

UBR7 supports nucleotide biosynthesis and NOTCH1-driven T cell acute lymphoblastic leukemia through regulation of PRPS enzymes.
Abstract
Ubiquitin protein ligase E3 component N-recognin 7 (UBR7) is the most divergent member of UBR box–containing E3 ubiquitin ligases/recognins that mediate the proteasomal degradation of its substrates through the N-end rule. Here, we used a proteomic approach and found phosphoribosyl pyrophosphate synthetases (PRPSs), the essential enzymes for nucleotide biosynthesis, as strong interacting partners of UBR7. UBR7 stabilizes PRPS catalytic subunits by mediating the polyubiquitination-directed degradation of PRPS-associated protein (PRPSAP), the negative regulator of PRPS. Loss of UBR7 leads to nucleotide biosynthesis defects. We define UBR7 as a transcriptional target of NOTCH1 and show that UBR7 is overexpressed in NOTCH1-driven T cell acute lymphoblastic leukemia (T-ALL). Impaired nucleotide biosynthesis caused by UBR7 depletion was concomitant with the attenuated cell proliferation and oncogenic potential of T-ALL. Collectively, these results establish UBR7 as a critical regulator of nucleotide metabolism through the regulation of the PRPS enzyme complex and uncover a metabolic vulnerability in NOTCH1-driven T-ALL.
INTRODUCTION
T cell acute lymphoblastic leukemia (T-ALL) is an aggressive blood cancer that accounts for ~15% of pediatric and ~25% of adult ALL cases (1, 2). The NOTCH signaling pathway is an evolutionary conserved pathway and NOTCH1 is absolutely required for the differentiation of hematopoietic progenitors into specific lineages and facilitates cell proliferation and survival (3). NOTCH1-activating mutations are found in ~60% T-ALL cases (4). The activation of NOTCH1 is marked by its proteolytic cleavage by γ-secretase that produces NOTCH1 intracellular domain (NICD1). Once released, NICD1 translocates to the nucleus and forms a transcription activation complex with DNA binding protein RBP-Jκ and coactivator mastermind-like family proteins to regulate the transcription of downstream targets (5). Several NOTCH1 target genes that mediate oncogenic program in T-ALL have been identified so far (5). While some of these targets, such as c-Myc, have been studied extensively (6–8), the role of many NOTCH1 targets in the context of T-ALL remains undetermined.
Ubiquitin protein ligase E3 component N-recognin 7 (UBR7) is a member of a family of ubiquitin protein ligases/recognins defined by the presence of a UBR box (9, 10). The UBR box generally recognizes an “N-degron” sequence present in its substrates and targets them for degradation. UBR7, however, does not recognize the canonical N-degron sequence (11). Moreover, it also does not have a canonical E3 ligase domain. Instead, it harbors a plant homeodomain (PHD) finger that makes it a unique UBR family member. UBR7 has been found to associate with chromatin and has been recently shown to monoubiquitinate histone H2B (12–14). Besides these observations, the cellular functions of UBR7 remain largely uncharacterized.
To better understand the biological roles of UBR7, we used the affinity purification–mass spectrometry (MS) approach and found phosphoribosyl pyrophosphate synthetase (PRPS) enzymes as robust interacting partners of UBR7. PRPS enzymes are a cornerstone to nucleotide synthesis because they synthesize phosphoribosyl pyrophosphate (PRPP) from ribose 5-phosphate and adenosine 5′-triphosphate (ATP) to produce purine and pyrimidine nucleotides (15, 16). The human PRPS protein family has three isoforms with very high sequence similarity (15, 17–19). Whereas PRPS3 isoform is specific to testis, PRPS1 and PRPS2 are ubiquitously expressed (15, 17–19). Both PRPS1 and PRPS2 have been found to exist in a complex with PRPS-associated proteins (PRPSAP1 and PRPSAP2), which have been described as negative regulators of PRPS catalytic subunits (18–22).
PRPS1 and PRPS2 have been implicated in cancer cell growth. One mechanism is through Ser180 or Ser183 dephosphorylation that converts PRPS1 or PRPS2, respectively, from monomer to hexamer and promotes glioblastoma (23). Although PRPS1 and PRPS2 are significantly redundant with 95% sequence identity, Ser103 and Thr225 phosphorylation of PRPS1 promotes colorectal cancer and hepatocellular carcinoma development, respectively (24, 25). In addition, specific mutations in PRPS1 lead to its constitutive activation via suppression of feedback inhibition, whereas PRPS2 largely remains resistant to such inhibitions (26, 27). Several activating mutations in PRPS1 have been found in relapsed childhood B cell ALL patients and confer resistance to anti-purine drug, which can be overcome by anti-pyrimidine drugs (28, 29). On the other hand, PRPS2 promotes nucleotide production in Myc-driven tumors (30).
Metabolic reprogramming is the hallmark of cancer that allows cancer cells to meet their biosynthetic requirement for cell growth (31–33). For instance, in the context of T-ALL, glutaminolysis has been described as a key anabolic pathway that supports leukemia cell growth downstream to NOTCH1; however, a subset of T-ALL with PTEN-null mutation exhibits a metabolic shunt toward glycolysis (34). While this tour de force study provides strong evidence to uncover the role of glutaminolysis in NOTCH1-driven T-ALL, the regulatory mechanisms underlying other fundamental metabolic processes, such as nucleotide biosynthesis, remain largely unknown.
In the present study, we establish a connection between NOTCH1 signaling and nucleotide metabolism. We define UBR7 as a novel transcriptional target of NOTCH1 and a key regulator of nucleotide biosynthesis. Using NOTCH1-driven T-ALL tumors and cell lines as models, our findings reveal a novel pathway involving the upstream regulation of UBR7 by NOTCH1, which, in turn, stabilizes essential nucleotide biosynthesis enzymes, PRPS1 and PRPS2, through the degradation of PRPSAP and facilitates T-ALL cell proliferation and oncogenic potential.
RESULTS
Interactome screen for identifying UBR7-interacting partners
To understand the biological roles of UBR7, we conducted an unbiased affinity purification coupled with a liquid chromatography–MS (LC-MS) approach to determine the binding partners of UBR7. FLAG affinity purification followed by MS from the whole-cell extracts of parental or FLAG-tagged UBR7-expressing stable human embryonic kidney (HEK) 293T (HEK293T) cell line revealed several enriched proteins with UBR7 (Fig. 1A and table S1). The high-confidence interactome of UBR7 was screened on the basis of a strict filtration scheme (see Materials and Methods) and reproducibility across independent biological experiments (table S1 and Fig. 1, B and C). PRPS1, PRPS2, and PRPS3 were found to be among top hits as they qualified our strict screening criteria (Fig. 1, A and C). UBR7-FLAG affinity purification and MS from the whole-cell extracts of a neuroblastoma cell line, SH-SY5Y, also revealed a similar interactome of UBR7 (fig. S1A and table S2).
Fig. 1. Identification of UBR7-interacting proteins.
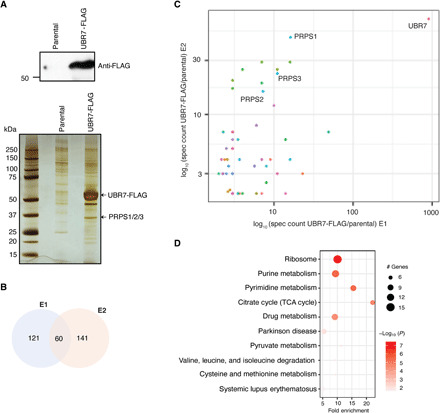
(A) UBR7-FLAG expression in stable HEK293T cells and silver stain showing the elutes from FLAG immunoprecipitation (IP) in parental and UBR7-FLAG–expressing HEK293T cells. (B) Venn diagram representing the number of common and exclusive FLAG-UBR7–associated proteins between two independent experiments E1 and E2. (C) Scatter plot from two independent MS runs (E1 and E2) showing the UBR7-FLAG–associated proteins. The axes represent the log of spec counts of UBR7-FLAG normalized to parental. A curated list of UBR7-interacting proteins can be found in table S1. (D) KEGG pathway analysis representing top 10 significantly enriched pathways associated with UBR7-interacting partners from E1.
UBR7 has been reported to be a chromatin-associated protein (12–14). Therefore, we determined the UBR7-interacting partners following anti-FLAG purification from soluble and nuclear fractions derived from HEK293T cells. Consistent with a report by Kleiner et al. (13), UBR7 was associated with histone H3 in both soluble and nuclear fractions (fig. S1B and table S3). On the other hand, the enrichment of PRPS1 was limited to the soluble fraction (fig. S1B and table S3).
In addition, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis revealed several enriched pathways associated with the UBR7 interactome (Fig. 1D and table S4). Of these enriched pathways, purine and pyrimidine metabolism, which involve PRPS enzymes, were highly significant (Fig. 1D). Given the essential role of these enzymes in nucleotide metabolism and that PRPS1 and PRPS2 play key roles in tumor progression of various cancer types (23–25, 28, 30), we focused our further analysis on PRPS1 and PRPS2 to investigate the functional consequence of UBR7-PRPS interaction.
UBR7 interacts with PRPS1 and PRPS2
The interaction of UBR7 with PRPS proteins was validated using coimmunoprecipitation (Co-IP) in HEK293T cells using antibodies specific to PRPS1 and PRPS2. Co-IP assays demonstrated that exogenously expressed UBR7-FLAG strongly associates with endogenous PRPS1 and PRPS2 (Fig. 2, A and B). Likewise, ectopically expressed FLAG-PRPS1 or PRPS2 coimmunoprecipitated endogenous UBR7 (Fig. 2, C and D). Furthermore, the interaction of endogenous UBR7 with endogenous PRPS1 and PRPS2 was also observed (Fig. 2, E and F).
Fig. 2. UBR7 interacts with PRPS1 and PRPS2.
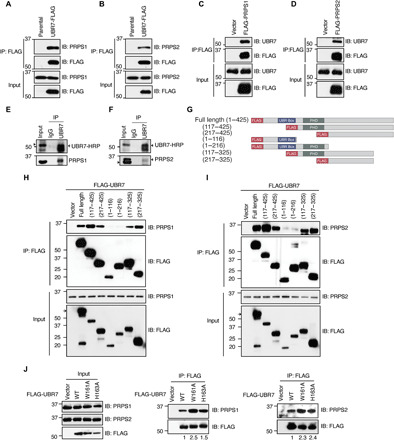
(A and B) Co-IP from the whole-cell extracts of HEK293T parental or stably expressing UBR7-FLAG showing the interaction of UBR7-FLAG with endogenous PRPS1 (A) or PRPS2 (B). (C and D) FLAG-PRPS1 (C) or FLAG-PRPS2 (D) interaction with endogenous UBR7 in HEK293T cells transfected with either empty vector, FLAG-PRPS1, or FLAG-PRPS2. (E and F) Co-IP showing the interaction of endogenous UBR7 with PRPS1 (E) or PRPS2 (F) in HEK293T cells. (G) Schematic representing N-terminally FLAG-tagged UBR7 full length and various truncation mutants. (H to J) A Co-IP assay from the whole-cell extracts of HEK293T cells transiently transfected with FLAG-UBR7 wild-type, various truncation (H and I), or point mutants (J), with endogenous PRPS1 and PRPS2. The numbers below the IP blots in (J) represent the band intensities of PRPS relative to FLAG-UBR7.
Truncation mutants were generated to determine the UBR7 domain required for the interaction with PRPS enzymes. Mutants were designed to eliminate recognized domains and the C terminus (Fig. 2G). Anti-FLAG IP from the lysates of cells transiently transfected with FLAG-tagged UBR7 full length or mutants followed by immunoblotting with PRPS1 or PRPS2 antibodies showed that N-terminal deletion mutants (117–425) and (217–425) retain the ability to interact with both PRPS1 and PRPS2 (Fig. 2, H and I). These data imply that the UBR box and PHD are dispensable for PRPS interaction. No interaction with UBR7 mutants (1–116) and (1–216) was observed, highlighting the C terminus (217–425) as the interaction site with PRPS1 and PRPS2 (Fig. 2, H and I). Furthermore, to identify a more refined region of UBR7 required for PRPS interaction, the last hundred amino acids from the C terminus were deleted. The mutants (117–325) and (217–325) were able to interact with both PRPS1 and PRPS2 (Fig. 2, H and I). This pattern of interaction reveals a confined region in UBR7, spanning from (217–325) required for PRPS interaction.
PHDs are known to be involved in mediating the association with chromatin (35). Given that UBR7 is also bound to chromatin though its PHD (12, 13), one possibility is that the PHD mediates the UBR7 chromatin association in such a manner that PRPS binding sites are no longer accessible. In agreement with this idea, UBR7 PHD point mutants, W161A and H163A, known to be deficient in chromatin binding (12, 13), showed enhanced ability to interact with both PRPS1 and PRPS2. (Fig. 2J). In addition, UBR7 pulled down PRPS exclusively in soluble nonchromatin fraction (fig. S1C). Together, the domain analysis data suggest a separate chromatin- and nonchromatin-associated roles of UBR7 that are regulated by distinct domains.
UBR7 stabilizes PRPS enzymes by promoting the degradation of PRPSAP
To determine the influence of UBR7 on PRPS1 and PRPS2 function, we asked whether the loss of UBR7 has an effect on the levels of these enzymes. Immunoblot analyses showed that knockdown of UBR7 by a constitutive shRNA leads to a decrease in both PRPS1 and PRPS2 (Fig. 3, A and B). The reduction in PRPS1 and PRPS2 protein levels was not due to alterations in transcription, as UBR7 knockdown did not cause a decrease in PRPS1 and PRPS2 transcripts (Fig. 3C). A moderate (~1.5-fold) yet statistically significant increase was seen in PRPS1 transcript after UBR7 knockdown. PRPS2 mRNA, on the other hand, remained unaltered (Fig. 3C). The reduction in PRPS1 and PRPS2 protein levels was also seen when UBR7 was depleted using a doxycycline-inducible shRNA (Fig. 3, D and E) with unaltered PRPS1/2 transcript levels (Fig. 3F). Last, a CRISPR-Cas9–mediated UBR7 ablation also led to a notable decrease in PRPS1 and PRPS2 protein levels compared to the parental counterparts without changes in the levels of DNA replication licensing factor protein MCM3, a protein that was not identified in UBR7 interactome screen and used as a negative control in immunoblot experiment (fig. S2, A to C). Consistent with our results using shRNA, CRISPR-Cas9–mediated UBR7 knockout also did not cause any significant change in the transcripts of PRPS1 and PRPS2 (fig. S2, D and E). Together, these data suggest that UBR7 regulates the stability of PRPS enzymes through posttranscriptional mechanisms.
Fig. 3. UBR7 stabilizes PRPS enzymes through the degradation of PRPSAP.
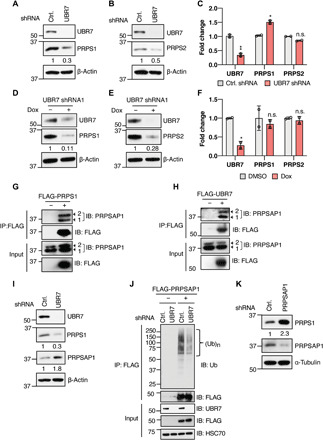
(A and B) Immunoblot showing the PRPS1 (A) and PRPS2 (B) levels in HEK293T cells transduced with control (Ctrl.) or UBR7 shRNA. (C) UBR7, PRPS1, and PRPS2 mRNA fold change in UBR7 knockdown cells normalized to control as measured from qPCR. (D and E) PRPS1 and PRPS2 levels in doxycycline-inducible UBR7 shRNA stable HEK293T cells treated with doxycycline (1 μg/ml) or DMSO for 48 hours. (F) UBR7, PRPS1, and PRPS2 mRNA fold change in doxycycline-inducible UBR7 knockdown cells normalized to control as measured from qPCR. (G and H) Co-IP demonstrating the interaction of FLAG-tagged PRPS1 (G) or UBR7 (H) with endogenous PRPSAP1 in HEK293T cells 1 and 2, represented by arrows, indicate two isoforms of PRPSAP1. (I) PRPS1 and PRPSAP1 levels in control or UBR7 shRNA HEK293T cells. (J) Ubiquitination of transiently transfected vector or FLAG-PRPSAP1 in control or UBR7 shRNA–transduced HEK293T cells treated with 10 μM MG132 for 6 hours before anti-FLAG IP. (K) PRPS1 levels in control or PRPSAP1 shRNA transduced HEK293T cells. Data points in (C) and (F) represent independent biological replicates. Each biological replicate is the mean of at least three technical replicates. Error bars are means ± SD, and P values were computed from Student’s t test and shown with respect to control shRNA (n.s.P > 0.05, *P ≤ 0.05, **P ≤ 0.01). Numbers below the immunoblots represent the relative band intensities.
Given that UBR7 is a putative E3 ubiquitin recognin/ligase, the reduction in PRPS enzymes upon UBR7 loss is counterintuitive. To understand the underlying mechanism of PRPS regulation by UBR7, we hypothesized that UBR7 may negatively regulate an inhibitor of PRPS and thus stabilizes PRPS enzymes. In line with this idea, affinity purification and MS of UBR7-FLAG also showed the enrichment of PRPSAP1 and PRPSAP2, albeit not as abundantly as PRPS1 and PRPS2 (table S1). The PRPSAP proteins have been described as negative regulators of PRPS enzymes via unknown mechanisms (21, 22), and it is possible that UBR7 stabilizes PRPS through the degradation of PRPSAPs. To test that the potential involvement of PRPSAP in UBR7 mediated the PRPS stabilization, first, the interaction of PRPSAP1 with PRPS1 and UBR7 was examined. Co-IP assays revealed a strong interaction of FLAG-tagged PRPS1 or UBR7 with endogenous PRPSAP1 (Fig. 3, G and H). Next, PRPSAP1 levels were tested in response to UBR7 depletion. We observed an accumulation of PRPSAP1 upon shRNA-mediated knockdown of UBR7 (Fig. 3I). The accumulation of PRPSAP1 was concomitant with decrease in PRPS1 levels (Fig. 3I). The increase in PRPSAP1 was also seen when UBR7 was depleted through doxycycline-inducible shRNA (fig. S2F). If UBR7 is involved in proteasomal degradation of PRPSAP1, the increase in PRPSAP1 upon UBR7 knockdown should be reflected in reduced polyubiquitination. Consistent with this rationale, the FLAG-tagged PRPSAP1 polyubiquitination was profoundly decreased when UBR7 was depleted (Fig. 3J). Collectively, these data suggest that UBR7 promotes the proteasomal degradation of PRPSAP1.
PRPSAP1 has been reported as a negative regulator of PRPS enzymes (21, 22); however, it remains unknown whether alteration in PRPSAP1 affects PRPS protein levels. Our results demonstrating the PRPSAP1 degradation and PRPS1 stabilization by UBR7 together with previously reported role of PRPSAP1 as an inhibitor of catalytic activity of PRPS (21, 22) suggest a correlation between PRPS enzymatic activity and protein levels. In substantiation to this idea, we observed enhanced PRPS1 levels in response to PRPSAP1 knockdown (Fig. 3K). The abundance of the other regulatory subunit of PRPS enzyme complex, PRPSAP2, is extremely scarce, and the negative regulation of PRPS enzymes is mainly ascribed to PRPSAP1 (20, 21). Together, these data provide a mechanistic evidence that UBR7 stabilizes PRPS enzymes by promoting the degradation of their negative regulator PRPSAP.
Given that both PRPS1 and PRPS2 are key enzymes for the maintenance of nucleotide synthesis, reduction in PRPS1 and PRPS2 levels caused by the loss of UBR7 should also result in decreased cellular nucleotide levels. To test the effects of UBR7 loss on nucleotide pools, HEK293T parental or UBR7 knockout cell lines were subjected to steady-state metabolite profiling through LC-MS. Consistent with the levels of PRPS1 and PRPS2, both purine [adenosine 5′-monophosphate (AMP) and guanosine 5′-monophosphate (GMP)] and pyrimidine [cytidine 5′-monophosphate (CMP) and uridine 5′-monophosphate (UMP)] nucleotide levels were reduced in UBR7-null cells as compared to parental counterpart (fig. S2G). The reduction in pyrimidine nucleotides was consistent with the accumulation of metabolites upstream to PRPS, including glutamine, N-carbamoyl-l-aspartate, and orotic acid (fig. S2G). Some intermediates involved in purine synthesis pathway such as 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) and inosine 5′-monophosphate (IMP) were increased in UBR7 knockout cells compared to parental (fig. S2G). It is important to note that this analysis was performed at the steady-state levels, which does not provide any information on the direct activity of the purine synthesis pathway. A possible explanation for the increase in AICAR and IMP levels could stem from the activation of the PRPS or phosphoribosylpyrophosphate amidotransferase (PPAT) that is involved in the purine synthesis pathway downstream to PRPS, through the loss of feedback inhibition induced by the end products of the de novo purine synthesis pathway. In support to this idea, adenosine 5′-diphosphate (ADP) and guanosine diphosphate (GDP) levels were decreased in the absence of UBR7 (fig. S2G), which have been described to inhibit PRPS or PPAT activity via direct binding mechanisms (26–28, 36–38).
In addition to nucleotide synthesis, PRPS is also involved in cofactor nicotinamide adenine dinucleotide (NAD), tryptophan, and histidine metabolism (15). Consistent with the reduced levels of PRPS enzymes upon UBR7 loss, a decrease in NAD, tryptophan, and histidine syntheses was also observed (fig. S2G). Overall, the metabolic profiling in HEK293T cells indicates a potential role of UBR7 in various metabolic processes through the regulation of PRPS enzymes.
UBR7 is highly expressed in T-ALL and is regulated by NOTCH1
Nucleotide synthesis is vital for cell proliferation, and cancer cells often rely on increased nucleotide synthesis for their growth (32, 39). On the basis of our results that UBR7 interacts with PRPS1 and PRPS2 and regulates nucleotide synthesis, we posited that UBR7 might be crucial for cancer cell proliferation. To understand the possible role of UBR7 in cancer, we compared the expression of UBR7 transcripts across the cancer cell line encyclopedia (CCLE) sample set (40). Among the several cancer types represented in the CCLE, UBR7 was most abundant in T-ALL compared to other cancer types (Fig. 4A). Gain-of-function mutations affecting the oncogenic transcription factor NOTCH1 play a critical role in T-ALL pathogenesis (4, 5). Given the high transcript levels of UBR7 in T-ALL, we hypothesized that UBR7 might be a transcriptional target of NOTCH1. To test this, a NOTCH1 ChIP-seq dataset from a NOTCH1 mutant T-ALL cell line, CUTLL1, was analyzed (41). NOTCH1 was found to be recruited to the UBR7 promoter, and this recruitment was decreased by 30% when NOTCH1 was inhibited pharmacologically using a gamma secretase inhibitor (GSI), compound E (Fig. 4B). We observed NOTCH1 binding to the UBR7 promoter through ChIP followed by quantitative PCR as well, which was comparable to that of HES1 binding, a known NOTCH1 target (42) and significantly higher than a distal site at UBR7 locus (fig. S3A). To assess whether high expression of UBR7 in T-ALL cell lines can also be observed in T-ALL patients, we analyzed UBR7 and PRPS1/2 expression in samples from the pediatric cancer (PeCan) genome project portal. While a significant up-regulation in UBR7 and PRPS2 was observed in NOTCH1-mutated T-ALL as compared to NOTCH1 wild-type T-ALL, no significant change was seen in PRPS1 transcripts in the same dataset (fig. S3B). The up-regulation in PRPS2 transcripts may or may not be dependent on UBR7, the latter being more likely as our findings suggest a posttranscriptional regulation of PRPS1/2 by UBR7. In addition, the analyses of gene expression profiling of T-ALL patients and mouse models (42, 43) revealed an up-regulation of UBR7 mRNA in NOTCH1 overexpressed T-ALL as compared to normal thymocyte counterparts (fig. S3, C and D).
Fig. 4. UBR7 is highly expressed in T-ALL and is regulated by NOTCH1.
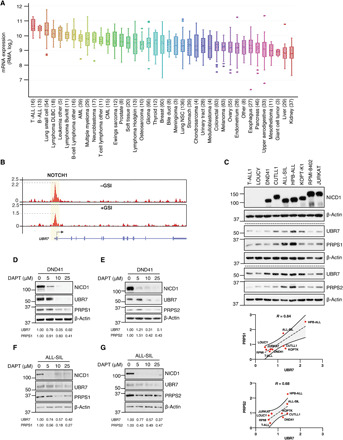
(A) UBR7 mRNA expression from CCLE dataset. (B) Tracks showing NOTCH1 ChIP-seq signal enrichment at the UBR7 locus in CUTLL1 cells treated with GSI (+) or GSI washed off (−). These data are accessible through GSE51800 (41). (C) Immunoblot showing UBR7, PRPS1, and PRPS2 levels in two NICD1-negative cell lines, T-ALL1 and Loucy, and various NICD1-positive cell lines. The band intensities (arbitrary unit) of UBR7, PRPS1, and PRPS2 normalized to β-actin and correlation curve with 95% confidence interval and Pearson correlation value (R) are shown below immunoblot. (D to G) UBR7, PRPS1, and PRPS2 levels in DND41 or ALL-SIL cells treated with increasing doses of GSI, DAPT, for 24 hours. The table below immunoblots shows the relative levels of UBR7, PRPS1, and PRPS2 normalized to β-actin.
Given the UBR7 mRNA overexpression in various NOTCH1-driven T-ALL datasets described above, we next determined whether UBR7 protein level is correlated with activated NOTCH1 in T-ALL. Immunoblotting in a wide range of NICD1-expressing T-ALL cells along with two NICD1-negative cell lines T-ALL1 and Loucy, demonstrated that UBR7 is highly abundant in NICD1-positive cell lines as compared to negative ones (Fig. 4C). The up-regulation in UBR7 proteins levels was highly correlated with both PRPS1 and PRPS2 levels with correlation coefficients of 0.84 and 0.68, respectively (Fig. 4C). Lastly, the inhibition of activating cleavage of NOTCH1 by a GSI, DAPT in two different NICD1-driven T-ALL cell lines, DND41 and ALL-SIL, caused down-regulation of UBR7, PRPS1, and PRPS2 (Fig. 4, D to G). A small yet statistically significant reduction in the transcripts of UBR7, PRPS1, and PRPS2, which was comparable to a decrease in NOTCH1, upon GSI DBZ treatment was also observed through the analysis of gene expression profiling from the mouse models of primary NOTCH1-induced leukemia (fig. S3E) (34). These data suggest that the reduction in protein levels of PRPS enzymes in response to NOTCH1 inhibition could be the combinatorial effect of UBR7 decrease and effect of reduced NOTCH1 activity that does not involve UBR7. Overall, these data establish UBR7 as a transcriptional target of NOTCH1 and indicate the potential role of UBR7 in T-ALL.
UBR7 loss leads to impaired nucleotide metabolism in T-ALL cells
The upstream regulation of UBR7 by NOTCH1 and its downstream effect on PRPS enzymes indicate a connection between NOTCH1 signaling and nucleotide metabolism in NOTCH1-activated T-ALL. As a proof of principle, we first asked whether the interaction of UBR7 with PRPS enzymes holds true in T-ALL cell lines as well. The Co-IP assays demonstrated that endogenous UBR7 pulls down both endogenous PRPS1 and PRPS2 in two T-ALL cell lines, CUTLL1 and Jurkat (Fig. 5, A and B). Moreover, consistent with our observation in HEK293T cells, UBR7 knockdown in CUTLL1 and Jurkat resulted in reduction of PRPS proteins but not mRNA (Fig. 5, C to F).
Fig. 5. UBR7 regulates nucleotide metabolism in T-ALL cells.
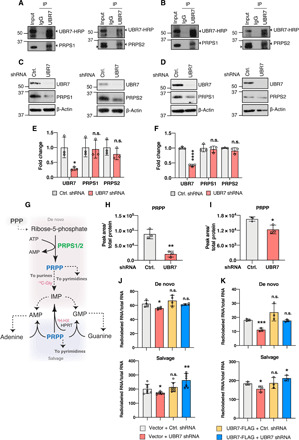
(A and B) Co-IP demonstrating the interaction of endogenous UBR7 with PRPS1 or PRPS2 in CUTLL1 (A) and Jurkat (B) cells. (C and D) Immunoblot showing the PRPS1 and PRPS2 levels in CUTLL1 (C) and Jurkat (D) cells transduced with control or UBR7 shRNA. (E and F) Fold change in UBR7, PRPS1, and PRPS2 mRNA in UBR7 shRNA CUTLL1 (E) or Jurkat (F) normalized to their control counterparts as measured from qPCR. (G) Schematic representing the involvement of PRPP in nucleotide synthesis by de novo and salvage pathway. 14C-glycine and 3H-hypoxanthine are highlighted as they are used in (J) and (K) to show their involvement in de novo and salvage purine synthesis pathway. Dashed lines represent the multiple steps. PPP, pentose phosphate pathway. (H and I) PRPP levels per unit total protein in control or UBR7 shRNA–treated CUTLL1 (H) and Jurkat (I) cells as measured from LC-MS. (J and K) De novo and salvage purine synthesis measured from 14C-glycine or 3H-hypoxanthine incorporation, respectively, in total RNA in CUTLL1 (J) and Jurkat (K) vector or UBR7-FLAG–expressing cells transduced with control or UBR7 shRNA. Data points represent independent biological replicate. Error bars are means ± SD, and P values were computed from Student’s t test and shown only with respect to control or vector + control shRNA as indicated (n.s.P > 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001).
PRPS enzymes catalyze the synthesis of 5′phosphoribosylpyrophosphate (PRPP) from ribose-5-phosphate, which is generated through pentose phosphate pathway (PPP) and used for nucleotide synthesis through two major metabolic routes: the de novo and salvage pathways (Fig. 5G) (15). Therefore, the reduction in PRPS protein levels upon UBR7 loss should be reflected in the cellular levels of PRPP. To test whether UBR7 knockdown affects PRPP levels, LC-MS–based steady-state metabolite profiling was conducted in CUTLL1 and Jurkat cells. In both cell lines, the levels of PRPP were markedly decreased when UBR7 was depleted (Fig. 5, H and I). PRPP is used as a sugar backbone for both purine and pyrimidine nucleotides (39), and the reduced PRPP levels that resulted from UBR7 deficiency were consistent with the decrease in purine and pyrimidine nucleotides (fig. S4, A and B). In addition, a varied degree of changes in the intermediates of PPP was also seen upon UBR7 loss (fig. S4, A and B). For instance, while UBR7 knockdown in CUTLL1 cells caused no notable changes in the levels of 6-phospho-d-gluconate, an intermediate in oxidative branch of the PPP, the same was reduced in UBR7-depleted Jurkat cells (fig. S4, A and B). Among the metabolites involved in the nonoxidative branch of PPP, UBR7-deficient CUTLL1 cells exhibited a decrease in ribose-5-phosphate, sedoheptulose-7-phosphate, and glyceraldehyde-3-phosphate and an increase in erythrose-6-phosphate, glyceraldehyde-3-phosphate, and fructose-6-phosphate (fig. S4A). A similar trend was observed in Jurkat cells, with the exception of erythrose-4-phosphate and fructose-6-phosphate showing no considerable change (fig. S4B). Together, while these observations are correlated with previously described diminished glucose consumption when PRPS1 was depleted (44), it is critical to point out that the changes in the levels of these metabolites upon UBR7 loss represent the steady-state level and cannot be interpreted as rate of their consumption or production.
To further dissect the role of UBR7 in the control of the nucleotide metabolic pathways, metabolic tracing analyses from radiolabeled 14C-glycine and 3H-hypoxanthine were used to specifically measure the activity of the de novo and salvage purine pathways, respectively. Note that the shRNA used here targets the 3′ untranslated region (3′UTR) and does not affect exogenous UBR7, allowing to assess the potential of exogenous UBR7 to reverse the metabolic defects caused by the knockdown of its endogenous counterpart. While cellular uptake rates of glycine and hypoxanthine were not affected by the depletion of UBR7 (fig. S4, C and D), a significant reduction in purine synthesis through both de novo and salvage pathways was observed upon UBR7 knockdown in CUTLL1 and Jurkat cells (Fig. 5, J and K). The decrease in purine synthesis after UBR7 depletion was similar to those caused by PRPS2 depletion (30). Purine synthesis activity was restored to a level comparable to that of the controls when exogenous UBR7 was introduced in endogenously depleted UBR7 background (Fig. 5, J and K), further reinforcing the idea that the decrease in purine synthesis was due to the loss of UBR7 specifically. Not only the reduced purine synthesis was consistent with the down-regulation of PRPS1/2, but also the rescue in purine syntheses coincided with the restoration in PRPS1/2 levels (fig. S4E). Because PRPP is also involved in pyrimidine synthesis, we tested the effect of UBR7 depletion on pyrimide synthesis as well through metabolic tracing using 14C-aspartate and 3H-uridine for de novo and salvage pathways, respectively. While the salvage pathway in CUTLL1 cells remained unaltered in response to UBR7 knockdown, de novo pyrimidine synthesis was significantly reduced (fig. S4F). On the other hand, pyrimidine synthesis through both de novo and salvage pathways was down-regulated in Jurkat cells when UBR7 was knocked down (fig. S4G). Similar to 14C-glycine and 3H-hypoxanthine, the cellular uptakes of 14C-aspartate and 3H-uridine were not affected by UBR7 knockdown (fig. S4, H and I). Overall, these results demonstrate that UBR7 regulates nucleotide metabolism in T-ALL by maintaining the levels of PRPS enzymes.
UBR7 is critical for NOTCH1-driven T-ALL cell proliferation and oncogenic potential
Nucleotide synthesis is essential for cell proliferation, and given that UBR7 supports nucleotide synthesis through the regulation of PRPS enzymes, UBR7 deficiency should negatively affect T-ALL cell proliferation. In agreement with this hypothesis, the knockdown of UBR7 significantly reduced the proliferation of both CUTLL1 and Jurkat cells (Fig. 6, A and B). To further substantiate the finding that reduced cell proliferation is specifically caused by the loss of UBR7 and not resulted from any off-target effect of UBR7 shRNA, the ability of exogenous UBR7 to rescue the proliferation defects was evaluated. In the UBR7 knockdown background, exogenous UBR7 in both the cell lines restored the proliferation rates (Fig. 6, A and B). Similar to the effect of constitutive UBR7 shRNA, a doxycycline-induced UBR7 shRNA also inhibited T-ALL cell proliferation, which was rescued upon reintroduction of UBR7 (fig. S5A). The rescue in proliferation coincided with enhanced PRPS levels (fig. S5B), further substantiating the idea that UBR7 regulates T-ALL cell proliferation through PRPS enzymes.
Fig. 6. UBR7 knockdown attenuates T-ALL cell proliferation and oncogenic potential.
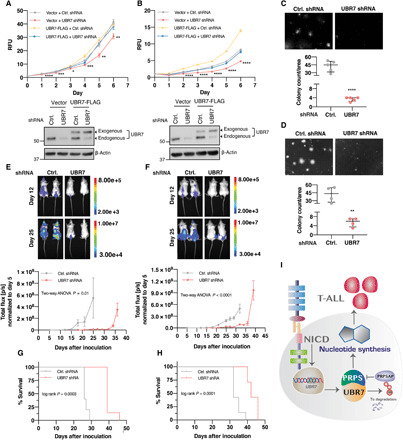
(A and B) Cell proliferation curves of CUTLL1 (A) and Jurkat (B), vector and UBR7-FLAG cells transduced with a constitutive UBR7 shRNA targeting 3′UTR as measured by relative fluorescence unit (RFU) of reduced alamarBlue. Data points represent mean of five technical replicates. Error bars are mean normalized to day 0 ± SD. P values are computed from Student’s t test and shown only with respect to vector + control shRNA (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001). Western blot analysis below the proliferation curve shows the knockdown of endogenous UBR7 and the expression of exogenous UBR7 from the CUTLL1 (A) and Jurkat (B) lysates of day 4. (C and D) Representative images and colony counts from colony formation assay of control or UBR7 shRNA–infected CUTLL1 (C) or Jurkat (D) cells cultured in methylcellulose-based medium for 3 weeks. Data points represent the independent biological replicate. P values are computed from Student’s t test (**P ≤ 0.01, and ****P ≤ 0.0001). (E and F) Representative IVIS images at days 12 and 25 after intravenous transplantation of mice with control or UBR7 shRNA–transduced CUTLL1-luciferase (E) and Jurkat-luciferase cells (F). The plots below represent the tumor progression measured from mean normalized IVIS intensity from all the mice in the group (CUTLL1-luciferase control shRNA, n = 5; CUTLL1-luciferase UBR7 shRNA, n = 7; Jurkat-luciferase control shRNA, n = 7; Jurkat-luciferase UBR7 shRNA, n = 7). (G and H) Mouse survival curves of CUTLL1 (G) or Jurkat (H) control and UBR7 shRNA groups. (I) Model showing the upstream regulation of UBR7 by NOTCH1 signaling and T-ALL promotion through UBR7-PRPS–mediated nucleotide synthesis. PRPSAP1 polyubiquitination–mediated degradation by UBR7 and inhibition of PRPS activity are also depicted.
If the reduced proliferation rate upon UBR7 loss is caused by decreased PRPS and thereby nucleotide biosynthesis, administration of nucleosides should also reinstate the cell proliferation. In line with this hypothesis, the proliferation rates of UBR7-depleted cells were enhanced when supplemented with ribose nucleosides (fig. S5C), again reinforcing the concept that UBR7 promotes T-ALL proliferation through the positive regulation of nucleotide biosynthesis. The role of UBR7 in T-ALL is further highlighted by an additive inhibitory effect of UBR7 silencing in conjunction with NOTCH1 inhibitor DAPT on cell viability (fig. S5D).
On the basis of the inhibitory effects of UBR7 depletion on T-ALL cell proliferation, we determined whether UBR7 also affects T-ALL oncogenic potential. To test the effect of UBR7 on oncogenicity of T-ALL cell lines, control or UBR7 shRNA–expressing CUTLL1 and Jurkat cells were grown in methylcellulose-based three-dimensional culture medium. Consistent with the cell proliferation defects, UBR7 loss resulted in significantly fewer (~10-fold) and smaller colonies as compared to control shRNA in both the cell lines (Fig. 6, C and D).
Given the reduced T-ALL cell proliferation and their colony formation ability caused by UBR7 knockdown, we assessed whether UBR7 loss has similar mitigatory effect on leukemogenesis in mouse xenograft models. CUTLL1 and Jurkat cells expressing a luciferase reporter transduced with control or UBR7 shRNA were injected in immunodeficient mice intravenously, and tumor progression was monitored through in vivo imaging system (IVIS). In concordance with our findings in cultured cells, silencing of UBR7 led to significant reduction in tumor growth in mice derived from both the cell lines as compared to their control counterparts (Fig. 6, E and F). Consequently, mice injected with reduced UBR7 expressing cells showed prolonged survival relative to control. Specifically, the median survival of mice injected with CUTLL1 cells expressing UBR7 shRNA was 39 days compared to 26 days for control injected mice. Likewise, mice injected with Jurkat cells expressing UBR7 shRNA survived for a median of 42 days as compared to 32 days in controls (Fig. 6, G and H). Together, these data demonstrate a vital role of UBR7 in cell proliferation and oncogenic potential of NOTCH1-driven T-ALL.
DISCUSSION
Nucleotide biosynthesis ensures the ample supply of nucleotide pools for nucleic acid synthesis to fulfill the high proliferative demand of cancer cells (32, 33, 39). Therefore, unraveling the novel molecular determinants of nucleotide metabolism will not only provide deeper mechanistic insight into oncogenesis but also offer solutions to therapeutic challenges. In the present study, we demonstrate UBR7 as a critical regulator of nucleotide metabolism in T-ALL. Proteomic and biochemical studies reveal that UBR7 robustly interacts with PRPS enzyme complexes including catalytic subunits PRPS1 and PRPS2 and regulatory subunits PRPSAP1 and PRPS2. PRPS enzymes are key to nucleotide metabolism and catalyze the first and rate-limiting step in the nucleotide biosynthesis pathway (15). We demonstrate that UBR7 promotes the ubiquitination-mediated degradation of PRPSAP1, a negative regulator of PRPS enzymes (21, 22), and the net result of the interaction of UBR7 with the PRPS enzyme complex is to maintain nucleotide biosynthesis. Furthermore, NOTCH1 activation directly up-regulates UBR7 expression to support T-ALL cell proliferation and leukemogenesis by promoting nucleotide biosynthesis (Fig. 6I).
While we demonstrate a perspicuous role of UBR7 in nucleotide biosynthesis, the effect of UBR7 on cellular metabolism could be broader. Affinity purification of UBR7 also identified several other proteins involved in various metabolic processes, including succinate dehydrogenase and lactate dehydrogenase (table S1). The interaction of UBR7 with these enzymes may affect the tricarboxylic acid (TCA) cycle and energy metabolism, as also indicated by the enrichment of TCA cycle and pyruvate metabolism associated with UBR7 interactome (Fig. 1D). Moreover, the steady-state metabolic profiling in HEK293T cells shows that UBR7 loss leads to a reduction in malic acid and fumaric acid (fig. S2G). It is possible that UBR7, owing to its E3 ubiquitin ligase/recognin activity, is involved in negative regulation of succinate dehydrogenase and affects the TCA cycle. Similarly, we observed a decrease in pyruvate and lactate (fig. S2G), which may again be due to the negative regulation of lactate dehydrogenase by UBR7. In addition, UBR7 was found to have an effect on glucose and amino acid metabolism as well (fig. S2G). Clearly, the effect of UBR7 on nucleotide metabolism is the tip of the iceberg, and future in-depth studies are required to fully delineate the role of UBR7 in cellular metabolism.
UBR7 has been found to be a chromatin-associated protein (12–14). In contrast, the role of UBR7 in nucleotide metabolism through PRPS regulation appears to be chromatin independent. This is supported by the observation that the UBR7-PRPS interaction occurs in soluble nonchromatin fraction and that UBR7 PHD finger mutants deficient in chromatin association have enhanced ability to interact with PRPS. UBR7 contains both a ubiquitin recognition box (UBR) and a putative E3 ligase domain, PHD (10). However, the potential involvement of UBR7 in ubiquitin-mediated proteasomal degradation pathway is yet to be determined. The only known ubiquitination substrate of UBR7 is histone H2B, which undergoes monoubiquitination (12). However, H2B monoubiquitination does not affect its stability. Here, we show that UBR7 promotes the polyubiquitination of PRPSAP1, resulting in stabilization of PRPS1. PRPSAP1 has been demonstrated to serve as a negative regulator of PRPS enzymes through classical biochemical approaches (21, 22); however, the underlying mechanisms governing PRPSAP1-mediated suppression of PRPS catalytic activity are not understood completely. Our results demonstrating posttranscriptional regulation of PRPS enzymes by UBR7, together with an accumulation of PRPS1 levels in the absence of PRPSAP1, indicate the regulation of PRPS at the level of translation. It is possible that the PRPS activity and the protein levels are mutually dependent, and the reduced activity of PRPS in the presence of PRPSAP1 may finally lead to impaired PRPS translation. PRPSAP2, which may have a function tantamount to PRPSAP1, also exists (21, 22). However, the abundance of PRPSAP2 is extremely low (1:20 per catalytic subunits) and its regulatory function may not be as prominent as that of PRPSAP1 (20, 21). These ideas warrant future studies to provide more mechanistic insight establishing the role PRPSAPs in the regulation of the PRPS enzyme complex.
PRPS has been reported to undergo arginylation-dependent ubiquitination (45). ATE1-mediated arginylation of proteins is thought to serve as a degron signal that is recognized by UBR box, which subsequently facilitates the ubiquitination-directed proteolysis (46). This concept and the findings that UBR7 does not recognize the canonical arginylated marks (11) are in agreement with our results that UBR7 stabilizes PRPS proteins.
Our work contributes to the emerging appreciation of an important role of UBR7 in cancers. UBR7 has been recently reported as a tumor suppressor in triple-negative breast cancer (12). UBR7 is lowly expressed in this breast cancer subtype, and its tumor-suppressive role is ascribed to its ability to monoubiquitinate histone H2B, which, in turn, inhibits metastasis (12). In contrast, our study establishes UBR7 as a positive regulator of NOTCH1-driven T-ALL. These two disparate results suggest that UBR7 has a context-dependent role in cancers, and future efforts should be directed to further consolidate the role of UBR7 in a wide range of cancers.
The efficacy of GSI therapy against NOTCH1-driven T-ALL is compromised because many patients become resistant to the treatment, presenting significant therapeutic hurdles (47). In the current study, both a GSI-sensitive cell line (CUTLL1) and a GSI-resistant cell line (Jurkat) were used to study the influence of UBR7 on their oncogenic potential. Our data suggest that UBR7 is critical for cell proliferation and clonogenic potential of both GSI-sensitive and GSI-resistant cell lines. With the growing evidence of metabolic pathways as druggable targets for leukemia therapy (48, 49), our data have the potential to provide alternate therapy options for GSI-resistant tumors, although a deeper mechanistic insight is required. Anti-purine drugs also have antiproliferative effect against ALL; however, certain activating mutations in PRPS1 limit the effectiveness of these drugs and lead to relapse of disease (28). The effect of these mutations on PRPS interaction with UBR7 is currently unknown, and whether disrupting UBR7-PRPS can be exploited for therapy is a subject of future study.
MATERIALS AND METHODS
Cell lines, plasmids, and transient transfections
HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS)–optima (Atlanta Biologicals) and 1% penicillin/streptomycin (Gibco, Thermo Fisher Scientific). T-ALL1, Loucy, CUTLL1, Jurkat, DND41, HPB-ALL, KOPT-K1, RPMI-8402, and ALL-SIL were cultured in 10% heat-inactivated FBS-optima, 1% penicillin/streptomycin, and 1× GlutaMAX (Gibco, Thermo Fisher Scientific). CUTLL1 and Jurkat cells expressing luciferase reporter have been described earlier (50). A C-terminal FLAG-tagged UBR7 gateway entry clone was generated by PCR amplification of human UBR7 using FLAG sequence overhang in the primer followed by BP Gateway recombination reaction with pDONR221 (Invitrogen, 12536017) vector. An expression clone was further generated in pLenti CMV Puro DEST w(118-1) (Addgene, 17452) through LR Gateway reaction. N-terminal FLAG-tagged UBR7 full-length and truncation mutants were cloned into pcDNA5/FRT/TO FLAG vector (Addgene, V652020) between Bam HI and Xho I sites. UBR7 W161A and H163A mutants were generated from site-directed mutagenesis as entry clones through BP recombination reaction with pDONR221 vector (Invitrogen, 12536017) and further subcloned in pcDNA5/FRT/TO FLAG vector (Addgene, V652020) between Bam HI and Xho I sites. FLAG-PRPS1(HG17214-NF) and PRPS2(HG15128-NF) were purchased from Sino Biological. PRPSAP1 (isoform 1) entry clone (HsCD00288768) was purchased from DNASU, and FLAG-PRPSAP1 expression clone was generated through LR reaction with pLenti6.2-3xFLAG-V5 (Addgene, 87072). All the plasmids were verified by sequencing. For transient transfection, 3 μg of DNA was transfected in 6-cm plate using Lipofectamine 3000 reagent (Thermo Fisher Scientific, L3000008) following the manufacturer’s protocol.
Lentivirus production and transduction
For lentivirus production, desired transfer plasmid (12 μg) was cotransfected with 3 μg of VSV-G coat protein vector, pMD2.G (Addgene, 12259), and 6 μg of psPAX2 (Addgene, 12260) in HEK293T cells using Lipofectamine 3000 reagent in 10-cm plate. After overnight transfection, fresh medium was replenished, and 24 hours later, the first batch of viral supernatant was collected and stored in 4°C. Cells were replenished with fresh medium again, and the final batch of lentivirus supernatant was collected 24 hours later. Both the viral batches were pooled and filtered through 0.45-μm surfactant-free cellulose acetate membrane filter. As needed, lentiviral particles were either stored in −80°C or used immediately for transduction. For transducing adherent cells, 1 × 105 cells were seeded a day before transduction. Cells were incubated with lentiviral particles with polybrene (6 μg/ml) for 6 hours. Cells were then replenished with fresh medium and grown for 24 hours followed by appropriate antibiotic selection. To transduce suspension cells, 8 × 105 cells per well of a 12-well plate were spinoculated with lentiviral particles with polybrene (6 μg/ml) at 800g for 1 hour at 32°C. Lentiviral supernatants were discarded as per Northwestern University Biological Safety manual, and cells were replenished with fresh medium. Remaining steps were the same as those of adherent cells.
shRNA, gRNA, and genome editing by CRISPR-Cas9
Doxycycline-inducible UBR7 shRNA1 targeting coding region was purchased from Horizon Discovery (clone ID V2THS_203155-CDS). UBR7 constitutive shRNA targeting 3′UTR (clone ID TRCN0000307300) and PRPSAP1 shRNA (clone ID TRCN0000315730) were purchased from MilliporeSigma. Doxycycline-inducible UBR7 shRNA2 targeting 3′UTR was cloned in pTRIPZ vector between Eco RI and Xho I sites using the 21-mer sense strand 5′-ggaagtcctttcagttgatat-3′ and antisense strand 5′-atatcaactgaaaggacttcc-3′. UBR7 KO1 and PRPS2 clones were generated in 293T cells using CRISPR-Cas9, as described previously (51). Briefly, two independent guides were designed to target exon 1 of human UBR7 and cloned into dual Cas9–enhanced green fluorescent protein (EGFP)/single-guide RNA (sgRNA)–encoding vector PX458 (Addgene, 48138). Single Cas9-EGFP 293T cells were fluorescence-activated cell sorting (FACS)–sorted into individual wells of 96-well plate 24 hours after transfection with PX458 containing sgRNA targeting UBR7. Successful single-cell sorting was confirmed using a light microscope. Single-cell clones were expanded and screened for UBR7 protein expression by Western blot using parental 293T cells as a positive control. Genomic edits for the clonal lines were determined by cloning and Sanger sequencing of PCR products spanning the edited region of UBR7. For HEK293T KO3 cell line, the sgRNA targeting the N terminus of UBR7 was cloned into pX330-U6-Chimeric_BB-CBh-hSpCas9 plasmid from F. Zang laboratory (Addgene, 42230) to make a double-stranded break. A linear repair PCR product was generated by amplifying plasmid with a puromycin-resistant gene followed by poly-A sites used for knocking out UBR7 gene. The KO3 cell line was used as pooled population.
Antibodies, IP, and Western blot
Anti-UBR7 antibodies (Cocalico) raised against the full-length human UBR7 recombinant protein were affinity-purified by coupling 5 mg of HST-NusA-UBR7 antigen to HiTrap-NHS column (GE Healthcare, 17071601) in Coupling Buffer [0.2 M NaHCO3, 0.5 M NaCl (pH 8.3)]. The activity and specificity of the antibody were determined by immunoblot of the lysate from parental and UBR7 KO 293T cells. PRPS1 (Proteintech, 15549-1), PRPS2 (Proteintech, 27024-1), α-tubulin (Proteintech, 66031-1), PRPS1/2 (Santa Cruz Biotechnology, 100822), PRPSAP1 (Santa Cruz Biotechnology, 398422), HSC70 (Santa Cruz Biotechnology, 7298), cleaved NOTCH1 (Cell Signaling Technology, 4147), ubiquitin (Cell Signaling Technology, 3933S), β-actin (Sigma-Aldrich, A2228), FLAG (Sigma-Aldrich, F3165), HSC90 (Enzo Life Sciences, ADI-SPA-846), and NPM1 (Sigma-Aldrich, B0556) were purchased commercially. Co-IPs were carried out in NETN lysis buffer containing 20 mM tris-HCl (pH 7.5), 150 mM NaCl, 0.5 mM EDTA, 0.5% NP-40, and protease inhibitor cocktail (Roche, 11697498001). For IP of FLAG-tagged proteins, whole-cell extracts were subjected to anti-FLAG M2 magnetic beads (Sigma-Aldrich, M8823) overnight at 4°C. Beads were then washed five times with lysis buffer. Unless otherwise indicated, the immunoprecipitated FLAG-tagged proteins were eluted with 3× FLAG peptide (0.25 μg/μl; Sigma-Aldrich, 4799) into lysis buffer. The elutes were subjected to SDS–polyacrylamide gel electrophoresis (PAGE) and analyzed by immunoblotting as indicated. For ubiquitination assay, cells were treated with 10 μM MG132 for 6 hours and lysed in buffer containing 1% SDS, 150 mM NaCl, 10 mM tris-HCl (pH 8), 2 mM Na2VO3, 50 mM NaF, protease inhibitor cocktail, and deubiquitinase inhibitor 5 mM N-ethylmaleimide (Sigma-Aldrich, E3876). The lysates were vortexed vigorously and heated for 10 min at 95°C and diluted by 10 times in buffer containing 10 mM tris-HCl (pH 8), 150 mM NaCl, 2 mM EDTA, 1% Triton X-100 (now the final SDS concentration is 0.1%), 2 mM Na2VO3, 50 mM NaF, protease inhibitor cocktail, and 5 mM N-ethylmaleimide. Lysates were then incubated with rotation at 4°C for 45 min and spun down at maximum speed for 15 min. The supernatants were transferred to fresh tubes and subjected for IP as described above. For co-IP of endogenous UBR7 with endogenous PRPS1 and PRPS2, whole-cell extracts were incubated with 0.6 μg of anti-UBR7 or normal rabbit immunoglobulin G (IgG) (Cell Signaling Technology, 2729) overnight followed by incubation with protein A + G magnetic beads (Thermo Fisher Scientific, 88802). Beads were washed five times in lysis buffer, and proteins bound to bead were eluted in 2× Laemmli sample buffer before analysis through SDS-PAGE. UBR7 antibody used in Western blot involving endogenous IP of UBR7 was labeled with horseradish peroxidase (HRP) using a Lightning-Link HRP antibody labeling kit (Novus Biologicals, 701-0030) following the manufacturer’s protocol. For immunoblots not involving IP, whole-cell lysates were prepared in radioimmunoprecipitation assay buffer (Thermo Fisher Scientific, 89900). Total protein content was quantified using bicinchoninic acid reagent (Thermo Fisher Scientific, 23225), and equal amount of cell lysate was subjected to SDS-PAGE and analyzed by desired antibodies. Western blots were quantified by densitometry using ImageJ program. Each immunoblot experiment was repeated at least twice, and one representative is shown in the article.
Biochemical fractionation
HEK293T cell pellets were resuspended in buffer A [10 mM Hepes (pH 7.9), 1.5 mM MgCl2, 10 mM KCl, 0.34 M sucrose, 10% glycerol, 1 mM dithiothreitol (DTT), and protease inhibitor cocktail]. After incubation on ice for 10 min, NP-40 was added to a final concentration of 0.3%, and cell suspensions were vortexed. Following this, lysates were centrifuged at maximum speed (16,000g) for 10 min at 4°C and supernatants were collected as soluble fraction. The remaining pellet was then lysed in 500 μl of lysis buffer B [50 mM tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 0.5% Triton X-100, 1 mM DTT, and protease inhibitor cocktail] and rotated for 30 min in cold room. Lysates were centrifuged at maximum speed (16,000g) for 15 min at 4°C, and the supernatants were collected as nuclear extract.
MS and data analysis
Equal amount of whole-cell lysates or nuclear and soluble fractions from parental or UBR7-FLAG–expressing cells was subjected to IP and eluted with 3xFLAG peptide as described above. The elutes were then TCA (20%)/acetone (100%) precipitated. The precipitates were then processed for MS as described previously (52). To generate a refined list of UBR7-FLAG interacting partners, spec count = 0 was changed to 1 in parental. A cutoff spec count of ≥3 (for experiment 1) or >2 (for experiment 2) for UBR7-FLAG was set, and proteins that did not satisfy this criterion were removed. Following this filtration step, UBR7-FLAG/parental spec count fold change was calculated. Last, only the accession number with highest spec count fold change was included in the list if a protein was associated with multiple accession numbers. Enriched KEGG pathways were identified using EnrichR (53). Bubble charts representing enrichment analysis were generated using the pathfindR package in R.
Steady-state metabolite profiling
To determine the steady-state levels of intracellular metabolites, cellular extracts from at least three independent biological replicates were prepared with 80% methanol and analyzed by LC-MS/MS. The peak area was normalized to protein concentration. In brief, culture medium was removed completely, and 4 ml of 80% methanol was added to the cells on dry ice followed by at least 1 hour of incubation in −80°C, as described previously (54). The cell suspension in 80% methanol was transferred to 15-ml tubes and centrifuged at 4000 rpm, 5 min, 4°C. The supernatant was carefully transferred to a fresh 50-ml tube on dry ice. To the remaining pellet, 0.5 ml of 80% methanol was added and transferred to 1.5-ml tubes followed by centrifugation at 15,000 rpm, 5 min, 4°C. This step was repeated once, and supernatants were pooled and dried with nitrogen gas using N-EVAP (Organomation Inc., Associates).
Incorporation of radiolabeled substrates to measure the activity of nucleotide synthesis
Actively proliferating CUTLL1 or Jurkat cells were switched to 10% dialyzed FBS-containing medium and incubated for 1 hour. Cells were then labeled with 2 μCi of the specific activity of either 14C-glycine (for de novo purine pathway), 3H-hypoxanthine (for salvage purine pathway), 14C-aspartate (for de novo pyrimidine pathway), or 3H-uridine (for pyrimidine salvage pathway) for 6 hours. RNA was isolated using an RNA extraction kit (Qiagen, 74106). For each sample, out of 100 μl of eluted RNA, 70 μl was mixed with 3 ml of scintillation fluid and counts per minute (CPM) was taken in a Beckman LS6500 scintillation counter. CPM was normalized with the total RNA from each sample.
To measure the uptake of glycine, hypoxanthine, aspartate, and uridine, the cells were labeled with 1 μCi of the specific activity of 14C-glycine, 3H-hypoxanthine, 14C-aspartate, or 3H-uridine for 5 min. Plates were then kept on ice to stop the uptake followed by two washes with chilled phosphate-buffered saline (PBS) and lysed in 0.5 ml of protein lysis buffer. 400 μl lysate was mixed with 3 ml of scintillation fluid, and the count was taken in a Beckman LS6500 scintillation counter. CPM was normalized with respective protein concentrations. All the conditions were analyzed with at least three independent biological replicates.
Analysis of publicly available datasets
UBR7 mRNA expression data in various cancer cell lines are available at Broad Institute Cancer Cell Line Encyclopedia. RNA sequencing (RNA-seq) data of 254 pediatric T-ALL patients from St. Jude PeCan data portal were used for the analysis. Similarly, NOTCH1, UBR7, PRPS1, and PRPS2 expressions represented in the article were evaluated by analyzing the gene expression profiling published earlier (34, 42, 43). Briefly, the samples were divided by their NOTCH1 mutation or expression status and plotted for gene expression levels. Welch’s t test was applied to calculate the statistic values between the NOTCH1 wild-type and mutated groups.
Real-time quantitative PCR, RNA-seq library preparation, and differential gene expression analysis through RNA-seq
Total RNA was isolated using an RNA extraction kit (Qiagen, 74106), and complementary DNA (cDNA) was prepared form 1 μg of RNA using an iScript cDNA synthesis kit (Bio-Rad, 1708890). cDNA was diluted 10 times, mixed with iTaq universal SYBR green supermix (Bio-Rad, 1725121), and subjected to real-time quantitative PCR using standard procedure. Gene expression analysis was performed using 2−ΔΔCT method, and for ChIP, percent input method was used for enrichment measurement to evaluate NOTCH1 binding at UBR7 or HES1 locus. The primers used are listed in Table 1.
Table 1. List of primers used in the study.
| qPCR primers for gene expression | |
| UBR7 | 5′-GAACAGGGAAAGGATGATGTCCG-3′ (forward) |
| 5′-AGCTCCTGAAGTTTGCAGCCAG-3′ (reverse) | |
| PRPS1 | 5′-GGCTGACACTTGTGGCACAATC-3′ (forward) |
| 5′-GATGCGAGAAATAGCAGGACCG-3′ (reverse) | |
| PRPS2 | 5′-GGTCACGAAGAAGTTCAGCAACC-3′ (forward) |
| 5′-GAGGAGTTCCATCAGGTTGTCG-3′ (reverse) | |
| GAPDH | 5′-TTCAACAGCGACACCCACTC-3′ (forward) |
| 5′-TGACAAAGTGGTCGTTGAGGG-3′ (reverse) | |
| qPCR primers for ChIP | |
| HES1 | 5′-AAGTTTCACACGAGCCGTTC-3′ (forward) |
| 5′-GCTGTTATCAGCACCAGCTC-3′ (reverse) | |
| UBR7 | 5′- AGCTTCCAGAACACGACACC-3′ (forward) |
| 5′-TCCGACAAACGGATGTCACT-3′ (reverse) | |
| UBR7 distal site | 5′-GGCTGACACTTGTGGCACAATC-3′ (forward) |
| 5′-GATGCGAGAAATAGCAGGACCG-3′ (reverse) | |
To analyze the PRPS1 and PRPS2 transcripts through RNA-seq, RNA was isolated from parental and UBR7 KO1 293T cells using the RNeasy Plus Mini Kit (Qiagen, 74134) according to the manufacturer’s protocol. RNA quality and concentration were validated on a bioanalyzer with the Agilent RNA 6000 Nano Kit (Agilent, 5067-1511). Total RNA (1 μg) was used as input for RNA-seq library preparation according to the Low Sample (LS) protocol for the Illumina TruSeq Stranded Total RNA Preparation Kit with Ribo-Depletion (Illumina, RS-122-2201). RNA-seq libraries were single-end sequenced with the NextSeq 550 Sequencing System. Raw BCL output files were processed using bclfastq (Illumina, version 2.17.1.14) and quality-trimmed with trimmomatic (55). Trimmed reads were aligned to the human genome (UCSC hg19) with TopHat (56), with gene annotations sourced from Ensembl release 72 assigned using Python package HTSeq 0.6.1 (57). Differential gene expression was determined using EdgeR (58).
Chromatin IP
1.5 × 108 CUTLL1 cells were cross-linked in fixation buffer (1% formaldehyde, 1× PBS, and 1% FBS in H2O) for 10 min at room temperature followed by quenching by 200 mM glycine for 5 min. 1 ml fixation buffer was used per million cells. In parallel, Magna ChIP protein A + G magnetic beads (MilliporeSigma, 16-663) were washed for preclearing with 1× PBS. Per 1.5 × 108 cells, 100 μl of beads was used. Washed beads were hybridized with a cocktail of 30 μg of anti-NOTCH1 (Santa Cruz Biotechnology, 376403) and 30 μg of anti–cleaved NOTCH1 (Cell Signaling Technology, 4147) or equivalent amount of normal mouse or rabbit IgG at 4°C with rotation for 8 hours. Per pellet of 1 × 107, cross-linked cells were resuspended in 1 ml of buffer containing 50 mM Hepes, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100 freshly supplemented with 1× protease inhibitor cocktail (Roche, 11697498001), 1 mM Na3VO5, and 1 mM NaF and incubated at 4°C while rotating for 10 min. Cells were then spun down at 1350g for 10 min at 4°C and resuspended in 1 ml of TES buffer [10 mM tris-Cl (pH 8.0), 1 mM EDTA, 0.1% SDS] supplemented with 1× protease inhibitor cocktail (Roche, 11697498001), 1 mM Na3VO5, and 1 mM NaF per 2.5 × 107 cells. Samples were sonicated in Covaris tube (520530) at 140-W peak incidence power, 10% duty cycle/duty factor, 200 cycles per burst, and 360-s treatment time. Following sonication, samples were spun down at maximum speed at 4°C for 10 min. Supernatant was transferred to new tubes, and 100 μl of 10× buffer containing 100 mM tris (pH 8.0), 1 M NaCl, 10 mM EDTA, 5 mM EGTA, 1% sodium deoxycholate, 5% N-laurylsarcosine, and 110 μl of 10% Triton X-100 was added. Soluble chromatin was precleared for 30 min at 4°C with 60 μl of protein A + G magnetic beads washed with 1× PBS. A proportion of precleared soluble chromatin was separated as input and rest of the sample was rotated overnight at 4°C with antibody hybridized to protein A + G magnetic beads. Supernatant was removed using magnetic stand, and beads were washed in the following order: low-salt buffer [0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM tris-Cl (pH 8.1), 150 mM NaCl]—twice, high-salt buffer [0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM tris-Cl (pH 8.1), 500 mM NaCl]—once, LiCl [10 mM tris-Cl (pH 8.0), 250 mM LiCl, 1% NP-40, 1% sodium deoxycholate, 1 mM EDTA]—once, and TE buffer [10 mM tris-Cl (pH 8.0), 1 mM EDTA]—once. To elute bead-bound complexes, 50 μl of elution buffer (100 mM NaHCO3, 1% SDS) was added to each sample, and samples were incubated at 65°C for 15 min, shaking at 1000 rpm on a thermomixer (Thermo Fisher Scientific). Elution was repeated a second time, and then 100 μl of ribonuclease (RNase) buffer [12 μl of 5 M NaCl, 0.2 μl of RNase (30 mg/ml), and 88 μl of TE] was added to each ChIP and input sample. Samples were incubated at 37°C for 20 min, followed by the addition of PK buffer [2.5 μl of proteinase K (20 mg/ml), 5 μl of 20% SDS, and 92.5 μl of TE] overnight at 65°C. An equal volume of phenol chloroform solution was added to the samples, which were vortexed for 1 min and transferred to MaXtract High Density tubes (Qiagen). Samples were centrifuged for 8 min at maximum speed, and the upper phase was transferred to new tubes and 1.5 μl of glycogen (20 mg/ml) was added. Then, 30 μl of 3 M sodium acetate (pH 5.2) and 800 μl of 100% ethanol were added, and samples were incubated at −80°C overnight. DNA pellets were washed in 70% ethanol, air-dried, and resuspended in 35 μl of nuclease-free water.
Cell proliferation assays
Two days after lentiviral transduction and antibiotic selection, 2 × 103 cells per well were seeded at least five replicates into a Corning 96-Well Clear Bottom Black plate (Corning, #3603). The day of cell plating was considered as day 0. AlamarBlue cell viability reagent (10%) (Thermo Fisher Scientific, #Y00-100) was added to each well followed by 4-hour incubation at 37°C. Cell proliferation was measured in terms of reduction of alamarBlue as measured through fluorescence or absorbance following the manufacturer’s protocol. Medium without cells but with alamarBlue was used as blank. Wherever indicated, cells were supplemented with EmbryoMax Nucleoside (Sigma-Aldrich, ES-008-D) to a final concentration of 1× or various doses of DAPT (Selleckchem, S2215). For experiments where doxycycline-inducible shRNA was used, stable cell lines were seeded on day 0 as described above and treated with 1 μM doxycycline or dimethyl sulfoxide (DMSO). Loss in medium volume due to evaporation was taken into consideration, and 10% fresh medium was supplemented each day. To calculate the proliferation from fluorescence-based readings, the mean fluorescence of the blank replicates was subtracted from the test samples, and each day, reading was normalized to respective day 0 reading to obtain relative fluorescence unit (RFU). To calculate the proliferation from absorbance-based readings, % reduction of alamarBlue Reagent was calculated using the following formula: [(Eoxi600 × A570) − (Eoxi570 × A600)] × 100/[(Ered570 × C600) − (Ered600 × C570)], where Eoxi570 = molar extinction coefficient (E) of oxidized alamarBlue Reagent at 570 nm = 80,586, Eoxi600 = E of oxidized alamarBlue Reagent at 600 nm = 117,216, A570 = absorbance of test wells at 570 nm, A600 = absorbance of test wells at 600 nm, Ered570 = E of reduced alamarBlue at 570 nm = 155,677, Ered600 = E of reduced alamarBlue at 600 nm = 14,652, C570 = absorbance of negative control well (medium, alamarBlue Reagent, no cells) at 570 nm, and C600 = absorbance of negative control well (medium, alamarBlue Reagent, no cells) at 600 nm. The % reduction in alamarBlue was normalized to day 0.
Colony formation assay
CUTLL1 (1 × 103) or Jurkat cells per 100 μl of RPMI medium were mixed with 1 ml of Methocult 4100–based (STEMCELL Technologies, 04100) RPMI medium and plated in 35-mm petri dish using luer-lock syringe (STEMCELL Technologies, 28230) and blunt-end needle (STEMCELL Technologies, 28110). Colonies were allowed to grow for 3 weeks at 37°C and counted using counting grid (STEMCELL Technologies, 27000). Bright-field images were acquired on a Zeiss LSM800 microscope in 20 × 20 tile mode at 10× objective and assembled using stitch function.
Mouse xenograft studies
All mice were housed in a barrier facility, and procedures were performed as approved by the Northwestern University Institutional Animal Care and Use Committee (IS000005877). Female NSG mice (6 to 7 weeks old; JAX 005557, NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) were acclimated for 10 days and randomized into four groups with similar mean average body weights. On day 0, each mouse received an intravenous injection of 1 × 106 cells in 0.15 ml of PBS via the tail vein (for CUTLL1-luciferase control shRNA, n = 5; for all the other groups, n = 7). Starting 5 days after inoculation, mice were imaged once per week for bioluminescent signal; once an IVIS signal was detected, mice were imaged at least twice weekly. On imaging day, approximately 10 min before imaging, mice were injected intraperitoneally with luciferase (2 mg per animal, suspended in 0.1 ml of saline). Mice were then anesthetized with approximately 2 to 2.5% isoflurane in oxygen, which was maintained during the procedure. Images were captured using the IVIS Spectrum In Vivo Imaging System (PerkinElmer) with the following image settings: emission filter open, exposure time 5 s, or automatically determined by software, Binning 8 (medium), FOV 22.6 cm, f-Stop 1. Data collection and analysis were performed using Living Image Software (Perkin Elmer), and total flux vales (photons/second) were determined by manually drawing rectangular regions of interest over identical regions of each mouse (from the nose to the tail base, encompassing all four limbs out to at least the wrist/ankle). Photo credit for the representative images shown in Fig. 6 (E and F): Iwona Stepien, Northwestern University. These representative IVIS images were finally processed using Aura imaging software. Body weights were measured at least twice per week on wake animals. Mice were observed daily, Monday to Friday, for signs of abnormal behavior and distress. Mice were euthanized if they lost 20% of their body weight or if they developed hind limb paralysis and became moribund. Euthanasia was induced by inhalation of CO2 followed by bilateral thoracotomy. Statistical analysis was performed using GraphPad Prism 8 software. For IVIS total flux, groups were compared over time using two-way analysis of variance (ANOVA) followed by Sidak’s multiple comparison tests. For survival, groups were compared using a log-rank (Mantel-Cox) test.
Acknowledgments
We thank N. Birch for help with colony formation assays, S. A. Marshall and A. Shilatifard for their support with RNA-seq library preparation, and E. Bartom for help with RNA-seq analysis via the Ceto pipeline. The mouse xenograft studies were conducted by Developmental Therapeutics Core, Northwestern University. We thank N. Ghoreishi-Haack, E. Dempsey, and I. Stepien for their help in mouse xenograft experiment. Metabolomics services were performed by the Metabolomics Core Facility at Robert H. Lurie Comprehensive Cancer Center of Northwestern University and Beth Israel Deaconess Medical Center Mass Spectrometry Facility of Harvard Medical School. Funding: D.R.F. was supported by Zell Scholarship from the Robert H. Lurie Comprehensive Cancer Center, Northwestern University, and the work was supported by the NICHD (1R21HD078946). Studies in the Ntziachristos laboratory are supported by NCI (CA 248770). Author contributions: S.S., U.S., A.K.H., K.M.S., J.B., J.H., K.A.W., and N.K. conducted the experiments or generated the reagents. S.S., Y.Z., A.K.H., J.N.S., P.N., I.B.-S., and D.R.F. analyzed the data. S.S., J.N.S., P.N., I.B.-S., and D.R.F. designed the experiments. S.S. and D.R.F. wrote the manuscript. P.N., I.B.-S., S.S., and D.R.F. edited the manuscript. The study was supervised by D.R.F. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/5/eabc9781/DC1
REFERENCES AND NOTES
- 1.Terwilliger T., Abdul-Hay M., Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 7, e577 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pui C.-H., Relling M. V., Downing J. R., Acute lymphoblastic leukemia. N. Engl. J. Med. 350, 1535–1548 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Hori K., Sen A., Artavanis-Tsakonas S., Notch signaling at a glance. J. Cell Sci. 126, 2135–2140 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weng A. P., Ferrando A. A., Lee W., Morris J. P. IV, Silverman L. B., Sanchez-Irizarry C., Blacklow S. C., Look A. T., Aster J. C., Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306, 269–271 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Ferrando A. A., The role of NOTCH1 signaling in T-ALL. Hematology Am. Soc. Hematol. Educ. Program 2019, 353–361 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palomero T., Lim W. K., Odom D. T., Sulis M. L., Real P. J., Margolin A., Barnes K. C., O’Neil J., Neuberg D., Weng A. P., Aster J. C., Sigaux F., Soulier J., Look A. T., Young R. A., Califano A., Ferrando A. A., NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. U.S.A. 103, 18261–18266 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weng A. P., Millholland J. M., Yashiro-Ohtani Y., Arcangeli M. L., Lau A., Wai C., Bianco C. D., Rodriguez C. G., Sai H., Tobias J., Li Y., Wolfe M. S., Shachaf C., Felsher D., Blacklow S. C., Pear W. S., Aster J. C., c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 20, 2096–2109 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herranz D., Ambesi-Impiombato A., Palomero T., Schnell S. A., Belver L., Wendorff A. A., Xu L., Castillo-Martin M., Llobet-Navás D., Cordon-Cardo C., Clappier E., Soulier J., Ferrando A. A., A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat. Med. 20, 1130–1137 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kwon Y. T., Reiss Y., Fried V. A., Hershko A., Yoon J. K., Gonda D. K., Sangan P., Copeland N. G., Jenkins N. A., Varshavsky A., The mouse and human genes encoding the recognition component of the N-end rule pathway. Proc. Natl. Acad. Sci. U.S.A. 95, 7898–7903 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tasaki T., Mulder L. C. F., Iwamatsu A., Lee M. J., Davydov I. V., Varshavsky A., Muesing M., Kwon Y. T., A family of mammalian E3 ubiquitin ligases that contain the UBR box motif and recognize N-degrons. Mol. Cell. Biol. 25, 7120–7136 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tasaki T., Zakrzewska A., Dudgeon D. D., Jiang Y., Lazo J. S., Kwon Y. T., The substrate recognition domains of the N-end rule pathway. J. Biol. Chem. 284, 1884–1895 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adhikary S., Chakravarti D., Terranova C., Sengupta I., Maitituoheti M., Dasgupta A., Srivastava D. K., Ma J., Raman A. T., Tarco E., Sahin A. A., Bassett R., Yang F., Tapia C., Roy S., Rai K., Das C., Atypical plant homeodomain of UBR7 functions as an H2BK120Ub ligase and breast tumor suppressor. Nat. Commun. 10, 1398 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kleiner R. E., Hang L. E., Molloy K. R., Chait B. T., Kapoor T. M., A chemical proteomics approach to reveal direct protein-protein interactions in living cells. Cell Chem. Biol. 25, 110–120.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foltz D. R., Jansen L. E. T., Black B. E., Bailey A. O., Yates J. R. III, Cleveland D. W., The human CENP-A centromeric nucleosome-associated complex. Nat. Cell Biol. 8, 458–469 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Hove-Jensen B., Andersen K. R., Kilstrup M., Martinussen J., Switzer R. L., Willemoës M., Phosphoribosyl Diphosphate (PRPP): Biosynthesis, enzymology, utilization, and metabolic significance. Microbiol. Mol. Biol. Rev. 81, e00040-16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lane A. N., Fan T. W.-M., Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 43, 2466–2485 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taira M., Iizasa T., Yamada K., Shimada H., Tatibana M., Tissue-differential expression of two distinct genes for phosphoribosyl pyrophosphate synthetase and existence of the testis-specific transcript. Biochim. Biophys. Acta 1007, 203–208 (1989). [DOI] [PubMed] [Google Scholar]
- 18.Tatibana M., Kita K., Taira M., Ishijima S., Sonoda T., Ishizuka T., Iizasa T., Ahmad I., Mammalian phosphoribosyl-pyrophosphate synthetase. Adv. Enzyme Regul. 35, 229–249 (1995). [DOI] [PubMed] [Google Scholar]
- 19.Becker M. A., Phosphoribosylpyrophosphate synthetase and the regulation of phosphoribosylpyrophosphate production in human cells. Prog. Nucleic Acid Res. Mol. Biol. 69, 115–148 (2001). [DOI] [PubMed] [Google Scholar]
- 20.Kita K., Otsuki T., Ishizuka T., Tatibana M., Rat liver phosphoribosyl pyrophosphate synthetase: Existence of the purified enzyme as heterogeneous aggregates and identification of the catalytic subunit. J. Biochem. 105, 736–741 (1989). [DOI] [PubMed] [Google Scholar]
- 21.Kita K., Ishizuka T., Ishijima S., Sonoda T., Tatibana M., A novel 39-kDa phosphoribosylpyrophosphate synthetase-associated protein of rat liver. Cloning, high sequence similarity to the catalytic subunits, and a negative regulatory role. J. Biol. Chem. 269, 8334–8340 (1994). [PubMed] [Google Scholar]
- 22.Ishijima S., Asai T., Kita K., Sonoda T., Tatibana M., Partial reconstitution of mammalian phosphoribosylpyrophosphate synthetase in Escherichia coli cells. Coexpression of catalytic subunits with the 39-kDa associated protein leads to formation of soluble multimeric complexes of various compositions. Biochim. Biophys. Acta 1342, 28–36 (1997). [DOI] [PubMed] [Google Scholar]
- 23.Qian X., Li X., Tan L., Lee J.-H., Xia Y., Cai Q., Zheng Y., Wang H., Lorenzi P. L., Lu Z., Conversion of PRPS hexamer to monomer by AMPK-mediated phosphorylation inhibits nucleotide synthesis in response to energy stress. Cancer Discov. 8, 94–107 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jing X., Wang X.-j., Zhang T., Zhu W., Fang Y., Wu H., Liu X., Ma D., Ji X., Jiang Y., Liu K., Chen X., Shi Y., Zhang Y., Shi M., Qiu W., Zhao R., Cell-Cycle–Dependent phosphorylation of PRPS1 fuels nucleotide synthesis and promotes tumorigenesis. Cancer Res. 79, 4650–4664 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Li X., Qian X., Peng L.-X., Jiang Y., Hawke D. H., Zheng Y., Xia Y., Lee J.-H., Cote G., Wang H., Wang L., Qian C.-N., Lu Z., A splicing switch from ketohexokinase-C to ketohexokinase-A drives hepatocellular carcinoma formation. Nat. Cell Biol. 18, 561–571 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nosal J. M., Switzer R. L., Becker M. A., Overexpression, purification, and characterization of recombinant human 5-phosphoribosyl-1-pyrophosphate synthetase isozymes I and II. J. Biol. Chem. 268, 10168–10175 (1993). [PubMed] [Google Scholar]
- 27.Li S., Lu Y., Peng B., Ding J., Crystal structure of human phosphoribosylpyrophosphate synthetase 1 reveals a novel allosteric site. Biochem. J. 401, 39–47 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li B., Li H., Bai Y., Kirschner-Schwabe R., Yang J. J., Chen Y., Lu G., Tzoneva G., Ma X., Wu T., Li W., Lu H., Ding L., Liang H., Huang X., Yang M., Jin L., Kang H., Chen S., Du A., Shen S., Ding J., Chen H., Chen J., von Stackelberg A., Gu L., Zhang J., Ferrando A., Tang J., Wang S., Zhou B.-B. S., Negative feedback–defective PRPS1 mutants drive thiopurine resistance in relapsed childhood ALL. Nat. Med. 21, 563–571 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang D., Chen Y., Fang H., Zheng L., Li Y., Yang F., Xu Y., du L., Zhou B.-B. S., Li H., Increase of PRPP enhances chemosensitivity of PRPS1 mutant acute lymphoblastic leukemia cells to 5-fluorouracil. J. Cell. Mol. Med. 22, 6202–6212 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cunningham J. T., Moreno M. V., Lodi A., Ronen S. M., Ruggero D., Protein and nucleotide biosynthesis are coupled by a single rate-limiting enzyme, PRPS2, to drive cancer. Cell 157, 1088–1103 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanahan D., Weinberg R. A., Hallmarks of cancer: The next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Vander Heiden M. G., Lunt S. Y., Dayton T. L., Fiske B. P., Israelsen W. J., Mattaini K. R., Vokes N. I., Stephanopoulos G., Cantley L. C., Metallo C. M., Locasale J. W., Metabolic pathway alterations that support cell proliferation. Cold Spring Harb. Symp. Quant. Biol. 76, 325–334 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Vander Heiden M. G., DeBerardinis R. J., Understanding the intersections between metabolism and cancer biology. Cell 168, 657–669 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herranz D., Ambesi-Impiombato A., Sudderth J., Sánchez-Martín M., Belver L., Tosello V., Xu L., Wendorff A. A., Castillo M., Haydu J. E., Márquez J., Matés J. M., Kung A. L., Rayport S., Cordon-Cardo C., De Berardinis R. J., Ferrando A. A., Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in T cell acute lymphoblastic leukemia. Nat. Med. 21, 1182–1189 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanchez R., Zhou M.-M., The PHD finger: A versatile epigenome reader. Trends Biochem. Sci. 36, 364–372 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou G., Smith J. L., Zalkin H., Binding of purine nucleotides to two regulatory sites results in synergistic feedback inhibition of glutamine 5-phosphoribosylpyrophosphate amidotransferase. J. Biol. Chem. 269, 6784–6789 (1994). [PubMed] [Google Scholar]
- 37.Chen S., Tomchick D. R., Wolle D., Hu P., Smith J. L., Switzer R. L., Zalkin H., Mechanism of the synergistic end-product regulation of Bacillus subtilis glutamine phosphoribosylpyrophosphate amidotransferase by nucleotides. Biochemistry 36, 10718–10726 (1997). [DOI] [PubMed] [Google Scholar]
- 38.Sonoda T., Kita K., Ishijima S., Ishizuka T., Ahmad I., Tatibana M., Kinetic and regulatory properties of rat liver phosphoribosylpyrophosphate synthetase complex are partly distinct from those of isolated recombinant component catalytic subunits. J. Biochem. 122, 635–640 (1997). [DOI] [PubMed] [Google Scholar]
- 39.Villa E., Ali E. S., Sahu U., Ben-Sahra I., Cancer cells tune the signaling pathways to empower de novo synthesis of nucleotides. Cancers (Basel) 11, 688 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A. A., Kim S., Wilson C. J., Lehár J., Kryukov G. V., Sonkin D., Reddy A., Liu M., Murray L., Berger M. F., Monahan J. E., Morais P., Meltzer J., Korejwa A., Jané-Valbuena J., Mapa F. A., Thibault J., Bric-Furlong E., Raman P., Shipway A., Engels I. H., Cheng J., Yu G. K., Yu J., Aspesi P., de Silva M., Jagtap K., Jones M. D., Wang L., Hatton C., Palescandolo E., Gupta S., Mahan S., Sougnez C., Onofrio R. C., Liefeld T., Conaill L. M., Winckler W., Reich M., Li N., Mesirov J. P., Gabriel S. B., Getz G., Ardlie K., Chan V., Myer V. E., Weber B. L., Porter J., Warmuth M., Finan P., Harris J. L., Meyerson M., Golub T. R., Morrissey M. P., Sellers W. R., Schlegel R., Garraway L. A., The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang H., Zang C., Taing L., Arnett K. L., Wong Y. J., Pear W. S., Blacklow S. C., Liu X. S., Aster J. C., NOTCH1-RBPJ complexes drive target gene expression through dynamic interactions with superenhancers. Proc. Natl. Acad. Sci. U.S.A. 111, 705–710 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ntziachristos P., Tsirigos A., Van Vlierberghe P., Nedjic J., Trimarchi T., Flaherty M. S., Ferres-Marco D., da Ros V., Tang Z., Siegle J., Asp P., Hadler M., Rigo I., De Keersmaecker K., Patel J., Huynh T., Utro F., Poglio S., Samon J. B., Paietta E., Racevskis J., Rowe J. M., Rabadan R., Levine R. L., Brown S., Pflumio F., Dominguez M., Ferrando A., Aifantis I., Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat. Med. 18, 298–302 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Vlierberghe P., Ambesi-Impiombato A., Perez-Garcia A., Haydu J. E., Rigo I., Hadler M., Tosello V., Gatta G. D., Paietta E., Racevskis J., Wiernik P. H., Luger S. M., Rowe J. M., Rue M., Ferrando A. A., ETV6 mutations in early immature human T cell leukemias. J. Exp. Med. 208, 2571–2579 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qiu Z., Guo W., Wang Q., Chen Z., Huang S., Zhao F., Yao M., Zhao Y., He X., MicroRNA-124 reduces the pentose phosphate pathway and proliferation by targeting PRPS1 and RPIA mRNAs in human colorectal cancer cells. Gastroenterology 149, 1587–1598.e11 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Zhang F., Patel D. M., Colavita K., Rodionova I., Buckley B., Scott D. A., Kumar A., Shabalina S. A., Saha S., Chernov M., Osterman A. L., Kashina A., Arginylation regulates purine nucleotide biosynthesis by enhancing the activity of phosphoribosyl pyrophosphate synthase. Nat. Commun. 6, 7517 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Varshavsky A., The N-end rule pathway and regulation by proteolysis. Protein Sci. 20, 1298–1345 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sorrentino C., Cuneo A., Roti G., Therapeutic targeting of notch signaling pathway in hematological malignancies. Mediterr. J. Hematol. Infect. Dis. 11, e2019037 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luengo A., Gui D. Y., Vander Heiden M. G., Targeting metabolism for cancer therapy. Cell Chem. Biol. 24, 1161–1180 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rashkovan M., Ferrando A., Metabolic dependencies and vulnerabilities in leukemia. Genes Dev. 33, 1460–1474 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jin Q., Martinez C. A., Arcipowski K. M., Zhu Y., Gutierrez-Diaz B. T., Wang K. K., Johnson M. R., Volk A. G., Wang F., Wu J., Grove C., Wang H., Sokirniy I., Thomas P. M., Goo Y. A., Abshiru N. A., Hijiya N., Peirs S., Vandamme N., Berx G., Goosens S., Marshall S. A., Rendleman E. J., Takahashi Y.-h., Wang L., Rawat R., Bartom E. T., Collings C. K., Van Vlierberghe P., Strikoudis A., Kelly S., Ueberheide B., Mantis C., Kandela I., Bourquin J.-P., Bornhauser B., Serafin V., Bresolin S., Paganin M., Accordi B., Basso G., Kelleher N. L., Weinstock J., Kumar S., Crispino J. D., Shilatifard A., Ntziachristos P., USP7 Cooperates with NOTCH1 to drive the oncogenic transcriptional program in T-Cell Leukemia. Clin. Cancer Res. 25, 222–239 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Doench J. G., Fusi N., Sullender M., Hegde M., Vaimberg E. W., Donovan K. F., Smith I., Tothova Z., Wilen C., Orchard R., Virgin H. W., Listgarten J., Root D. E., Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 34, 184–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hickox A. E., Wong A. C. Y., Pak K., Strojny C., Ramirez M., Yates J. R. III, Ryan A. F., Savas J. N., Global analysis of protein expression of inner ear hair cells. J. Neurosci. 37, 1320–1339 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen E. Y., Tan C. M., Kou Y., Duan Q., Wang Z., Meirelles G. V., Clark N. R., Ma’ayan A., Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yuan M., Kremer D. M., Huang H., Breitkopf S. B., Ben-Sahra I., Manning B. D., Lyssiotis C. A., Asara J. M., Ex vivo and in vivo stable isotope labelling of central carbon metabolism and related pathways with analysis by LC-MS/MS. Nat. Protoc. 14, 313–330 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Trapnell C., Pachter L., Salzberg S. L., TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Anders S., Pyl P. T., Huber W., HTSeq––A Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robinson M. D., McCarthy D. J., Smyth G. K., edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/5/eabc9781/DC1