the 77Se(F1) projection of a 2D 1H,77Se CPMG‐HSQMBC [11] spectrum (Figure 2 B) recorded in half the time of the direct 77Se spectrum (Figure 2 A). Yet, dissipation of the transferable magnetisation through competing homonuclear n J H,H coupling and transverse R 2 relaxation reduces the gains and complicates the calculation of an optimal 1H→77Se transfer delay, Δ opt, that is otherwise expected to be Δ calc=0.5/n J H,Se. We, therefore, measured the Δ dependence of all SeDG correlation signals for the CPMG‐HSQMBC experiment [11] that implements 1H→77Se polarisation transfer by CPMG‐INEPT to suppress the phase modulation from evolving homonuclear n J H,H couplings. Yet, CPMG‐INEPT does not suppress the spread of magnetisation by in‐phase 1H→1H’ HoHaHa transfer that produces a strong indirect correlation signal for H3 despite no direct 4 J H3,Se coupling (Figure 3 A and Figure S1 in the Supporting Information). In contrast, the correlation signals for H1 (2 J H1,Se=7.0 Hz), H2 (3 J H2,Se=7.4 Hz), and H4 (5 J H1,Se=3.2 Hz) derive from direct 1H→77Se along with some contributing indirect 1H→1H’ HoHaHa transfer that may cause differences between experimental (Δ opt) and expected (Δ calc=(2⋅n J H,Se)−1) optimal transfer delays. Thus (as shown in Figure S2), for the H1 signal we observe the shortest Δ opt=70 ms (coinciding with Δ calc), followed by Δ opt=85 ms for the indirect H3 correlation signal, and Δ opt >100 ms for H2 and H4 (longer Δ were not tested to avoid excessive sample heating and duty cycles from CPMG pulsing). Table 1 summarises all extracted 3 J H,H’ and n J H,Se coupling constants along with the experimental and expected optimal transfer delays for SeDG.
Figure 2.
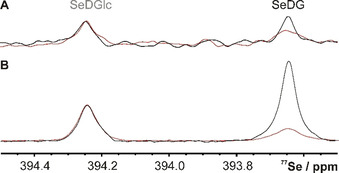
77Se NMR spectrum (1H decoupled) of SeDG and SeDGlc (4 mm) in the absence (black) and presence (red, acquired with double number of scans) of hGal‐3 (0.125 mm). A) Direct detection with 1024 scans, 3 s interscan delay, 52 min experiment time. B) Indirect detection by 2D 1H,77Se CPMG‐HSQMBC (positive F1 projection) with 70 ms 1H→77Se transfer delay, 8 scans, 1.5 s interscan delay, 26 min experiment time. Spectra were processed with 3 Hz line broadening. Signal‐to‐noise ratios for same experiment time: 5 (A) vs. 97 (B).
Figure 3.
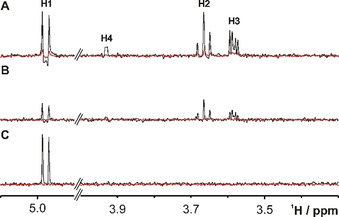
1D 1H traces from 2D 1H,77Se correlation spectra taken at the 77Se chemical shift (393.7 ppm) of SeDG (4 mm) in the absence (black) or presence (red) of hGal‐3 (0.125 mm). A) CPMG‐HSQMBC (Δ opt(H1)=70 ms, B CPMG=1143 Hz). The indirect H3 correlation signal (4 J H3,Se=0) derives from HoHaHa (TOCSY) transfer. B) HeHaHa (B DIPSI2=1450 Hz, Δ opt(H1)=80 ms). The average signal intensity is −60 % lower than in the HSQMBC spectrum. C) H1 selective HeHaHa (B DIPSI2=285 Hz, Δ opt(H1)=100 ms). The H1 signal now shows pure in‐phase splitting (3 J H1,H2=10 Hz) and 10 % (175 %) sensitivity gain relative to the corresponding CPMG‐HSQMBC (HeHaHa) spectrum. Corresponding 1H traces for the nonbinding SeDGlc are shown in Figure S4.
Table 1.
Scalar coupling constants (cf. Figure S1) and optimal 1H→77Se transfer delays (cf. Figure S2) for SeDG and different polarisation transfer schemes.
|
SeDG signal |
H1 |
H2 |
H3 |
H4 |
|---|---|---|---|---|
|
3 J H,H′ [Hz] |
10.0 |
10.0/9.45 |
9.45/3.4 |
3.4/1.0 |
|
n J H,Se [Hz] |
7.0 (n=2) |
7.4 (n=3) |
0 (n=4) |
3.2 (n=5) |
|
Δ calc [ms][a] |
71.4 |
67.6 |
– |
156 |
|
Δ opt [ms][b] |
70 |
>100 |
85 |
>100 |
|
Δ opt,HeHaHa [ms][c] |
70 |
80 |
100 |
140 |
|
Δ opt,selHeHaHa [ms][d] |
100 |
– |
– |
– |
|
Δ calc,selHeHaHa [ms][e] |
101 |
– |
– |
– |
[a] Theoretical transfer delay Δ calc = 1/(2⋅n J H,Se) in the absence of competing couplings and relaxation. [b] Optimal transfer delay for the 1H,77Se CPMG‐HSQMBC experiment; duty cycle limitations precluded sampling for Δ > 100 ms. [c] Optimal transfer delay for the broadband 1H,77Se HeHaHa experiment. [d] Optimal transfer delay for the selective H1,77Se HeHaHa experiment. [e] Theoretical transfer delay Δ calc,selHeHaHa = 1/(√2⋅2 J H1,Se) in the absence of HoHaHa.
By focusing on the H1,77Se correlation signal of SeDG and using the pertaining optimal transfer delay, Δ opt=70 ms, we may compare the sensitivity of direct and indirect 77Se detection via 1H↔77Se out‐and‐back transfer by CPMG‐HSQMBC. With comparable experimental and processing parameters, the signal‐to‐noise ratios relative to a 1H spectrum were 0.077 % for direct versus 1.763 % for indirect 77Se detection (Table 2). Thus, indirect 77Se detection yields a 23‐fold sensitivity enhancement over direct 77Se detection, while a factor of 62.5=(γ H/γ Se)2.5 is expected theoretically. This underperformance of the HSQMBC experiment is due to the mentioned dissipation of transferable magnetisation by homonuclear Hartmann–Hahn transfer and transverse R 2(H) relaxation during its two CPMG‐INEPT steps. Their transfer efficiency each is the square root of the signal‐to‐noise ratio for this 1H↔77Se out‐and‐back transfer experiment, relative to a direct 1H experiment without 77Se selection, after taking into account the 13.1‐fold lower natural abundance of 77Se (7.63 %) versus 1H (100 %). Thus, with the optimised experimental parameters (Δ opt=70 ms, B CPMG=1371 Hz, δ CPMG=157.3 μs), each INEPT step for H1,77Se correlation reaches maximal 48 % efficiency. Finally, compensation for the inherently lower 77Se sensitivity from a lower gyromagnetic ratio and isotope abundance than 1H (by multiplication with factors 62.5 and 13.1, respectively) yields a reduced detection sensitivity of 63.5 % for 77Se relative to 1H. This reduction is largely due to lower detection sensitivity for 77Se on the outer coil versus 1H detection on the inner coil of our TBI probehead, but with bias from further factors (e.g., higher salt effect losses for 1H). In summary, the presented deconvolution of gain and loss factors in 77Se NMR confirms the most important sensitivity enhancement from indirect 1H[77Se] detection, although it falls short of the theoretical maximum and may offer margin for improvement. A further important and general 13‐fold gain from 77Se isotope enrichment has so far remained unexploited, but would be vital to enable highly informative yet insensitive NMR experiments, for example, to elucidate binding epitopes and specific protein‐ligand contacts.
Table 2.
Signal‐to‐noise (SNrel) ratios (relative to 1H=100 %) and gain and loss factors for indirect (by CPMG‐HSQMBC) versus direct 77Se NMR detection.
|
77Se detection method |
77Se direct |
1H[77Se] CPMG‐HSQMBC |
|---|---|---|
|
signal halfwidth |
3.0 Hz |
1.5 Hz |
|
SNrel=SN/√number‐of‐scans |
0.077 % |
1.763 % |
|
f gain=SNrel(1H[77Se])/SNrel(77Se)[a] |
– |
23 |
|
f theor=(γ H/γ Se)⋅(γ H/γ Se)1.5[b] |
– |
62.5 |
|
f 100 %=100 %/7.63 %[c] |
13.1 |
13.1 |
|
SN100 %=SNrel⋅f 100 % [d] |
1.0 % |
23.1 % |
|
f INEPT=√SN100 %(1H[77Se])[e] |
– |
48.1 % |
|
f coil,Se =SN100 %⋅f theor [f] |
63.5 % |
– |
Spectra were recorded with the same FID resolution (0.5 Hz), receiver gain, long total interscan delay (6.3 s>3⋅T1), and were processed identically after exponential line broadening with the indicated natural signal halfwidth. For the CPMG‐HSQMBC experiment, the SN of H1 was multiplied by 2 to compensate a 50 % loss from gradient coherence selection. [a] f gain is the experimentally achieved gain with the CPMG‐HSQMBC indirect versus direct 77Se detection scheme. [b] f theor is the theoretical gain for indirect 77Se detection with an 1H↔77Se out‐and‐back transfer scheme, relative to direct 77Se detection. [c] f 100 % is the gain from 100 % enrichment of 77Se. [d] SN100=SNrel compensated for natural 77Se isotope abundance. [e] f INEPT is the transfer efficiency of each CPMG‐INEPT step in the CPMG‐HSQMBC experiment (with Δ opt=70 ms, BCPMG=1371 Hz, δ CPMG=157.3 μs). [f] f coil,Se is the outer‐coil 77Se detection sensitivity relative to inner‐coil 1H detection (BRUKER 600 MHz TBI probehead).
We next explored ways to improve the 1H↔77Se transfer efficiency fundamental for indirect 1H[77Se] detection, which must aim at reducing the dissipation of transferable magnetisation from homonuclear n J H,H coupling evolution and transverse R 2(1H) relaxation. The latter may be exacerbated by line broadening from chemical or conformational exchange processes, as often seen for reversible ligand binding. This can be suppressed by efficient spin locking which, in the presence of homonuclear coupling, also facilitates homonuclear Hartmann–Hahn transfer between the locked spins. If continuous spin lock pulsing is applied synchronously to distinct isotopes with heteronuclear coupling, analogous heteronuclear Hartmann‐Hahn transfer is likewise facilitated. Thus, we replaced both CPMG‐INEPT steps of the HSQMBC experiments with simultaneous DIPSI2 [14] spin locking for both 1H and 77Se to compose the 1H,77Se HeHaHa experiment (all pulse program codes are given in the Supporting Information). Again, we experimentally derived optimal transfer delays, Δ opt,HeHaHa, for each correlation signal in SeDG (Table 1 and Figure S2). Except for H1 (Δ opt,HeHaHa=70 ms), these differ somewhat from the corresponding Δ opt delays for the CPMG‐HSQMBC experiment, indicating a distinct superposition of undesired HoHaHa with HeHaHa or CPMG‐INEPT transfer, respectively. Yet, 1H,77Se HeHaHa (Figure 3 B) shows only about a third of the transfer efficiency of CPMG‐INEPT (Figure 3 A), mainly due to two general problems with HeHaHa. First, the distinct 1H inner and 77Se outer probehead coils produce different spatial variations of the BRF field strength, causing locally varying dephasing between locked 1H and 77Se spins that diminishes the net Hartmann‐Hahn transfer across the sampled volume. Such losses could be mitigated with a probehead using a single coil for 1H and 77Se. Second, while 77Se magnetisation during CPMG‐INEPT only alternates along z and, therefore, is only subject to slow longitudinal R 1(77Se) relaxation, it spends about half the DIPSI2 spin lock time in the transverse plane [15] and then accumulates substantial losses from fast transverse R 2(77Se) relaxation.
Despite these shortcomings, the HeHaHa experiment offers an easy way to suppress any dissipation of transferable H1 magnetisation by 1H,1H′ HoHaHa in selenoglycosides and instead force it exclusively into heteronuclear H1→77Se HeHaHa transfer: As the anomeric H1 is well separated by Δν H1,Hi≥1 ppm from all other glycoside protons Hi, it suffices to set the HeHaHa spin lock offset, ν SL(1H), to H1 and reduce its power to B SL<Δν H1,Hi [Hz], that is, to B SL<600 Hz on a 600 MHz spectrometer. Only H1 is then spin‐locked while all 1H resonating outside ν SL(1H)± ⋅B SL dephase and can, therefore, not partake in any Hartmann–Hahn transfer, be it homonuclear (with H1) or heteronuclear (with 77Se). To still enable the desired H1→77Se HeHaHa, however, the same low BSL must be applied at the selenoglycoside's 77Se signal offset to similarly spin‐lock a narrow frequency range ν SL(77Se)± ⋅B SL. The SeDG spectrum of such selective H1→77Se HeHaHa experiment should show only one correlation signal, as indeed observed (Figure 3 C). Full HoHaHa suppression is furthermore confirmed by the pure in‐phase appearance of the H1 doublet signal (with 3 J H1,H2=10 Hz) while its notable antiphase distortion in both the CPMG‐HSQMBC (Figure 3 A) and broadband HeHaHa spectrum (Figure 3 B) derives from antiphase HoHaHa transfer via the [H1yH2z−H1zH2y] Hamiltonian. With the obvious elimination of any HoHaHa (and HeHaHa other than for H1) transfer in the selective H1→77Se HeHaHa experiment, the relevant spin system is reduced to three coupled nuclei, that is, two H1 connected by one 77Se in the symmetric SeDG molecule, where both H1 do not couple directly (4 J H1,H1’≈0). The amplitude for planar HeHaHa transfer by DIPSI2x then simplifies to H1x→Sex⋅sin2(π√2⋅Δ⋅J/2) [Eq. (202) in ref. [16]] and is maximal for the optimal transfer delay Δ calc,sel HeHaHa=(√2⋅2 J H1,Se)−1=101 ms. Also this prediction is confirmed experimentally (Table 1).
With the corresponding optimal H1→77Se transfer delay, the selective HeHaHa spectrum (Figure 3 C) shows threefold higher intensity than unselective (broadband) HeHaHa, illustrating the loss from H1 magnetisation dissipation by undesired HoHaHa transfer. While this loss should be similar for the CPMG‐HSQMBC experiment, its spectrum (Figure 3 A) shows only about 10 % reduced H1 signal intensity. Apparently, the threefold sensitivity gain from HoHaHa suppression in the selective HeHaHa experiment is nearly nullified by the abovementioned losses from local Hartmann–Hahn mismatch and contributing transverse 77Se relaxation. Yet, this sensitivity to transverse 77Se relaxation can be turned into an important advantage for the selective 1H,77Se HeHaHa experiment when applied in a competitive NMR screening setup. There, a single selenoglycoside compound is used as spy ligand to identify, by clean 77Se NMR, other ligands competing for binding at the same site. The NMR experiment should then detect the spy ligand signal specifically and with maximal sensitivity, that is, by indirect 77Se detection through 1H,77Se correlation, as demonstrated in principle before. [11] Furthermore, the detected signal should change maximally between bound and free states of the spy ligand to indicate its displacement with maximal sensitivity. As confirmed by Figures 1 and 2, both 1H and 77Se signal intensities are very sensitive indicators of molecular binding, due to their inverse dependence on the transverse relaxation rates that strongly increase for small molecules binding to a large target. By depending also on the 77Se transverse relaxation, the 1H,77Se HeHaHa (which may be thought of as a 77Se relaxation filtered 1H experiment) should then indicate spy ligand displacement more sensitively than the CPMG‐HSQMBC experiment. Indeed, this is indirectly confirmed by the 1D 1H[77Se] traces for SeDG in the absence and presence of hGal‐3, where the H1 signal drops by a factor of 6 for CPMG‐HSQMBC (Figure 3 A) and more than 27.5 (below the noise level) for selective H1,77Se HeHaHa (Figure 3 C). Thus, a selective H1,77Se HeHaHa spectrum would indicate spy ligand displacement with the largest amplitude, overall sensitivity, and spectral simplicity (showing just the purely in‐phase H1 doublet signal), however, at the price of covering only a minimal 77Se (and 1H) bandwidth. Yet, this drawback becomes obsolete when working with only one selenium containing compound (with a single 77Se signal), where 77Se frequency editing in a second spectral dimension is likewise irrelevant. For these reasons, a competitive screening assay with a selenoglycoside spy ligand should be the ideal application for a selective 1D H1[77Se] HeHaHa experiment, and vice versa. As an important added benefit, competitive screening assays can directly yield the affinity constant (K A) of a ligand relative to that of the spy ligand, as previously demonstrated by analogous 19F NMR observation of a fluorine containing spy ligand. [17] In preparation for such applications, we proceeded to characterise the affinity and thermodynamics of SeDG binding to hGal‐1 and hGal‐3, in comparison with related ligands.
Affinity and thermodynamics of O‐, S‐ and Se‐glycoside binding to hGal‐1 and hGal‐3
We started our comparative analysis of SeDG and TDG binding to hGal‐1 and hGal‐3 with fluorescence anisotropy (FA) measurements, taking advantage of the conserved single Trp residue in the carbohydrate recognition domain (CRD) of all galectins. As this Trp engages in CH–π interactions with the β‐face of galactose, FA measurements also confirm specific ligand contact at this site. The expected galectin FA dependence on added SeDG or TDG concentration was indeed observed and indicated a K D of some 100 μm for their binding to both hGal‐1 and hGal‐3 (Figure 4).
Figure 4.
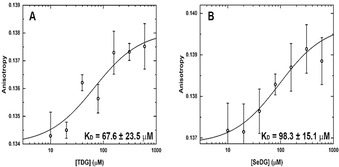
Interaction of hGal‐1 with TDG and SeDG monitored by intrinsic Trp FA. Binding isotherms for increasing concentrations of A) TDG and B) SeDG at 25 °C. Equilibrium K D values were derived by data fitting to a nonlinear regression model for single site‐specific binding using GraphPad Prism 8 and assuming one binding site per monomer. Error bars represent standard deviations from five repeated FA measurements. K D errors are estimates from the fitting algorithm.
Since full‐length hGal‐3 has two further Trp residues in its N‐terminal tail (Trp22, Trp26), the FA measurements were repeated for a proteolytically truncated hGal‐3 form and for a hGal‐1‐like variant of two covalently connected hGal‐3 CRDs. Both yielded K D values quite similar to hGal‐3 (Figure S5) confirming that the two N‐terminal Trp residues in hGal‐3 do not sense the binding event and, therefore, do not bias the FA changes by the Trp residue in the binding site. Thus, we may conclude that Trp sensitive FA spectroscopy is a robust and reliable method to measure ligand affinities for galectins and confirm their recognition in the canonical binding site. [18] We then verified that CD measurements likewise respond to ligand addition, yielding similar K D values as FA except for TDG binding to all hGal‐3 forms, where CD indicates somewhat higher K D values (Figure S6). Overall, both FA and CD confirm that TDG and SeDG are well recognised in the canonical binding site of galectins. [8a]
We then used isothermal titration calorimetry (ITC) to determine the enthalpic and entropic contributions in the thermodynamics of S‐ or Se‐digalactoside binding (Table 3; Figures S8a and S9a). Normalised to the number of CRD domains in the galectin (n/n 0), the derived stoichiometry consistently reached values around 1, proving full galectin activity. Throughout, our thermodynamic data shows the common pattern of an enthalpy driven binding process, as also reported for the natural ligand LacNAc. [19] For both hGal‐1 and hGal‐3, the affinities for TDG and SeDG are rather similar while TDG shows larger free binding enthalpies, balanced by a higher entropic penalty. When the glycosidic linkage is extended from one to two selenium atoms, the entropic penalty for DSeDG binding to hGal‐3 increases strongly and reduces the affinity by three orders of magnitude (Figure 5 A, B), in line with previous findings showing that neither disulfide nor diselenide linked glycans are recognised by galectins.[ 8a , 20 ]
Table 3.
Thermodynamic data (from ITC) for ligand binding to human hGal‐1 and ‐3 (at 25 °C).
|
Ligand[a] |
K D [μm] |
ΔG°obs [kcal mol−1] |
ΔH°obs [kcal mol−1] |
−TΔS°obs [kcal mol−1] |
Stoichiometry n/n 0 |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
H2O |
D2O |
H2O |
D2O |
H2O |
D2O |
H2O |
D2O |
H2O |
D2O |
|
hGal‐1 (n 0 =2) | ||||||||||
|
TDG |
82.7±2.9 |
69.7±1.4 |
−5.57 |
−5.67 |
−11.6±0.2 |
−11.9±0.1 |
6.07 |
6.24 |
1.00 |
1.05 |
|
SeDG |
108.0±1.4 |
126.0±2.7 |
−5.41 |
−5.32 |
−10.7±0.1 |
−8.9±0.1 |
5.27 |
3.62 |
0.99 |
1.04 |
|
LacNAc |
81.7±1.7 |
67.0±1.7 |
−5.57 |
−5.69 |
−10.6±0.1 |
−10.8±0.1 |
5.03 |
5.14 |
1.00 |
0.98 |
|
DiLacNAc |
94.7±2.2 |
101.0±1.4 |
−5.49 |
−5.45 |
−10.0±0.2 |
−8.2±0.1 |
4.53 |
2.70 |
0.97 |
1.00 |
|
LNT |
175.0±3.2 |
157.0±6.4 |
−5.13 |
−5.18 |
−5.5±0.1 |
−4.3±0.1 |
0.34 |
−0.92 |
1.01 |
0.93 |
|
hGal‐3 (n 0 =1) | ||||||||||
|
TDG |
67.6±2.0 |
51.4±1.9 |
−5.69 |
−5.85 |
−11.1±0.2 |
−11.1±0.6 |
5.39 |
5.27 |
1.01 |
0.98 |
|
SeDG |
93.1±2.6 |
96.3±4.5 |
−5.50 |
−5.48 |
−9.7±0.2 |
−9.1±1.1 |
4.18 |
3.65 |
1.12 |
0.94 |
|
DSeDG |
2800.0±180 |
– |
−3.48 |
– |
−9.7±0.5 |
– |
6.23 |
– |
0.95 |
– |
|
LacNAc |
38.6±0.5 |
32.0±2.2 |
−6.02 |
−6.13 |
−12.9±0.1 |
−11.9±1.2 |
6.88 |
5.80 |
0.99 |
0.96 |
|
DiLacNAc |
5.6±0.1 |
5.8±0.4 |
−7.17 |
−7.14 |
−9.7±0.0 |
−13.5±0.3 |
2.57 |
6.31 |
0.96 |
0.99 |
|
LNT |
28.1±0.5 |
27.4±0.8 |
−6.21 |
−6.23 |
−8.3±0.1 |
−9.0±0.1 |
2.04 |
2.80 |
1.10 |
1.00 |
[a] TDG=thiodigalactoside, SeDG=selenodigalactoside, DSeDG=diselenodigalactoside, LacNAc=N‐acetyllactosamine, DiLacNAc=di‐N‐acetyllactosamine, LNT=lacto‐N‐tetraose.
Figure 5.
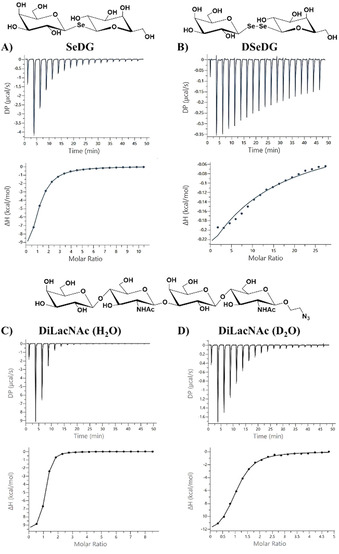
ITC titration profile of A) hGal‐3 (110 μm) with SeDG (6.0 mm), B) hGal‐3 (70 μm) with DSeDG (10.0 mm), C) hGal‐3 (135 μm) with DiLacNAc (6.0 mm), and D) hGal‐3 (40 μm) with DiLacNAc (1.0 mm) in phosphate buffer (pH 7.2) containing 20 mm phosphate, 10 mm NaCl, and 10 mm BME in water (A–C) or D2O (D). Ligand was injected every 150 s at 25 °C. The top panels show the experimental ITC data and bottom panels a fit to a one‐site model of the binding data using MicroCal PEAQ‐ITC analysis software. Resulting values for the stoichiometry (n), binding affinity (K A), dissociation constant (K D), enthalpy (ΔH), and change in entropy with respect to temperature (TΔS) are shown in Table 3.
Solvent impact on binding
Since the observed enthalpy driven binding with an accompanying decrease in entropy is still considered an “enigma”, [21] we repeated our measurements in D2O to identify a possible solvent isotope effect (SIE) as an indicator for the solvent's involvement in the binding event. [12a] After previous application to two leguminous lectins, [12] this is the first such study on human galectins.
The CD measurements in D2O (Figure S7) largely reproduce the K D values derived in H2O and, thus, indicate no significant SIE for TDG, SeDG, or LacNAc binding by hGal‐1 and the three hGal‐3 proteins. The same rough similarity of K D values is observed by ITC that enables, however, a more precise and comprehensive analysis of the binding thermodynamics (Table 3; Figures S8b and S9b). A very small net change in enthalpy and entropy was observed for each of the three disaccharides to both galectins. The enthalpy of binding was less favourable in D2O only for SeDG binding to hGal‐1, but is balanced by a similar reduction of the entropic penalty (Table 3).
This observation indicates a possible difference in galectin and/or ligand dissolution upon binding of SeDG versus LacNAc or TDG. The differential hydration of protein and ligand atoms is known to contribute strongly to an observed enthalpic destabilisation in D2O versus H2O, as reported for FK506 or rapamycin binding to FKBP‐12, [22] and could involve rather slow water exchange between the protein's hydration shell and bulk solvent. For Lac bound hGal‐3 CRD, a combination of molecular dynamics simulations and deuterium NMR relaxation dispersion experiments demonstrated such exchange of bound and bulk water molecules on a (sub)nanosecond timescale at room temperature. [23] Ongoing crystallographic attempts to characterise the water network for hGal‐3 [24] may provide further important insight. Moreover, Lac binding was found to cause subtle water reorganisation around the central Trp indole ring in the hGal‐1 CRD, [25] which must be considered to fully understand the detrimental impact on Lac binding caused by Trp mutation not only to Leu, but also to Phe or Tyr that both preserve a capacity for CH–π interaction with the ligand. Finally, a dissolution effect on the binding thermodynamics was likewise reported for other types of CRD and for oligosaccharides longer than the essential core.[ 12 , 26 ]
To further substantiate the indicated water involvement in galectin binding, we included two chain extended O‐galactosides (Scheme S1), DiLacNAc (1) and lacto‐N‐tetraose (LNT, 2), in our ITC studies (Table 3; Figures S8 and S9). Of note, mammalian Gal‐1 and Gal‐3 are known to show different affinity with dependence on the oligosaccharide chain length (more strongly for Gal‐3 than for Gal‐1) and positional preference for the primarily recognised galactose moiety (i.e., the nonreducing end LacNAc for Gal‐1 vs. reducing‐end LacNAc for Gal‐3).[ 7 , 19a , 27 ] In line with this and previous studies on bovine Gal‐1, [19a] our ITC data (Table 3) shows that the chain length extension slightly reduces the affinity (and more so for LNT) of human Gal‐1, relative to LacNAc. As expected, human Gal‐3 contrarily shows some affinity increase (and more so for DiLacNAc) (Figure 4 C, D). Most intriguingly, however, for both galectins the enthalpic and entropic contributions to tetrasaccharide binding change more notably than for SeDG or LacNAc, and differently in D2O versus H2O (Table 3; Figures S8 and S9), although these changes compensate each other in all cases to preserve similar affinities. Thus, hGal‐1 binding of DiLacNAc or LNT shows an enthalpic destabilisation with balancing entropic stabilisation in D2O, while the inverse is observed for hGal‐3. In the latter case, an enthalpy‐generating stabilisation of solvent structure is conceivable and could be provided either by direct glycan‐protein contact, as modelled for Gal‐3 in complex with LacNAc di‐ and trimers, [28] or by ligand induced interactions between Gal‐3 monomers, either via their CRD [29] or N‐terminal tail. [30]
Conclusions
Selenoglycosides offer unique opportunities for studying glycan recognition by target lectins due to a hydrolytic stability similar to that of thioglycosides and, particularly, due to the natural abundance of a stable isotope (77Se) enabling observation by selective high‐resolution NMR spectroscopy. Using selenodigalactoside (SeDG) and the human hGal‐1 and hGal‐3, we have confirmed and quantified the significant sensitivity gains achievable by indirect versus direct 77Se NMR detection. For the optimal 1H,77Se out‐and‐back correlation method, a 63‐fold enhancement is predicted, but dissipation of transferable 1H magnetisation by transverse relaxation and homonuclear 1H,1H′ coupling evolution reduces the gains to about in practice. To eliminate these losses, we introduced the selective heteronuclear H1→77Se Hartmann–Hahn transfer (HeHaHa) experiment which, compared with its nonselective variant, is shown to yield a threefold sensitivity gain from suppressing any concurring homonuclear H1→Hi Hartmann–Hahn (HoHaHa) transfer. Yet, spatial heterogeneity of the 1H and 77Se spin lock fields and the added sensitivity to transverse 77Se relaxation reduce these gains to 10 % relative to the benchmark CPMG‐HSQMBC experiment. This latter implicit “T2(77Se) filter” effect of the selective HeHaHa experiment, however, should favourably increase its indicator sensitivity in competitive NMR screening assays using a selenoglycoside spy ligand. For such applications, we also characterised the affinity and thermodynamics of SeDG binding to hGal‐1 and hGal‐3, which yielded a K D of about 100 μm and confirmed its equivalence, as a ligand, with both LacNAc and TDG. The tryptophan at the glycan binding site of hGal‐1 and hGal‐3 thereby allowed the site‐specific binding to be monitored by fluorescence anisotropy (FA) as a fast and robust alternative to CD or ITC. Finally, a comparison of data obtained in H2O versus D2O revealed no significant solvent isotope effect (SIE) on hGal‐1 and hGal‐3 binding of TDG or LacNAc, but hints at a small SIE for SeDG. A clear SIE with compensation of enthalpic and entropic contributions emerges, however, especially for hGal‐3 when binding LacNAc dimers, indicating some mayor solvent rearrangement at its binding site as the glycan chain length is extended.
Experimental Section
Galectins and their ligands
Human Gal‐1 and ‐3 were recombinantly produced, purified by affinity chromatography on lactosylated Sepharose 4B, and examined for purity by gel electrophoresis and mass spectrometry as described.[ 29b , 31 ] SeDG and DSeDG were synthesised and analysed for purity as described previously. [8a]
NMR spectroscopy
All NMR experiments were recorded at 298 K on a BRUKER AVANCE III 600 MHz spectrometer equipped with a TBI probehead with z‐gradients. All cited 2D 1H,77Se correlation experiments were recorded with the relevant NMR parameters given in the text and Figure legends, and with the pulse program codes printed in the Supporting Information.
CD spectroscopy
CD measurements were carried out with a JASCO J‐815 spectropolarimeter equipped with a Peltier‐type temperature controller, a thermostabilised cell holder, and interfaced with a thermostatic bath. Near‐UV spectra were recorded in a quartz cell of 1 cm path length at a protein concentration between 2.5 and 3.0 mg mL−1 in phosphate‐buffered saline (PBS), pH 7.0. Ten consecutive scans were accumulated and the average spectra were stored. Data was corrected for the baseline contribution from buffer and ligands. The observed ellipticities were converted into mean residue ellipticities [θ] based on a mean molecular per residue mass of 109 Da for hGal‐1, 112.6 Da for hGal‐3 CRD, and 112.1 Da for the hGal‐3 homodimer. K D values were derived by fitting the CD data to a nonlinear regression model for single site‐specific binding using GraphPad Prism 8, assuming one binding site for each domain of hGal‐1 and ‐3 and two binding sites for the hGal‐3 homodimer. Figures were generated with ORIGIN2018 (www.originlab.com).
Fluorescence anisotropy
FA was measured using a Fluoromax‐4 (Horiba Jobin Yvon) photon counting spectrofluorometer with excitation at 295 nm (5 nm band pass) and emission at 350 nm (5 nm band pass) using 5×10 mm cells at 25 °C. Measurements were carried out at a protein concentration of 0.15 mg mL−1. Freeze‐dried protein was dissolved in PBS (pH 7.0). FA levels were calculated from the excitation/emission polarisation combinations, taking into account the G correction factor and according to standard equations. [32] Equilibrium K D values were determined as described above for the CD data. Figures were likewise generated with ORIGIN2018 (www.originlab.com).
ITC measurements
ITC titrations were monitored in a PEAQ‐ITC calorimeter (Malvern, Westborough, MA, USA) using 200 μL galectin containing solution in 20 mm phosphate buffer (pH 7.2), 10 mm NaCl, 10 mm β‐mercaptoethanol, injected at 150 s intervals in aliquots of 2 μL (up to a total of 36.4 μL) at 25 °C, 750 rpm, as described before.[ 6j , 33 ] In each titration, an offset parameter was also fitted to account for potential background. Data was processed using the MicroCal PEAQ‐ITC Analysis software. Studies in D2O were carried out by dissolving lyophilised protein (originally dialysed in buffered H2O) in D2O.
2‐Azidoethyl (β‐d‐galactopyranosyl)‐(1→4)‐(2‐acetamido‐2‐deoxy‐β‐d‐glucopyranosyl)‐(1→3)‐(β‐d‐galactopyranosyl)‐(1→4)‐2‐acetamido‐2‐deoxy‐β‐d‐glucopyranoside (1)
2‐Azidoethyl (2,3,4,6‐tetra‐O‐acetyl‐β‐d‐galactopyranosyl)‐(1→4)‐(3,6‐di‐O‐acetyl‐2‐acetamido‐2‐deoxy‐β‐d‐glucopyranosyl)‐(1→3)‐(2,4,6‐tri‐O‐acetyl‐β‐d‐galactopyranosyl)‐(1→4)‐3,6‐di‐O‐acetyl‐2‐acetamido‐2‐deoxy‐β‐d‐glucopyranoside (4, 20.9 mg, 16.3 μmol) was dissolved in 0.35 mL dry DMSO under N2 at ambient temperature. Dry MeOH (0.10 mL) was added, followed by NaOMe (2.5 mg, 46 μmol), and the resulting reaction mixture was stirred overnight until TLC‐analysis (3:7, H2O/MeCN) of the reaction mixture showed full conversion. The mixture was neutralised with freshly washed DOWEX 8WX50 (H+), filtered, concentrated and lyophilised to remove remaining DMSO. The crude material (14 mg) was purified by size exclusion on Bio‐Gel P‐2 gel (H2O/tBuOH, 99:1). Collection of the appropriate fraction followed by concentration and lyophilisation yielded 1 as a fluffy white solid (12.6 mg, 94 %). [α] =−0.25 (c=0.30, H2O), 1H NMR (500 MHz, D2O): δ=4.58 (d, J=8.3 Hz, 1 H), 4.48 (d, J=8.3 Hz, 1 H), 4.35 (apparent t, J=8.1 Hz, 2 H), 4.04 (d, J=3.3 Hz, 1 H), 3.93 (ddd, J=11.4, J=5.6, J=3.1, 1 H), 3.87 (dd, J=12.4 Hz, J=2.2 Hz, 1 H), 3.83 (dd, J=12.3 Hz, J=2.3 Hz, 1 H), 3.81 (d, J=3.4 Hz, 1 H), 3.76–3.57 (m, 16 H), 3.55 (dd, J=10.0 Hz, J=3.5 Hz, 1 H), 3.51–3.44 (m, 3 H), 3.42 (dd, J=10.0 Hz, J=7.8 Hz, 1 H,), 3.37 (ddd, J=13.7 Hz, J=7.5, J=3.0 Hz, 1 H), 3.30 (ddd, J=13.8 Hz, J=5.7 Hz, J=3.1 Hz, 1 H), 1.92 (s, 3 H), 1.91 (s, 3 H). 13C NMR (126 MHz, D2O): δ=174 Hz.8, 174.6, 102.9, 102.8, 102.7, 100.9, 82.0, 78.4, 78.1,75.3, 74.8, 74.7, 74.5, 72.4, 72.4, 72.1, 70.9, 69.9, 68.7, 68.5, 68.3, 61.0, 60.9, 60.0, 59.8, 55.1, 54.9, 50.3, 22.2, 22.1. NMR spectroscopy characterisation of 1 is illustrated in Figure S13 A–C. HRMS (ESI) m/z calculated for C30H51N5O21: 840.2974 [M+Na]+; found: 840.2964. The data were in agreement with earlier published NMR data. [34]
3‐Aminopropyl (β‐d‐galactopyranosyl)‐(1→3)‐(2‐acetamido‐2‐deoxy‐β‐d‐glucopyranosyl)‐(1→3)‐(β‐d‐galactopyranosyl)‐(1→4)‐β‐d‐glucopyranoside (2)
EtOH (1.2 mL) was added to 3‐azidopropyl (3,4,6‐tri‐O‐benzyl‐β‐d‐galactopyranosyl)‐(1→3)‐(4,6‐O‐benzylidene‐2‐acetamido‐2‐deoxy‐β‐d‐glucopyranosyl)‐(1→3)‐(2,4,6‐tri‐O‐benzyl‐β‐d‐galactopyranosyl)‐(1→4)‐2,3,6‐tri‐O‐benzyl‐β‐d‐glucopyranoside (5, 50 mg, 30 μmol), followed by water (296 μL) and 0.1 m aq. HCl (296 μL, 29.6 μmol). EtOAc was then added dropwise until a homogeneous solution was formed. Pd/C (5 % wt., 10 mg, 4.7 μmol) and Pd(OH)2/C (20 % wt., 10 mg, 14 μmol) were added, and the resulting suspension was stirred under H2 (balloon) at room temperature for 19 hours. The suspension was then diluted with water, filtered through Celite, and concentrated in vacuo. The product was purified by size‐exclusion chromatography on Bio‐Gel P‐2 gel (H2O/tBuOH, 99:1) yielding a white solid after concentration and lyophilisation (2, 11 mg, 46 %). 1H NMR (400 MHz, D2O): δ=4.76 (d, J=8.3 Hz, 1 H), 4.53 (d, J=8.0 Hz, 1 H), 4.48–4.45 (m, 2 H), 4.18 (d, J=3.2 Hz, 1 H), 4.12–3.97 (m, 2 H), 3.94–3.90 (m, 3 H), 3.88–3.70 (m, 11 H), 3.70–3.46 (m, 8 H), 3.35 (m, 1 H), 3.15 (t, J=7.0 Hz, 2 H), 2.05–1.98 (m, 5 H). NMR‐spectroscopy characterisation of 2 is illustrated in Figure S14A, B. HRMS (ESI) m/z calculated for C29H52N2O21: 765.3141 [M+H]+; found: 765.3124. The data were in agreement with earlier published NMR data (2‐carbon spacer). [35]
Conflict of interest
The authors declare no conflict of interests.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We are much indebted to L. Szilágyi (University of Debrecen, Hungary) for kindly providing selenoglycosides for this study. We are grateful to Drs. B. Friday, A. Leddoz and A. W. L. Nose for inspiring discussions, as well as for generous funding by the Spanish Ministry of Economy and Competitiveness (grant BFU2016‐77835‐R; to A.R.), the Department of Industry, Tourism and Trade of the Government of the Autonomous Community of the Basque Country (Elkartek BG2019; to T.D.), the Severo Ochoa Excellence Accreditation from MCIU (SEV‐2016‐0644; to T.D.), the Science Foundation of Ireland (grants 13/IA/1959 and 16/RC/3889; to S.O.), National Institute of Health (NIH) grant CA242351 to M.C., and the COST Action CA 18103 (InnoGly; to H.‐J.G.).
T. Diercks, F. J. Medrano, F. G. FitzGerald, D. Beckwith, M. J. Pedersen, M. Reihill, A.-K. Ludwig, A. Romero, S. Oscarson, M. Cudic, H.-J. Gabius, Chem. Eur. J. 2021, 27, 316.
Contributor Information
Dr. Tammo Diercks, Email: tdiercks@cicbiogune.es.
Prof. Dr. Antonio Romero, Email: romero@cib.csic.es.
Prof. Dr. Stefan Oscarson, Email: stefan.oscarson@ucd.ie.
Prof. Dr. Maré Cudic, Email: mcudic@fau.edu.
Prof. Dr. Hans‐Joachim Gabius, Email: gabius@tiph.vetmed.uni-muenchen.de.
References
- 1.
- 1a. Winterburn P. J., Phelps C. F., Nature 1972, 236, 147–151; [DOI] [PubMed] [Google Scholar]
- 1b. Reuter G., Gabius H.-J., Cell. Mol. Life Sci. 1999, 55, 368–422; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. The Sugar Code: Fundamentals of Glycosciences (Ed.: Gabius H.-J.), Wiley-VCH, Weinheim, 2009; [Google Scholar]
- 1d. Schnaar R. L., J. Leukoc. Biol. 2016, 99, 825–838; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1e. Corfield A. P., Histochem. Cell Biol. 2017, 147, 119–147; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1f. Gabius H.-J., Roth J., Histochem. Cell Biol. 2017, 147, 111–117; [DOI] [PubMed] [Google Scholar]
- 1g. Cummings R. D., Glycoconjugate J. 2019, 36, 241–257; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1h. Kaltner H., Abad-Rodríguez J., Corfield A. P., Kopitz J., Gabius H.-J., Biochem. J. 2019, 476, 2623–2655. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Horton D., Hutson D. H., Adv. Carbohydr. Chem. 1963, 18, 123–199; [DOI] [PubMed] [Google Scholar]
- 2b. Driguez H., Top. Curr. Chem. 1997, 187, 85–116; [Google Scholar]
- 2c. Driguez H., ChemBioChem 2001, 2, 311–318; [DOI] [PubMed] [Google Scholar]
- 2d. Pachamuthu K., Schmidt R. R., Chem. Rev. 2006, 106, 160–187; [DOI] [PubMed] [Google Scholar]
- 2e. Szilagyi L., Varela O., Curr. Org. Chem. 2006, 10, 1745–1770. [Google Scholar]
- 3.
- 3a. Tvaroška I., Collect. Czech. Chem. Commun. 1984, 49, 345–354; [Google Scholar]
- 3b. Mazeau K., Tvaroška I., Carbohydr. Res. 1992, 225, 27–41; [DOI] [PubMed] [Google Scholar]
- 3c. Muñoz J. L., García-Herrero A., Asensio J. L., Auzennaeau F. J., Cañada F. J., Jiménez-Barbero J., J. Chem. Soc. Perkin Trans. 2001, 1, 867; [Google Scholar]
- 3d. Strino F., Lii J. H., Gabius H.-J., Nyholm P. G., J. Comput. Aid. Mol. Des. 2009, 23, 845–852. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Lis H., Sharon N., Chem. Rev. 1998, 98, 637–674; [DOI] [PubMed] [Google Scholar]
- 4b. Manning J. C., Romero A., Habermann F. A., García Caballero G., Kaltner H., Gabius H.-J., Histochem. Cell Biol. 2017, 147, 199–222; [DOI] [PubMed] [Google Scholar]
- 4c. Kaltner H., García Caballero G., Ludwig A.-K., Manning J. C., Gabius H.-J., Histochem. Cell Biol. 2018, 149, 547–568. [DOI] [PubMed] [Google Scholar]
- 5. Teichberg V. I., Silman I., Beitsch D. D., Resheff G., Proc. Natl. Acad. Sci. USA 1975, 72, 1383–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Barondes S. H., Science 1984, 223, 1259–1264; [DOI] [PubMed] [Google Scholar]
- 6b. Harrison F. L., J. Cell Sci. 1991, 100, 9–14; [DOI] [PubMed] [Google Scholar]
- 6c. Barondes S. H., Trends Glycosci. Glycotechnol. 1997, 9, 1–7; [Google Scholar]
- 6d. Gabius H.-J., Eur. J. Biochem. 1997, 243, 543–576; [DOI] [PubMed] [Google Scholar]
- 6e. Kasai K.-i., Trends Glycosci. Glycotechnol. 1997, 9, 167–170; [Google Scholar]
- 6f. Cooper D. N. W., Biochim. Biophys. Acta Gen. Subj. 2002, 1572, 209–231; [DOI] [PubMed] [Google Scholar]
- 6g. Yang R. Y., Liu F.-T., Cell. Mol. Life Sci. 2003, 60, 267–276; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6h. Kaltner H., Toegel S., García Caballero G., Manning J. C., Ledeen R. W., Gabius H.-J., Histochem. Cell Biol. 2017, 147, 239–256; [DOI] [PubMed] [Google Scholar]
- 6i. Trends in Glycoscience and Glycotechnology (Ed.: J. E. Hirabayashi), 2018, 30, SE1-223;
- 6j. Ludwig A.-K., Michalak M., Xiao Q., Gilles U., Medrano F. J., Ma H., FitzGerald F. G., Hasley W. D., Melendez-Davila A., Liu M., Rahimi K., Kostina N. Y., Rodriguez-Emmenegger C., Möller M., Lindner I., Kaltner H., Cudic M., Reusch D., Kopitz J., Romero A., Oscarson S., Klein M. L., Gabius H.-J., Percec V., Proc. Natl. Acad. Sci. USA 2019, 116, 2837–2842; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6k. Romero A., Gabius H.-J., Expert Opin. Ther. Targets 2019, 23, 819–828; [DOI] [PubMed] [Google Scholar]
- 6l. García Caballero G., Kaltner H., Kutzner T. J., Ludwig A.-K., Manning J. C., Schmidt S., Sinowatz F., Gabius H.-J., Histol. Histopathol. 2020, 35, 509–539. [DOI] [PubMed] [Google Scholar]
- 7. Sparrow C., Leffler H., Barondes S. H., J. Biol. Chem. 1987, 262, 7383–7390. [PubMed] [Google Scholar]
- 8.
- 8a. André S., Kövér K. E., Gabius H.-J., Szilágyi L., Bioorg. Med. Chem. Lett. 2015, 25, 931–935; [DOI] [PubMed] [Google Scholar]
- 8b. Kaltner H., Szabo T., Fehér K., André S., Balla S., Manning J. C., Szilágyi L., Gabius H.-J., Bioorg. Med. Chem. 2017, 25, 3158–3170; [DOI] [PubMed] [Google Scholar]
- 8c. Strino F., Lii J. H., Koppisetty C. A. K., Nyholm P. G., Gabius H.-J., J. Comput. Aid. Mol. Des. 2010, 24, 1009–1021. [DOI] [PubMed] [Google Scholar]
- 9. Buts L., Loris R., De Genst E., Oscarson S., Lahmann M., Messens J., Brosens E., Wyns L., De Greve H., Bouckaert J., Acta Crystallogr. Sect. D Biol. Crystallogr. 2003, 59, 1012–1015. [DOI] [PubMed] [Google Scholar]
- 10. Hamark C., Landström J., Widmalm G., Chem. Eur. J. 2014, 20, 13905–13908. [DOI] [PubMed] [Google Scholar]
- 11. Raics M., Timári I., Diercks T., Szilágyi L., Gabius H.-J., Kövér K. E., ChemBioChem 2019, 20, 1688–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Chervenak M. C., Toone E. J., J. Am. Chem. Soc. 1994, 116, 10533–10539; [Google Scholar]
- 12b. Dam T. K., Oscarson S., Sacchettini J. C., Brewer C. F., J. Biol. Chem. 1998, 273, 32826–32832; [DOI] [PubMed] [Google Scholar]
- 12c. Isbister B. D., St. Hilaire P. M., Toone E. J., J. Am. Chem. Soc. 1995, 117, 12877–12878. [Google Scholar]
- 13.
- 13a. Muñoz F. J., Santos J. I., Ardá A., André S., Gabius H.-J., Sinisterra J. V., Jiménez-Barbero J., Hernáiz M. J., Org. Biomol. Chem. 2010, 8, 2986–2992; [DOI] [PubMed] [Google Scholar]
- 13b. Solís D., Maté M. J., Lohr M., Ribeiro J. P., López-Merino L., André S., Buzamet E., Javier Cañada F., Kaltner H., Lensch M., Ruiz F. M., Haroske G., Wollina U., Kloor M., Kopitz J., Sáiz J. L., Menéndez M., Jiménez-Barbero J., Romero A., Gabius H.-J., Int. J. Biochem. Cell Biol. 2010, 42, 1019–1029. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Brown L. R., Sanctuary B. C., J. Magn. Reson. 1991, 91, 413–421; [Google Scholar]
- 14b. Shaka A. J., Lee C. J., Pines A., J. Magn. Reson. 1988, 77, 274–293. [Google Scholar]
- 15. Furrer J., Kramer F., Marino J. P., Glaser S. J., Luy B., J. Magn. Reson. 2004, 166, 39–46. [DOI] [PubMed] [Google Scholar]
- 16. Glaser S. J., Quant J. J. in Homonuclear and Heteronuclear Hartmann–Hahn Transfer in Isotropic Liquids, Vol. 19 (Ed.: Warren W. S.), Academic Press, San Diego, 1996, pp. 59–254. [Google Scholar]
- 17. André S., Cañada F. J., Shiao T. C., Largartera L., Diercks T., Bergeron-Brlek M., el Biari K., Papadopoulos A., Ribeiro J. P., Touaibia M., Solís D., Menéndez M., Jiménez-Barbero J., Roy R., Gabius H.-J., Eur. J. Org. Chem. 2012, 4354–4364. [Google Scholar]
- 18.
- 18a. Göhler A., Buchner C., André S., Doose S., Kaltner H., Gabius H.-J., Analyst 2011, 136, 5270–5276; [DOI] [PubMed] [Google Scholar]
- 18b. Göhler A., Buchner C., Doose S., Kaltner H., Gabius H.-J., Biochimie 2012, 94, 2649–2655; [DOI] [PubMed] [Google Scholar]
- 18c. Levi G., Teichberg V. I., J. Biol. Chem. 1981, 256, 5735–5740; [PubMed] [Google Scholar]
- 18d. Sindrewicz P., Li X., Yates E. A., Turnbull J. E., Lian L. Y., Yu L. G., Sci. Rep. 2019, 9, 11851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Ahmad N., Gabius H.-J., Kaltner H., André S., Kuwabara I., Liu F.-T., Oscarson S., Norberg T., Brewer C. F., Can. J. Chem. 2002, 80, 1096–1104; [Google Scholar]
- 19b. Bharadwaj S., Kaltner H., Korchagina E. Y., Bovin N. V., Gabius H.-J., Surolia A., Biochim. Biophys. Acta Gen. Subj. 1999, 1472, 191–196; [DOI] [PubMed] [Google Scholar]
- 19c. Dam T. K., Gabius H.-J., André S., Kaltner H., Lensch M., Brewer C. F., Biochemistry 2005, 44, 12564–12571; [DOI] [PubMed] [Google Scholar]
- 19d. Gupta D., Cho M., Cummings R. D., Brewer C. F., Biochemistry 1996, 35, 15236–15243; [DOI] [PubMed] [Google Scholar]
- 19e. Ramkumar R., Surolia A., Podder S. K., Biochem. J. 1995, 308, 237–241; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19f. Schwarz F. P., Ahmed H., Bianchet M. A., Amzel L. M., Vasta G. R., Biochemistry 1998, 37, 5867–5877. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a. André S., Pei Z., Siebert H.-C., Ramström O., Gabius H.-J., Bioorg. Med. Chem. 2006, 14, 6314–6323; [DOI] [PubMed] [Google Scholar]
- 20b. Martín-Santamaría S., André S., Buzamet E., Caraballo R., Fernández-Cureses G., Morando M., Ribeiro J. P., Ramirez-Gualito K., de Pascual-Teresa B., Cañada F. J., Menéndez M., Ramström O., Jiménez-Barbero J., Solís D., Gabius H.-J., Org. Biomol. Chem. 2011, 9, 5445–5455. [DOI] [PubMed] [Google Scholar]
- 21. Lemieux R. U., Acc. Chem. Res. 1996, 29, 373–380. [Google Scholar]
- 22. Connelly P. R., Thomson J. A., Fitzgibbon M. J., Bruzzese F. J., Biochemistry 1993, 32, 5583–5590. [DOI] [PubMed] [Google Scholar]
- 23. Saraboji K., Håkansson M., Genheden S., Diehl C., Qvist J., Weininger U., Nilsson U. J., Leffler H., Ryde U., Akke M., Logan D. T., Biochemistry 2012, 51, 296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.
- 24a. Su J., Zhang T., Wang P., Liu F., Tai G., Zhou Y., Acta. Biochim. Biophys. Sin. 2015, 47, 192–198; [DOI] [PubMed] [Google Scholar]
- 24b. Caldararu O., Manzoni F., Oksanen E., Logan D. T., Ryde U., Acta Crystallogr. Sect. D Biol. Crystallogr. 2019, 75, 368–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.
- 25a. Abbott W. M., Feizi T., J. Biol. Chem. 1991, 266, 5552–5557; [PubMed] [Google Scholar]
- 25b. Hirabayashi J., Kasai K.-i., J. Biol. Chem. 1991, 266, 23648–23653; [PubMed] [Google Scholar]
- 25c. Di Lella S., Ma L., Ricci J. C., Rabinovich G. A., Asher S. A., Alvarez R. M., Biochemistry 2009, 48, 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.
- 26a. Navarra G., Zihlmann P., Jakob R. P., Stangier K., Preston R. C., Rabbani S., Smiesko M., Wagner B., Maier T., Ernst B., ChemBioChem 2017, 18, 539–544; [DOI] [PubMed] [Google Scholar]
- 26b. Sager C. P., Eris D., Smiesko M., Hevey R., Ernst B., Beilstein J. Org. Chem. 2017, 13, 2584–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.
- 27a. Solomon J. C., Stoll M. S., Penfold P., Abbott W. M., Childs R. A., Hanfland P., Feizi T., Carbohydr. Res. 1991, 213, 293–307; [DOI] [PubMed] [Google Scholar]
- 27b. Sato S., Hughes R. C., J. Biol. Chem. 1992, 267, 6983–6990; [PubMed] [Google Scholar]
- 27c. Knibbs R. N., Agrwal N., Wang J. L., Goldstein I. J., J. Biol. Chem. 1993, 268, 14940–14947; [PubMed] [Google Scholar]
- 27d. Feizi T., Solomon J. C., Yuen C. T., Jeng K. C., Frigeri L. G., Hsu D. K., Liu F.-T., Biochemistry 1994, 33, 6342–6349; [DOI] [PubMed] [Google Scholar]
- 27e. Di Virgilio S., Glushka J., Moremen K., Pierce M., Glycobiology 1999, 9, 353–364; [DOI] [PubMed] [Google Scholar]
- 27f. Hirabayashi J., Hashidate T., Arata Y., Nishi N., Nakamura T., Hirashima M., Urashima T., Oka T., Futai M., Müller W. E. G., Yagi F., Kasai K.-i., Biochim. Biophys. Acta Gen. Subj. 2002, 1572, 232–254; [DOI] [PubMed] [Google Scholar]
- 27g. Stowell S. R., Dias-Baruffi M., Penttila L., Renkonen O., Nyame A. K., Cummings R. D., Glycobiology 2003, 14, 157–167; [DOI] [PubMed] [Google Scholar]
- 27h. Leppänen A., Stowell S., Blixt O., Cummings R. D., J. Biol. Chem. 2005, 280, 5549–5562; [DOI] [PubMed] [Google Scholar]
- 27i. Stowell S. R., Arthur C. M., Mehta P., Slanina K. A., Blixt O., Leffler H., Smith D. F., Cummings R. D., J. Biol. Chem. 2008, 283, 10109–10123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nagae M., Nishi N., Murata T., Usui T., Nakamura T., Wakatsuki S., Kato R., Glycobiology 2008, 19, 112–117. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Ippel H., Miller M. C., Vértesy S., Zheng Y., Canada F. J., Suylen D., Umemoto K., Romanò C., Hackeng T., Tai G., Leffler H., Kopitz J., André S., Kübler D., Jiménez-Barbero J., Oscarson S., Gabius H.-J., Mayo K. H., Glycobiology 2016, 26, 888–903; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29b. Flores-Ibarra A., Vértesy S., Medrano F. J., Gabius H.-J., Romero A., Sci. Rep. 2018, 8, 9835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Halimi H., Rigato A., Byrne D., Ferracci G., Sebban-Kreuzer C., ElAntak L., Guerlesquin F., PLoS One 2014, 9, e111836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.
- 31a. Kopitz J., Vértesy S., André S., Fiedler S., Schnölzer M., Gabius H.-J., Biochimie 2014, 104, 90–99; [DOI] [PubMed] [Google Scholar]
- 31b. Amano M., Eriksson H., Manning J. C., Detjen K. M., André S., Nishimura S.-I., Lehtiö J., Gabius H.-J., FEBS J. 2012, 279, 4062–4080. [DOI] [PubMed] [Google Scholar]
- 32. Lakowicz J. R., Principles of Fluorescence Spectroscopy, Springer, New York, 2006, Chapter 10, p. 353. [Google Scholar]
- 33. Kutzner T. J., Gabba A., FitzGerald F. G., Shilova N. V., García Caballero G., Ludwig A.-K., Manning J. C., Knospe C., Kaltner H., Sinowatz F., Murphy P. V., Cudic M., Bovin N. V., Gabius H.-J., Glycobiology 2019, 29, 593–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vasiliu D., Razi N., Zhang Y., Jacobsen N., Allin K., Liu X., Hoffmann J., Bohorov O., Blixt O., Carbohydr. Res. 2006, 341, 1447–1457. [DOI] [PubMed] [Google Scholar]
- 35. Sherman A. A., Yudina O. N., Mironov Y. V., Sukhova E. V., Shashkov A. S., Menshov V. M., Nifantiev N. E., Carbohydr. Res. 2001, 336, 13–46. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary