

Abstract
A semimechanistic physiologically based pharmacokinetic (PBPK) model for chloroquine (CQ), a highly lysosomotropic weak base, was applied to digitized rat and human concentration versus time data. The PBPK model in rat featured plasma and red blood cell (RBC) concentrations, extensive and apparent nonlinear tissue distribution, fitted hepatic and renal intrinsic clearances, and a plasma half-life of about 1 day. Tissue-to-plasma CQ ratios at 50 hours after dosing were highest in lung, kidney, liver, and spleen (182–318) and lower in heart, muscle, brain, eye, and skin (11–66). The RBC-to-plasma ratio of 11.6 was assumed to reflect cell lipid partitioning. A lysosome-based extended model was used to calculate subcellular CQ concentrations based on tissue mass balances, fitted plasma, interstitial and free cytosol concentrations, and literature-based pH and pKa values. The CQ tissue component concentrations ranked as follows: lysosome > > acidic phospholipid > plasma = interstitial = cytosol ≥ neutral lipids. The extensive lysosome sequestration appeared to change over time and was attributed to lowering pH values caused by proton pump influx of hydrogen ions. The human-to-rat volume of distribution (Vss) ratio of 7 used to scale rat tissue partitioning to human along with estimation of hepatic clearance allowed excellent fitting of oral-dose PK (150–600 mg) of CQ with a 50-day half-life in humans. The prolonged PK of chloroquine was well characterized for rat and human with this PBPK model. The calculated intratissue concentrations and lysosomal effects have therapeutic relevance for CQ and other cationic drugs.
SIGNIFICANCE STATEMENT
Sequestration in lysosomes is a major factor controlling the pharmacokinetics and pharmacology of chloroquine and other cationic drugs. This report provides comprehensive physiologic modeling of chloroquine distribution in tissues and overall disposition in rat and human that reveals expected complexities and inferences related to its subcellular association with various tissue components.
Introduction
Chloroquine (CQ) is a classic antimalarial agent that was identified in 1934 and approved by the US Food and Drug Administration in 1949 and that features additional immunomodulatory, antiviral, anticancer, and neurologic activities (Savarino et al., 2003; Plantone and Koudriavtseva, 2018; Schrezenmeier and Dorner, 2020). The general pharmacological and pharmacokinetic (PK) properties of this 4-aminoquinoline compound are shared by hydroxychloroquine (HCQ) and several newer compounds (White, 1985).
The clinical pharmacokinetics of CQ have been extensively reviewed (White, 1985; Ducharme and Farinotti, 1996). In humans, the drug is well absorbed after oral doses of 150–600 mg, exhibits modest plasma protein binding [fraction unbound (fu) is 0.40] (Walker et al., 1983), undergoes partial renal excretion with 70% of an oral dose excreted unchanged in urine, is partly metabolized by CYP2C8 and CYP3A4/5 enzymes to de-ethylated metabolites (McChesney et al., 1966, Kim, et al., 2003), and is enantiomeric with modest differences in disposition of its R- and S-forms. The monodesethyl metabolite is also active. Most notable is its extensive tissue distribution, which has a steady-state volume of distribution (Vss) of about 800 l/kg and terminal half-life of 30–60 days in human (Frisk-Holmberg et al., 1984; Moore et al., 2011). The strong tissue affinity and large Vss of CQ is attributed to lysosomal trapping of this lipophilic cation along with association with acidic phospholipids in cell membranes.
Although there are few experimental data for CQ distribution in human tissues, there is extensive evidence for CQ distribution into various tissues of rats. Lysosomal uptake of CQ has been directly studied (Allison and Young, 1964; MacIntyre and Cutler, 1988, 1993; Tietz et al., 1990; Daniel et al., 1995; Zheng et al., 2011). The drug has often been used as a positive control for assessing lysosomal function and uptake of other moderate to strong bases (Cramb, 1986; Myers et al., 1995; Ishizaki et al., 2000). The overall distribution of CQ into 10 tissues of rats over time was described (Adelusi and Salako, 1982a), but the data have not been analyzed by physiologically based PK (PBPK) modeling. On the other hand, more limited tissue data for HCQ in rats were subjected to PBPK (Collins et al., 2018).
Although it has long been recognized that lipophilic bases exhibit strong tissue binding and relatively large Vss values (Watanabe and Kozaki, 1978), it was not until 2002 that a PBPK perspective offered a tissue compositional concept for these drugs depicting the joint roles of ionization, lipid partitioning, pH differentials, and lysosomal trapping of ionized cations (Yokogawa et al., 2002). These ideas were later extended to consider diffusive movement of drug molecules within cell compartments (Trapp et al., 2008; Zheng et al., 2011). A more complete tissue composition–based model for such compounds was evolved (Assmus et al., 2017).
Software for operation of generic PBPK models (Simulations Plus, SimCyp, PK-Sim) includes general methods for estimating tissue-to-plasma partition coefficients (Kp) of various drugs based on chemical nature (acid, base, neutral), physicochemical properties (log P, pKa), the fu of the drug, and the partial tissue composition. Prediction methods for moderate to strong lipophilic bases include the lipid composition of various tissues (Rodgers et al., 2005). Such predictions are considered good when tissue-to-plasma ratios fall within 3-fold of known values. This approach was extended to include lysosomal trapping in lysosome-rich organs such as liver, kidney, and lung (Assmus et al., 2017). Although compounds considered included propranolol and imipramine, which are known (Cramb, 1986; Ishizaki et al., 2000) to trap in lysosomes of rats, none of the nine antimalarial agents in clinical use (White, 1985; Trapp et al., 2008) were included. CQ differs from many others in being a divalent rather than monovalent base. Equations for Kp that include lysosomal trapping in some tissues are in the current Simulations Plus (Lancaster, CA) software.
With the extensive preclinical and human PK data available for CQ and the absence of detailed modeling, it is of interest to apply PBPK modeling to explore how the extensive lysosomal trapping explains in vivo PK. This report 1) applies a generalized PBPK model to digitized data for CQ in various tissues of rats (Adelusi and Salako, 1982a); 2) scales the PBPK model to digitized data for CQ PK in humans (Frisk-Holmberg et al., 1984); 3) adapts published equations (Rodgers et al., 2005; Assmus et al., 2017) for calculating tissue drug concentrations with lysosomal trapping; 4) considers the relevance of lysosomotropic effects of CQ (lysosomal changes in pH, volume, and lipids) on its PK; and 5) relates these assessments to the pharmacology of CQ and related compounds.
Materials and Methods
Data Analysis.
The CQ concentration versus time data for plasma and tissues of rats (Adelusi and Salako 1982a) and blood CQ concentration versus time for humans (Frisk-Holmberg et al., 1984) were obtained by digitization (http://geocities.com/graph https://www.jcprg.org/gsys/2.4/). The blood from rats was obtained by cardiac puncture. Blood concentrations for humans were converted to plasma concentrations using the authors’ published relationship. The numerical data used are listed in Supplemental Tables 1 and 2 in the Supplemental Materials.
Measured tissue CQ concentration [Ct(meas)] data of rat were corrected for assumed residual blood by first converting measured red blood cell (RBC)-to-plasma (Cpl) concentration ratios (Kb) to whole-blood concentrations (Cbl):
| (1) |
The hematocrit (Hct) value of rat used was 0.4 (Lee and Blaufox, 1985). Then, adjustments to the corrected tissue concentrations (Ct) were made using
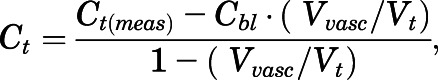 |
(2) |
where Vvasc and Vt are tissue vascular and total tissue volumes. Literature values for Vvasc were from Bernareggi and Rowland (1991).
Tissue partition coefficients (Kp) were obtained by parameter estimation through PBPK modeling as described in detail below. In addition, in silico Kp predictions were obtained for comparison using GastroPlus PBPK simulator (version 9.7.009; Simulations Plus, Inc.) based on published methods (Poulin and Theil, 2002; Berezhkovskiy, 2004; Rodgers and Rowland, 2006) and an adapted method (Assmus et al., 2017) in the software that includes lysosomal trapping for basic compounds.
PBPK Model.
Figure 1 shows the proposed general PBPK model structure for CQ. This model consists of RBCs, plasma, liver, kidney, lung, spleen, heart, brain, muscle, skin, eye, and a remainder compartment. The elimination pathway for CQ is partly hepatic metabolism and partly renal clearance in rodents. Only CQ in plasma (not RBC) was assumed to access tissue spaces. The renal clearance for CQ consisting of glomerular filtration rate (GFR) and active secretion was fixed to 2 times GFR. Physiologic parameter definitions and values are listed in Tables 1 and 2. Plasma flow and tissue volumes were obtained from Brown et al. (1997) and Bernareggi and Rowland (1991).
Fig. 1.

Schematic of the PBPK model structure for chloroquine. Parameters and symbols are defined in the text and Tables 1 and 2. Lines with arrows indicate plasma flows and drug transport and elimination.
TABLE 1.
Physiologic parameters of tissues for chloroquine in rat and human. Dashes, Not Available.
| Tissue | Volume (V) | Plasma Flow (Q) | Fractional Interstitial Spacea | |||
|---|---|---|---|---|---|---|
| Ratb,c | Manc | Ratc | Manc | |||
| ml/kg | ml/h per kilogram | % | ||||
| Liver | 32.3 | 25.7 | 2191 | 628.6 | 16.1 | |
| Kidney | 5.25 | 4.45 | 1385 | 484.6 | 27.3 | |
| Lung | 2.64 | 7.60 | 11,181d | 2769 | 33.6 | |
| Spleen | 0.973 | 2.60 | 679 | 138.6 | 20.7 | |
| Heart | 2.19 | 4.71 | 574 | 110.7 | 32.0 | |
| Brain | 4.64 | 20.1 | 248 | 315.7 | 16.2 | |
| Muscle | 422.7 | 400 | 3512 | 528.9 | 11.8 | |
| Skin | 174.5 | 37.1 | 758 | 160.6 | 38.2 | |
| Eye | 0.74d | 0.214d | 99.6d | 0.073d | — | |
| Blood | 105.3 | 79.1 | 18,641 | 4615 | — | |
| Plasma | Artery | 21.1 | 13.7 | 11,185d | 2769d | — |
| Vein | 42.1 | 32.1 | 11,185d | 2769d | — | |
| Remainder | 290.9 | 452.1 | 1735 | 401 | — | |
| GFR (ml/h per kilogram)e | Rat | 315 | ||||
| Human | 111 | |||||
From Rodgers et al. (2005).
Corrected for residual blood volume (Bernareggi and Rowland, 1991).
From Brown et al. (1997).
From Feke et al. (1989), Yu et al. (1991), and Geng et al. (2009).
From Davies and Morris (1993).
TABLE 2.
Summary of fitted and observed chloroquine pharmacokinetic parameters for rat (CV%). Dashes; Not Available.
| Tissue | Bmax | KD | PS | Ctissue/Cplasma | Estimated Kpa by GastroPlus | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 10 h | 50 h | 150 h | Method 1 | Method 2 | Method 3 | Method 4 | ||||
| µg/ml | µg/ml | ml/h per kilogram | ||||||||
| Liver | 36.8 (71.9) | KD1: 0.00452 (17.7) | PS1: 7297 (43.2) | 150 | 318 | 634 | 7.10 | 10.0 | 376 | 858 |
| Kidney | 14.2 (12.3) | KD1: 0.00452 (17.7) | PS2: 269 (18.6) | 81.8 | 182 | 421 | 4.26 | 6.04 | 403 | 750 |
| Lung | 15.6 (9.31) | KD1: 0.00452 (17.7) | PS2: 269 (18.6) | 129 | 275 | 625 | 8.44 | 14.7 | 319 | 597 |
| Spleen | 12.9 (8.58) | KD1: 0.00452 (17.7) | PS2: 269 (18.6) | 131 | 267 | 589 | 4.34 | 6.16 | 255 | 403 |
| Heart | 3.78 (8.68) | KD1: 0.00452 (17.7) | PS3: 148 (29.9) | 32.4 | 65.6 | 143 | 4.75 | 6.73 | 181 | 261 |
| Brain | 1.74 (8.89) | KD1: 0.00452 (17.7) | PS3: 148 (29.9) | 19.0 | 38.4 | 84.5 | 11.3 | 16.1 | 35.6 | 40.2 |
| Muscle | 0.525 (10.9) | KD2: 0.00544 (21.8) | PS4: 3798 (35.2) | 6.32 | 12.0 | 23.9 | 4.33 | 6.14 | 124 | 165 |
| Skin | 0.695 (10.6) | KD2: 0.00544 (21.8) | PS4: 3798 (35.2) | 5.77 | 10.9 | 21.4 | 5.29 | 7.50 | 107 | 139 |
| Eye | 1.49 (9.55) | KD2: 0.00544 (21.8) | — | 19.4 | 35.6 | 74.5 | NA | NA | NA | NA |
| Remainder | 7.04 (30.8) | KD1: 0.00452 (17.7) | — | — | — | NA | NA | NA | NA | |
| Kb | RBC-to-plasma partition coefficient of RBC | 11.6 (10.8) | ||||||||
| kaliver | Absorption rate constant into liver (h−1) | 0.0306 (9.40) | ||||||||
| kaplasma | Absorption rate constant into plasma (h−1) | 0.372 (20.6) | ||||||||
| CLu,int | Hepatic intrinsic clearance (ml/h per kilogram) | 11,600 (70.3) | ||||||||
| CLs,renal | Systemic renal clearance (ml/h per kilogram) | 630b | ||||||||
| fu | Free fraction of drug in plasma (%) | 40.0c | ||||||||
NA, not applicable.
Estimated partition coefficient (Kp) value using GastroPlus: method 1, (Poulin and Theil, 2002); method 2, (Berezhkovskiy, 2004); method 3, (Rodgers and Rowland, 2006); method 4, (SimulationsPlus).
Fixed to 2-fold GFR.
A nonlinear total cytosol (TC)-to-cytosol water partition coefficient for CQ (Kptissue) was applied because of variable concentration ratios between tissues and plasma. It was assumed that only the free fraction of CQ from the interstitial space (IS) was available to cytosol and available for apparent binding:
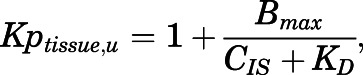 |
(3) |
where CIS is the free concentration of CQ in interstitial space, Bmax is the binding capacity for different tissues (micrograms per milliliter), and KD is the equilibrium dissociation constant (micrograms per milliliter). A different KD (KD2) was applied for skin (Olatunde, 1971), eye, and muscle because of melanin binding of CQ in skin and eye and because muscle has a similar curve shape as skin.
The differential equations for various compartments of the PBPK model are as follows:
- Artery:
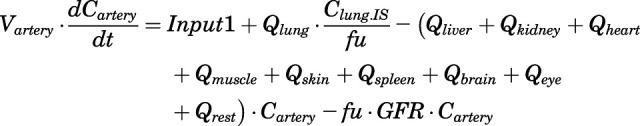
(4) - Vein:
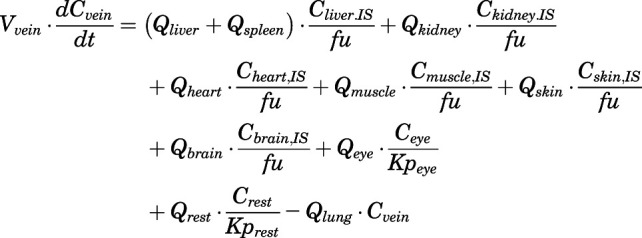
(5) - Liver:
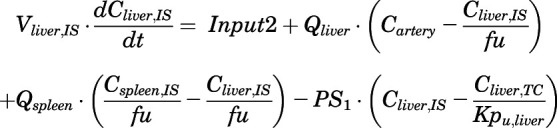
(6) 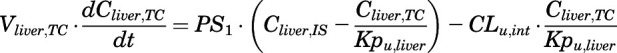
(7) - Kidney:
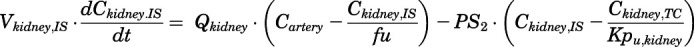
(8) 
(9) - Heart:
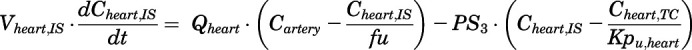
(10) 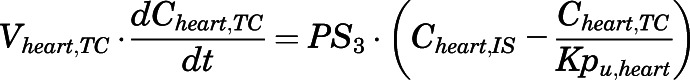
(11) - Lung:
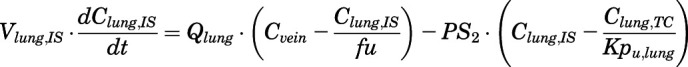
(12) 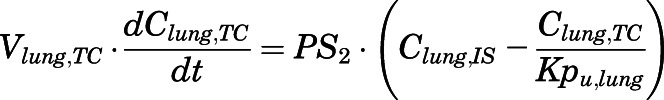
(13) - Spleen:

(14) 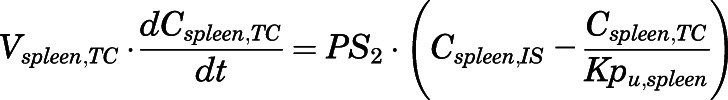
(15) - Brain:
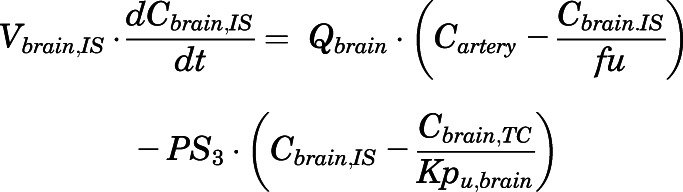
(16) 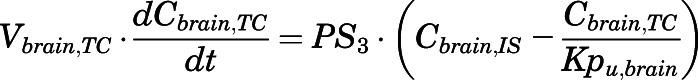
(17) - Muscle:
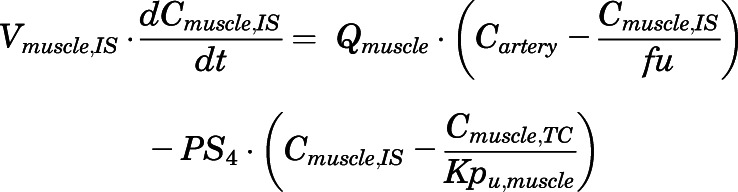
(18) 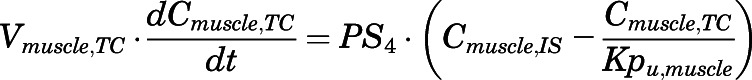
(19) - Skin:
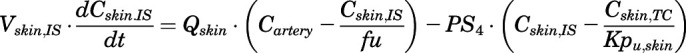
(20) 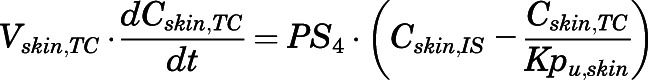
(21) - Eye:
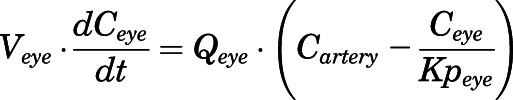
(22) - Remainder:
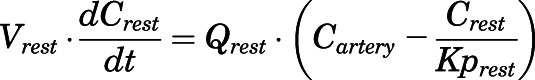
(23)
For the above equations, interstitial volume (Vi,IS), intracellular volume (Vi,TC), IS concentration (Ci,IS), TC concentration (Ci,TC), plasma flow (Qi), and partition coefficients (Kpu,i) are applied for tissue i; CLu,int and CLk,int are the unbound intrinsic clearances in liver and kidney; and PS1-4 are the permeability coefficients between interstitial and cell spaces. Assuming tissue density of 1 g/ml, Vrest = body weight − summation of volumes for listed tissues, plasma, and RBC; Qrest = cardiac plasma output − summation of plasma flows for listed tissues.
The CQ concentrations in RBC and plasma were related as the partition coefficient (Kb):
| (24) |
The dosing input for intraperitoneal (IP) dosing of CQ in rats was
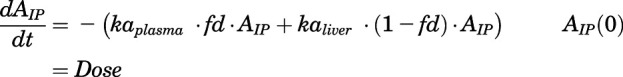 |
(25) |
| (26) |
| (27) |
where fd is the dose fraction directly entering plasma that was fixed to 0.1.
For human data, the systemic renal clearance (CLs,kidney) was set as 70% of total systemic clearance (CLs,total), calculated based on noncompartmental analysis of the plasma data, and intrinsic hepatic clearance (CLu,int) was fitted. The connectivity of systemic (CLs,total) and intrinsic clearances was assessed using the following equation:
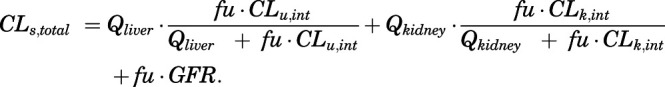 |
(28) |
The oral dosing input of CQ in humans in place of Input2 in eq. 6 is
| (29) |
where the bioavailability (F) of CQ was assumed to be 1.0 (Frisk-Holmberg et al., 1984), and free fraction of plasma (fu) for human is 0.4 (Ducharme and Farinotti, 1996).
The initial conditions for all differential equations are equal to 0 except for the dosing sites.
Extended Lysosome Model.
The lysosome model (Fig. 2) was adapted from Assmus et al. (2017) and Rodgers et al. (2005) and based on assumptions that 1) only neutral molecules diffuse through the lysosome membrane; 2) IS and cytosol concentrations are in PS-determined equilibrium with plasma concentrations; 3) neutral drug equilibrium occurs between lysosome and cytosol concentrations; 4) plasma and IS pH = 7.4, cytosol pH = 7.0, initial lysosome pH = 4.6, and lysosome pH = 5 at 10 hours; and 5) only neutral molecules bind to neutral lipids (NL) and neutral phospholipids (NP), whereas ionized drug binds to acidic phospholipids (AP).
Fig. 2.

Schematic of lysosomal distribution model structure for chloroquine. Parameters and symbols are defined in the text and tables. Lines with arrows indicate plasma flows and drug transfer. The lysosome model was applied to all tissues except eye and remainder. Fncyt is neutral CQ fraction in cytosol and Fnlys is neutral CQ fraction in lysosomes.
The equations for the extended lysosome model are based on the mass balance
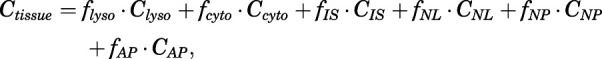 |
(30) |
where Ctissue and CIS are total tissue and interstitial CQ concentrations fitted using the PBPK model; Clyso, Ccyto, CNL, CNP, and CAP are CQ concentrations associated with lysosomes, cytosol, neutral lipids, neutral phospholipids, and acidic phospholipids; and fi values are volume fractions of total tissue for corresponding components. The following approach was applied for each tissue as adapted from Assmus et al. (2017) with assigned parameters from Rodgers et al. (2005).
The Ccyto values are calculated from the fitted CTC profiles using the partition coefficients:
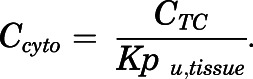 |
(31) |
The CNL and CNP concentrations are calculated using the published n-octanol-to-water partition coefficient (log P = 4.63) (Lullmann and Wehling, 1979):
| (32) |
| (33) |
where Fni values are neutral CQ fractions in cytosol calculated from the known pKa values for the divalent CQ (Trapp et al., 2008):
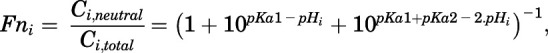 |
(34) |
where i is either plasma, lysosome, or cytosol.
The CAP concentrations of CQ were calculated using the association constant (Kap):
| (35) |
where AP values are tissue concentrations of acidic phospholipids, and Kap was calculated from the Kpu,RBC using (Rodgers et al., 2005)
 |
(36) |
where Kpu,RBC = Kb/fu = 29, and fu is the free fraction of drug in plasma.
The CQ concentrations in lysosomes in relation to Ccyto are governed by equilibration of the neutral molecules and pH partitioning:
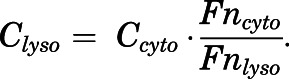 |
(37) |
It was assumed that the pH of lysosome is 5 at 10 hours for all tissues. With initial substitution of fAP = (1 – fNL – fIS – fNP – fcyto − flyso), eq. 30 was rearranged to calculate the effective volume fraction flyso for all tissues using
| (38) |
where Ctissue is the total CQ tissue concentration fitted with the PBPK model. Then, assuming flyso is constant, the pH of lysosomes for each time point was calculated using
| (39) |
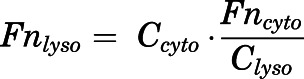 |
(40) |
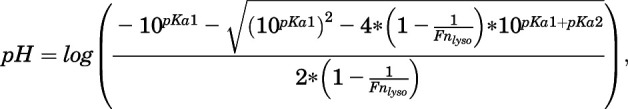 |
(41) |
where eq. 41 was derived by rearranging eq. 34.
The modeling was performed in stages. First, eqs. 3–27 were applied to fit the rat PBPK plasma and tissue data and generate Ctissue, CIS, and CTC concentrations over time. This assumes that the nonlinear tissue binding reflects overall lipid partitioning and lysosomal trapping of CQ. Then, the lysosome model (eqs. 30–39) was used to calculate the theoretical lipid and lysosome concentrations of CQ followed by generation of apparent pH values over time for each tissue (eqs. 39–41). Subsequently, the PBPK model was applied to fit the human PK data. Lastly, the tissue subcomponent model was applied to the human PK using literature values for lipid components in human (Rodgers et al., 2005).
Model Fitting.
For rat data, the following values were estimated: binding capacity (Bmax) and equilibrium dissociation constants (KD1 and KD2) comprising the apparent partition coefficients (Kp) for all tissues, permeability coefficients (PS1–4), two CQ absorption rate constants (kaplasma and kaliver), and the CLu,int. For human data, kaplasma, KD1, and CLu,int were estimated. All fittings and simulations were implemented using ADAPT 5 (Biomedical Simulations Resource, University of Southern California, Los Angeles, CA) using maximum-likelihood estimation. The model was evaluated based on visual inspection of the fitted profiles and CV% of parameter estimates. The variance model was 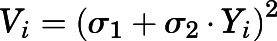 , where Vi represents the variance of the ith data point, Yi is the ith model prediction, and σ1 and σ2 are variance model parameters. Figures were created using GraphPad Prism 8.42 (GraphPad Software, La Jolla, CA). The PBPK model code is provided in the Supplemental Materials.
, where Vi represents the variance of the ith data point, Yi is the ith model prediction, and σ1 and σ2 are variance model parameters. Figures were created using GraphPad Prism 8.42 (GraphPad Software, La Jolla, CA). The PBPK model code is provided in the Supplemental Materials.
Results
Whole-Body Pharmacokinetics of CQ for Rat.
Figure 3 shows the measured CQ concentration in plasma and tissues from Adelusi and Salako (1982a) along with the PBPK model-fitted time-course profiles after single intraperitoneal dosing. The highest drug concentrations are found in liver, lung, spleen, and kidney. Heart, brain, and eye had intermediate concentrations, and the others were much lower. The plasma and RBC concentrations were parallel, which supported use of a linear Kb constant. Generally, the PBPK model captures the plasma, RBC, and tissue PK very well. Blood in rats was obtained by cardiac punctures and thus was a mixture of arterial and venous blood. It was necessary to assume that this represented arterial concentrations, but use of mixed venous blood concentrations is commonplace in many PBPK models. The sampling site is only likely to make a difference for drugs with much more rapid distribution kinetics than CQ (Huang and Isoherranen, 2020).
Fig. 3.

Chloroquine concentration-time profiles for all tissues after 10 mg/kg i.p. single dosing in rats. Measured chloroquine concentrations in plasma, RBCs, and tissues are indicated by different symbols, and black solid lines show the PBPK model fitting. Data are from Adelusi and Salako (1982a).
The fitted parameters for each tissue obtained from model fitting are listed in Table 2. Most parameters were estimated with good precision as indicated by the CV% values. The tissue-to-plasma ratios and Kp values for all tissues exceed 1.0 after the absorption/distribution phase and vary among the tissues. The tissue-to-plasma ratios increase over time, but at 50 hours, they are 318 for liver, 275 for lung, 267 for spleen, 182 for kidney, and 66 for heart, indicating extensive distribution of CQ. The Bmax and KD account for the variable Kp values that create the nonparallel decline of tissue versus plasma concentrations and produce higher tissue-to-plasma ratios as time progresses (Table 2). The Bmax values were specific for each tissue, but two groups of KD values provided good fittings across the array of tissues. These were optimized by trial-and-error seeking of the most parsimonious sets of parameters. The tissues with the highest CQ concentrations had the higher Bmax values, as expected. The permeability component of the model was applied to all tissues except eye and remainder; the inclusion of PS1–4 significantly improved the up-curve shapes of all organs at the early time periods compared with fittings without this parameter. These up-curves varied somewhat, and optimal fittings were obtained by using four different PS values, with liver exhibiting the highest PS value and brain the lowest as expected (Jeong et al., 2017). The KD2 values were associated with the tissues with the least CQ uptake.
For optimal fittings, it was necessary to separate the intraperitoneal dosing into two routes, 90% into liver and 10% into plasma with 100% bioavailability, so two different ka values were applied, slower into liver and faster into plasma. It seemed reasonable that a small part of the dose could be absorbed systemically. Renal clearance consisting of passive filtration (fu∙GFR) and active transport along with CLu,int are responsible for CQ clearance. The systemic renal clearance (630 ml/h per kilogram) was fixed to 2 times GFR (Grundmann et al., 1972). The GFR was handled as direct removal from arterial plasma (eq. 4), as the very high kidney concentrations complicated its attachment to eq. 8. The model-estimated hepatic intrinsic clearance was 11,600 ml/h per kilogram and exhibited the highest CV% (70.3) of all parameters. All other values were less than 36%. Based on systemic clearances obtained using eq. 28, the kidney accounts for 29.7% of CQ disposition, and the liver accounts for 70.3%, which is in agreement with findings that 26%–47% of the dose was excreted unchanged in urine of rats (Grundmann et al., 1972).
Table 2 lists the tissue-to-plasma ratios of CQ at times 10, 50, and 150 hours to demonstrate the range of values as well as their time dependence. The table also lists expected Kp values using three methods that do not include lysosomal uptake and one that does (method 4). Methods 1–3, which include lipid binding as the major factor, predict the early tissue-to-plasma ratios of CQ reasonably well only for those tissues with low lysosomal content. Method 4, which includes lysosomal uptake, reasonably predicts CQ values in most tissues except for muscle and skin. Methods such as eqs. 32–36 are theoretical and are based on reasonable physiologic and physicochemical principles and the measured composition of tissues. However, they are very general and supported only by use of predictions of tissue-to-plasma ratios (Kp) of a variety of drugs (Rodgers et al., 2005; Assmus et al., 2017). Agreement between measured and predicted Kp values with such equations is inexact, usually within 2- or 3-fold, as with the results in Table 2. Direct in vivo measurement of drug associated with subcellular components requires imaging, which is difficult with whole tissues and for drugs that neither are labeled nor fluoresce.
The basic PBPK model for CQ relies on digitized plasma concentrations and calculated tissue-to-plasma ratios (Adelusi and Salako, 1982a) and is thus close but inexact. However, the PBPK model–predicted descriptors of these concentrations such as Cmax, time to Cmax, and half-life are in reasonable concordance with the published values (see Supplemental Materials).
Extended Lysosome Model of CQ for Rat.
Figure 2 shows the structure of the multicomponent lysosome model based on Assmus et al. (2017) that was applied to the CQ tissue data. Table 3 lists the parameters and sources that were employed in eqs. 32–39 to calculate the subcellular concentrations of CQ. It was necessary to assume a pH of 5.0 at 10 hours, leading to a starting and eventual steady-state lysosomal pH of 4.6 for all tissues. It is cautioned that there could be a range of pH values in lysosomes distributed in cells and among various tissues (Schmitt et al., 2019). Figure 4 shows calculated lysosome, cytosol, IS, neutral lipid, neutral phospholipid, and acidic phospholipid CQ concentration versus time profiles in all of the eight tissues except eye along with the corresponding measured tissue concentrations. The lysosome concentrations are far higher than all others as governed by the pH gradient between lysosomes and cytosol. There is markedly greater ionization of CQ at the lower pH as indicated by the lower Fn values in Table 3. The APs as governed by the Kap of 8.52 g/mg and high degree of ionization of CQ have the next highest CQ concentrations. The NL and NP concentrations of CQ are very low in spite of the relatively high log P of 4.63 owing to their access to only the neutral form of CQ for which the Fn is extremely low. The free drug in plasma, IS concentrations, and cytosol water concentrations are generally similar, as these are equilibrating entities in the PBPK model. The cytosol and IS concentrations overlap in all tissues except liver and kidney, where the cytosol concentrations are lower because of the clearance processes.
TABLE 3.
Summary of parameters for lysosome distribution model. Dashes, Not Available.
| Tissue | Lysosome Volumea | Component Fractionb | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Interstitial Space | Cytosol | Neutral Lipid | Neutral Phospholipid | AP (mg/g)c | |||||||
| Rat | Human | Rat | Human | Rat | Human | Rat | Human | ||||
| % | |||||||||||
| Liver | 9.55 | 16.1 | 57.3 | 59.0 | 1.40 | 3.48 | 2.40 | 2.52 | 4.56 | 4.56 | |
| Kidney | 3.28 | 27.3 | 48.3 | 51.0 | 1.20 | 2.07 | 2.42 | 1.62 | 5.03 | 5.03 | |
| Lung | 2.77 | 33.6 | 44.6 | 47.5 | 2.20 | 0.30 | 1.28 | 0.80 | 3.91 | 3.91 | |
| Spleen | 3.18 | 20.7 | 57.9 | 58.1 | 0.77 | 0.20 | 1.13 | 1.98 | 3.18 | 3.18 | |
| Heart | 0.595 | 32.0 | 45.6 | 45.0 | 1.40 | 1.15 | 1.11 | 1.66 | 2.25 | 2.25 | |
| Brain | 0.481 | 16.2 | 62.0 | 60.8 | 3.90 | 5.10 | 0.15 | 5.65 | 0.40 | 9.60 | |
| Muscle | 0.126 | 11.8 | 63.0 | 64.2 | 1.00 | 2.38 | 0.72 | 0.72 | 1.53 | 1.53 | |
| Skin | 0.096 | 38.2 | 29.1 | 33.6 | 6.00 | 2.84 | 0.44 | 1.11 | 3.18 | 3.18 | |
| RBC | — | — | 60.3 | 0.17 | 0.29 | 0.50 | |||||
| pKa1 | 10.1 | ||||||||||
| pKa2 | 8.40 | ||||||||||
| Log P | 4.63 | ||||||||||
| pHd | Lysosome | 5.0 (at 10 h) | |||||||||
| Cytosol | 7.0 | ||||||||||
| IS and plasma | 7.4 | ||||||||||
| Fn | Lysosome | 3.16E-09 (at 10 h) | |||||||||
| Cytosol | 3.04E-05 | ||||||||||
| IS and plasma | 1.81E-04 | ||||||||||
Calculated based on 10-h fitted data.
From Rodgers et al. (2005) and Poulin and Theil (2002).
AP concentration.
From Assmus et al. (2017).
Fig. 4.

Model-predicted lysosome, cytosol, IS, NL, NP, and AP chloroquine concentrations vs. time after 10 mg/kg i.p. dosing in rats. Solid symbols are observed values, and black lines are PBPK-fitted total tissue concentrations.
Figure 5 shows the calculated pH values in lysosomes in various tissues over time. It is assumed that the lysosome pH starts at a low value before CQ dosing, initially rises to higher values owing to influx of CQ, and slowly returns toward a baseline value of 4.6 with influx of hydrogen ions by the proton pump mechanism (Ishizaki et al., 2000; Ishida et al., 2013). This diminishment in lysosomal pH over time causing more uptake of CQ is the model-assigned reason for the increasing tissue-to-plasma ratios of CQ (Table 2).
Fig. 5.

Model-predicted lysosome pH values vs. time in indicated tissues after 10 mg/kg i.p. dosing in rats. Broken line indicates the expected initial rise caused by influx of drug, and the dot-and-dash line indicates the presumed lysosomal baseline and steady-state pH.
An additional set of plasma and tissue data for CQ in rats (Adelusi and Salako, 1982b) was used to evaluate predictability of the basic PBPK model. Good concordance was found, as shown in the Supplementary Materials.
Pharmacokinetics of CQ in Human.
The human PBPK model employed physiologic parameters for human (Table 1), adjusted partition coefficients (from Table 2) based on the human-to-rat 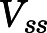 ratio, and fitted values of CLu,int, dissociation constant (KD1), and kaplasm (Table 4). Figure 6 shows excellent fittings of the 150-, 300-, and 600-mg doses of CQ over the full time course, and parameter CV% values were very small. Oral absorption was relatively rapid, producing the early high CQ concentrations.
ratio, and fitted values of CLu,int, dissociation constant (KD1), and kaplasm (Table 4). Figure 6 shows excellent fittings of the 150-, 300-, and 600-mg doses of CQ over the full time course, and parameter CV% values were very small. Oral absorption was relatively rapid, producing the early high CQ concentrations.
TABLE 4.
Summary of assigned and fitted chloroquine pharmacokinetic parameters for human (CV%)
| Parameter | Description | Estimated Value |
|---|---|---|
| CLu,int | Hepatic clearance (ml/h per kilogram) | 1060 (14.5) |
| kaoral | Absorption rate constant (h−1) | 0.0245 (13.2) |
| KD1 | Dissociation constant (µg/ml) | 0.0228 (8.5) |
| R | Adjustment factor for Kp | 7.0a |
| F | Bioavailability (%) | 100b |
| fu | Free fraction of drug in plasma (%) | 40.0c |
Based on multiplying PBPK Kptissue values by the ratio of human-to-rat Vss.
Fig. 6.

Plasma concentration-time profiles of chloroquine after single oral dosing in healthy humans. Black lines show the PBPK model fitting. Data were digitized from Frisk-Holmberg et al. (1984).
The noncompartmental Vss for humans is seven times that of rat, averaging 820 l/kg in human (Frisk-Holmberg et al., 1984) and (our calculated) 113 l/kg in rat (Adelusi and Salako, 1982a); thus, we set R equal to 7.0 as an adjustment factor for multiplying rat Kp values. It was reported that 70% unchanged CQ is excreted by kidney in human (McChesney et al., 1966); hence, the CLs,kidney was fixed to 70% of CLs,total that was estimated using noncompartmental analysis, and CLu,int was estimated by fitting the human data. The CLu,int is 1060 ml/h per kilogram, which results in CLs,total (722 ml/h per kilogram), which is similar to a reported value (Frisk-Holmberg et al., 1984).
Figure 7 shows the model-predicted CQ concentrations associated with all of the tissue components for four major tissues in human after the 600-mg dose. These were calculated based on published lipid contents for human (Table 3), but rat lysosomal fractions were employed. Although the rank order of concentrations appears similar to those in rat (Fig. 4), lysosomal concentrations were relatively higher in human. For example, in human, lysosome-to-plasma CQ ratios at 1000 hours were as follows: 25,054 for liver, 39,654 for kidney, 56,608 for lung, and 208,976 for muscle. Corresponding values for rat at 50 hours were 3380, 8110, 8160, and 8530.
Fig. 7.

Model-predicted lysosome, cytosol, IS, NL, NP, and AP chloroquine concentrations vs. time in four indicated tissues after oral dosing of 600 mg in human.
Discussion
Justification of the PBPK Model.
The properties of CQ are well appreciated, as it has been in clinical use since the 1950s and is a frequent probe for lysosomal functioning. Comprehensive reviews of its PK and pharmacology are available (White, 1985; Browning, 2014). The plasma and whole-blood PK of CQ have been studied in several species, and clearances were shown to scale allometrically (Moore et al., 2011).
The avid uptake of CQ into the liver, kidney, spleen, and lungs, which have abundant lysosomes, and lesser distribution to muscle and other tissues has been well appreciated from other studies in rats (McChesney et al., 1965, 1967; Grundmann et al., 1972; Osifo, 1980), but the present data provide the most comprehensive view of the overall PK and tissue distribution of CQ in any species. The present PBPK modeling utilizes a two-stage approach, with the first approach generally assessing the array of 10 tissues and blood with classic PBPK modeling concepts, including apparent nonlinear tissue distribution. This provided estimates of CQ concentrations in IS and cell cytosol with the nonlinear component assumed to reflect CQ concentrations associated with various lipids and lysosomes. A complex distribution model for lipid binding and lysosome distribution (Assmus et al., 2017) was then adapted to allocate the total tissue concentrations of CQ into its subcomponents.
In support of our PBPK modeling, there is considerable evidence for lysosomal tissue distribution of CQ in rat. The fluorescence of CQ facilitated early viewing of its high concentrations in intracellular lysosomes of cells (Allison and Young, 1964). Differential centrifugation and electrophoresis methods allowed measurement of the slow uptake of CQ into the hepatic lysosomes of rats dosed with CQ along with its inhibition of phospholipase A (Hostetler et al., 1985). Isolated rat hepatocytes demonstrated both uptake and metabolism of CQ, including marked reduction of uptake by ammonium chloride, a lysosomal inhibitor that alters lysosomal pH (MacIntyre and Cutler, 1993). Their application of a cellular PK model argued that the permeability of the lysosomal membrane is rate-limiting for hepatocyte uptake of CQ. A more complex model (similar to Fig. 2) for the lysosomal uptake of CQ was developed for cells in culture (Trapp et al., 2008; Zheng et al., 2011). This process is mimicked by the simpler diffusion step (PS) between IS and cell content applied in our PBPK model (Fig. 1) to account for the slow early rise in CQ concentrations in many tissues (Fig. 3). The in vitro binding of CQ to various individual polar phospholipids has been measured (Lullmann and Wehling, 1979). The apparent partition coefficient of CQ for phosphatidylcholine was about 77, which is consistent with our AP-to-plasma ratios, and a Kap of about 2.0 g/mg, which is close to our Kap of 8.52 g/mg. Such in vitro binding was nonlinear for CQ and other compounds studied.
The nonlinear Kp in our basic PBPK model was used initially to account for the increases in tissue-to-plasma concentrations of CQ over time (Fig. 3; Table 2). This may actually reflect a time-dependent process. Part of the lysosomotropic effects of CQ is the inhibition of phospholipid degradation (Hostetler et al., 1985). Use of isolated hepatocytes showed that acute exposure to CQ produces an increased lysosomal pH attributed to proton consumption (Tietz et al., 1990; Myers et al., 1995). Imaging of canine kidney cells has demonstrated phospholipidosis accompanied by altered vesicular pH and increased vesicle volume (Zheng et al., 2011). Our modeling (Fig. 5) assumed that CQ produced, within hours of dosing, a rise in lysosome pH that slowly returned to the baseline that was owing to both elimination of CQ and influx of H+ ions by the proton pump mechanism responsible for maintaining the normal low pH of 4–5. It is possible that phospholipidosis and increased vesicle volume also contribute to changing tissue-to-plasma ratios after CQ dosing (Table 2).
CQ exhibits strong binding to melanin, particularly in the eye (Schroeder and Gerber, 2014). This is implicated in ocular toxicity. The rat eye has modest concentrations of CQ (Fig. 3; Table 2) and a small Bmax. Melanin binding is saturable in vitro, and the eye accounts for a very small fraction of CQ in the body.
The present effort was partly inspired by a publication describing the PBPK modeling of HCQ using data obtained from mice, many similar stated concepts, and extrapolation to humans (Collins et al., 2018). However, their modeling is based on only four tissues (blood, liver, kidney, and gut), whereas CQ studies offer much richer data. The authors did not provide their full array of equations used, and their extrapolations to human do not cover the very long half-life known for HCQ (Tett et al., 1988).
Pharmacokinetics of Chloroquine in Human.
The plasma concentration versus time courses of CQ in human for the three dose levels were well captured with the PBPK model, with three parameters needing customization (Fig. 6; Table 4). These data are representative of many studies of CQ PK in human (Moore et al., 2011). Multiplying the rat Kp values by the human-to-rat Vss ratio of 7 was key. In turn, this implies that the total tissue and lysosomal concentrations of CQ are 7-fold higher in human than rat (Fig. 7). This is supported by measurements of CQ showing skin-to-plasma ratios of about 34 at 48 hours after an intravenous dose in patients (Olatunde, 1971), and the 10-hour skin ratio was 5.77 in rats (Table 2). Another cationic antimalarial quinoline, HCQ, exhibits a Vss value that is 5.7-fold higher in human (86 l/kg) than in rats (15 l/kg) based on blood concentrations (Tett et al., 1988; Emami et al., 1998). It is possible that lysosomal pH is lower in human to produce greater sequestration of these drugs. Larger lysosomal volumes and greater acidic phospholipid content may be contributory. On the other hand, Vss values for 10 other basic drugs are similar in human and rat (Sawada et al., 1984).
Therapeutic Implications of the PBPK Model.
There are many therapeutic dosing regimens for CQ, typically ranging from 100 to 600 mg per day (Browning, 2014). The human data in Fig. 6 reflect this range. The mechanism of action of CQ in malaria is thought to be its lysosomotropic effect on the acidic food vacuoles of the parasite that increases pH and interferes with the digestive degradation of hemoglobin in RBCs.
Drugs such as CQ and HCQ are also used for treatment of patients with rheumatoid arthritis and systemic lupus erythematosus. Their myriad effects are attributed to interference of antigen processing in macrophages, downregulation of immune responses, alteration of signaling pathways and transcriptional activity, and inhibition of cytokine production (Fox, 1993; Schrezenmeier and Dorner, 2020). Although the lysosomotropic effects of CQ are stated to be most important clinically, more than 20 additional actions contributing to both therapeutic and adverse effects have been cited (Browning, 2014). There is current interest in using HCQ for autophagy modulation, the natural metabolic digestion of cell proteins and other materials in lysosomes; upregulation of this process is a resistance mechanism for some tumors (Shi et al., 2017).
Our PK modeling predicts that CQ concentrations in the cytosol will be very low, similar to free drug concentrations in plasma and IS (Figs. 4 and 7). However, multiple actions of CQ appear connected to or are triggered by the changes in lysosomal pH and associated alterations in lysosomal and cellular functions that ensue. The pharmacology of CQ and other lysosomotropic drugs is far more complex than can be explained by the very low unbound concentrations that are commonly thought to drive actions of many drugs. Some in vitro screening systems for drug activity may not invoke the same lysosomal triggers. For example, the IC50 for CQ inhibition of mitogen-induced human lymphocyte proliferation is 19.5 µM or about 6.4 μg/ml (Kamal and Jusko, 2004), an in vitro system that is meaningful for immune effects of corticosteroids. This concentration is far above peak exposures of 0.1 µg/ml after 600-mg doses of CQ (Fig. 6). Yet CQ is effective at these doses for treatment of patients with rheumatoid arthritis and systemic lupus erythematosus.
Of current interest, CQ and HCQ were found active in inhibiting SARS-CoV-2 in vitro, with IC50 concentrations of around 6 µM (Liu et al., 2020). These too are well above typical therapeutic plasma concentrations. It can be questioned whether these in vitro responses are relevant in vivo and whether a lysosomotropic mechanism is present in some cell cultures. It has been argued that the lysosomotropic effects could partly make CQ effective as an antiviral agent (Savarino et al., 2003; Plantone and Koudriavtseva, 2018). Some viruses enter their target cells by endosomes that merge into lysosomes. The low pH and action of enzymes liberates infectious nucleic acids from virus particles. Raising the lysosomal pH thus interferes with this process. However, recent attempts to use CQ and HCQ to treat COVID-19 viral infections have not shown efficacy and risk various toxicities (Qaseem et al., 2020).
This report demonstrates application of state-of-the-art PBPK modeling concepts, methods, and insights for an old drug with highly interesting tissue distribution and mechanisms of action. The principles underlying this modeling approach will likely be relevant to other cationic drugs that sequester in lysosomes, although their physicochemical properties and degree of changes in lysosome pH and structure may require more specific adjustments.
Acknowledgments
We appreciate the review of this manuscript by Dr. Viera Lucakova from Simulations Plus.
Abbreviations
- AP
acidic phospholipid
- CLs,total
total systemic clearance
- CLu,int
intrinsic hepatic clearance
- CQ
chloroquine
- fu
fraction unbound
- GFR
glomerular filtration rate
- HCQ
hydroxychloroquine
- IS
interstitial space
- NL
neutral lipid
- NP
neutral phospholipid
- PBPK
physiologically based pharmacokinetic
- PK
pharmacokinetic
- RBC
red blood cell
- TC
total cytosol
- Vss
volume of distribution
Authorship Contributions
Participated in research design: Liu, Jusko.
Performed data analysis: Liu.
Wrote or contributed to the writing of the manuscript: Liu, Jusko.
Footnotes
This work was supported by National Institutes of Health National Institute of General Medical Sciences [Grant R35-GM131800].
No author has an actual or perceived conflict of interest with the contents of this article.
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Adelusi SA, Salako LA. (1982a) Kinetics of the distribution and elimination of chloroquine in the rat. Gen Pharmacol 13:433–437. [DOI] [PubMed] [Google Scholar]
- Adelusi SA, Salako LA. (1982b) The effect of protein-energy malnutrition on the absorption, distribution and elimination of chloroquine in the rat. Gen Pharmacol 13:505–509. [DOI] [PubMed] [Google Scholar]
- Allison AC, Young MR. (1964) Uptake of dyes and drugs by living cells in culture. Life Sci (1962) 3:1407–1414. [DOI] [PubMed] [Google Scholar]
- Assmus F, Houston JB, Galetin A. (2017) Incorporation of lysosomal sequestration in the mechanistic model for prediction of tissue distribution of basic drugs. Eur J Pharm Sci 109:419–430. [DOI] [PubMed] [Google Scholar]
- Berezhkovskiy LM. (2004) Volume of distribution at steady state for a linear pharmacokinetic system with peripheral elimination. J Pharm Sci 93:1628–1640. [DOI] [PubMed] [Google Scholar]
- Bernareggi A, Rowland M. (1991) Physiologic modeling of cyclosporin kinetics in rat and man. J Pharmacokinet Biopharm 19:21–50. [DOI] [PubMed] [Google Scholar]
- Brown RP, Delp MD, Lindstedt SL, Rhomberg LR, Beliles RP. (1997) Physiological parameter values for physiologically based pharmacokinetic models. Toxicol Ind Health 13:407–484. [DOI] [PubMed] [Google Scholar]
- Browning DJ. (2014) Pharmacology of chloroquine and hydroxychloroquine, in Hydroxychloroquine and Chloroquine Retinopathy, Springer, New York. [Google Scholar]
- Collins KP, Jackson KM, Gustafson DL. (2018) Hydroxychloroquine: a physiologically-based pharmacokinetic model in the context of cancer-related autophagy modulation. J Pharmacol Exp Ther 365:447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramb G. (1986) Selective lysosomal uptake and accumulation of the beta-adrenergic antagonist propranolol in cultured and isolated cell systems. Biochem Pharmacol 35:1365–1372. [DOI] [PubMed] [Google Scholar]
- Davies B, Morris T. (1993) Physiological parameters in laboratory animals and humans. Pharm Res 10:1093–1095. [DOI] [PubMed] [Google Scholar]
- Daniel W, Syrek M, Janczar L, Boksa J. (1995) The pharmacokinetics of promazine and its metabolites after acute and chronic administration to rats--a comparison with the pharmacokinetics of imipramine. Pol J Pharmacol 47:127–136. [PubMed] [Google Scholar]
- Ducharme J, Farinotti R. (1996) Clinical pharmacokinetics and metabolism of chloroquine. Focus on recent advancements. Clin Pharmacokinet 31:257–274. [DOI] [PubMed] [Google Scholar]
- Emami J, Pasutto FM, Jamali F. (1998) Effect of experimental diabetes mellitus and arthritis on the pharmacokinetics of hydroxychloroquine enantiomers in rats. Pharm Res 15:897–903. [DOI] [PubMed] [Google Scholar]
- Feke GT, Tagawa H, Deupree DM, Goger DG, Sebag J, Weiter JJ. (1989) Blood flow in the normal human retina. Invest Ophthalmol Vis Sci 30:58–65. [PubMed] [Google Scholar]
- Fox RI. (1993) Mechanism of action of hydroxychloroquine as an antirheumatic drug. Semin Arthritis Rheum 23 (Suppl 1):82–91. [DOI] [PubMed] [Google Scholar]
- Frisk-Holmberg M, Bergqvist Y, Termond E, Domeij-Nyberg B. (1984) The single dose kinetics of chloroquine and its major metabolite desethylchloroquine in healthy subjects. Eur J Clin Pharmacol 26:521–530. [DOI] [PubMed] [Google Scholar]
- Geng Y, Greenberg KP, Wolfe R, Gray DC, Hunter JJ, Dubra A, Flannery JG, Williams DR, Porter J. (2009) In vivo imaging of microscopic structures in the rat retina. Invest Ophthalmol Vis Sci 50:5872–5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundmann M, Mikulíkrová I, Vrublovský P. (1972) Tissue distribution of chloroquine in rats in the course of long-term application. Arch Int Pharmacodyn Ther 197:45–52. [PubMed] [Google Scholar]
- Hostetler KY, Reasor M, Yazaki PJ. (1985) Chloroquine-induced phospholipid fatty liver. Measurement of drug and lipid concentrations in rat liver lysosomes. J Biol Chem 260:215–219. [PubMed] [Google Scholar]
- Huang W, Isoherranen N. (2020) Sampling site has a critical impact on physiologically based pharmacokinetic modeling. J Pharmacol Exp Ther 372:30–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida Y, Nayak S, Mindell JA, Grabe M. (2013) A model of lysosomal pH regulation. J Gen Physiol 141:705–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizaki J, Yokogawa K, Ichimura F, Ohkuma S. (2000) Uptake of imipramine in rat liver lysosomes in vitro and its inhibition by basic drugs. J Pharmacol Exp Ther 294:1088–1098. [PubMed] [Google Scholar]
- Jeong Y-S, Yim CS, Ryu H-M, Noh C-K, Song Y-K, Chung S-J. (2017) Estimation of the minimum permeability coefficient in rats for perfusion-limited tissue distribution in whole-body physiologically-based pharmacokinetics. Eur J Pharm Biopharm 115:1–17. [DOI] [PubMed] [Google Scholar]
- Kamal MA, Jusko WJ. (2004) Interactions of prednisolone and other immunosuppressants used in dual treatment of systemic lupus erythematosus in lymphocyte proliferation assays. J Clin Pharmacol 44:1034–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K-A, Park J-Y, Lee J-S, Lim S. (2003) Cytochrome P450 2C8 and CYP3A4/5 are involved in chloroquine metabolism in human liver microsomes. Arch Pharm Res 26:631–637. [DOI] [PubMed] [Google Scholar]
- Lee HB, Blaufox MD. (1985) Blood volume in the rat. J Nucl Med 26:72–76. [PubMed] [Google Scholar]
- Liu J, Cao R, Xu M, Wang X, Zhang H, Hu H, Li Y, Hu Z, Zhong W, Wang M. (2020) Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov 6:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüllmann H, Wehling M. (1979) The binding of drugs to different polar lipids in vitro. Biochem Pharmacol 28:3409–3415. [DOI] [PubMed] [Google Scholar]
- MacIntyre AC, Cutler DJ. (1988) Role of lysosomes in hepatic accumulation of chloroquine. J Pharm Sci 77:196–199. [DOI] [PubMed] [Google Scholar]
- MacIntyre AC, Cutler DJ. (1993) Kinetics of chloroquine uptake into isolated rat hepatocytes. J Pharm Sci 82:592–600. [DOI] [PubMed] [Google Scholar]
- McChesney EW, Banks WF, Jr, Sullivan DJ. (1965) Metabolism of chloroquine and hydroxychloroquine in albino and pigmented rats. Toxicol Appl Pharmacol 7:627–636. [DOI] [PubMed] [Google Scholar]
- McChesney EW, Conway WD, Banks WF, Jr, Rogers JE, Shekosky JM. (1966) Studies of the metabolism of some compounds of the 4-amino-7-chloroquinoline series. J Pharmacol Exp Ther 151:482–493. [PubMed] [Google Scholar]
- McChesney EW, Banks WF, Jr, Fabian RJ. (1967) Tissue distribution of chloroquine, hydroxychloroquine, and desethylchloroquine in the rat. Toxicol Appl Pharmacol 10:501–513. [DOI] [PubMed] [Google Scholar]
- Moore BR, Page-Sharp M, Stoney JR, Ilett KF, Jago JD, Batty KT. (2011) Pharmacokinetics, pharmacodynamics, and allometric scaling of chloroquine in a murine malaria model. Antimicrob Agents Chemother 55:3899–3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers BM, Tietz PS, Tarara JE, LaRusso NF. (1995) Dynamic measurements of the acute and chronic effects of lysosomotropic agents on hepatocyte lysosomal pH using flow cytometry. Hepatology 22:1519–1526. [PubMed] [Google Scholar]
- Olatunde IA. (1971) Chloroquine concentrations in the skin of rabbits and man. Br J Pharmacol 43:335–340. [PMC free article] [PubMed] [Google Scholar]
- Osifo NG. (1980) Chloroquine pharmacokinetics in tissues of pyrogen treated rats and implications for chloroquine related pruritus. Res Commun Chem Pathol Pharmacol 30:419–430. [PubMed] [Google Scholar]
- Plantone D, Koudriavtseva T. (2018) Current and future use of chloroquine and hydroxychloroquine in infectious, immune, neoplastic, and neurological diseases: a mini-review. Clin Drug Investig 38:653–671. [DOI] [PubMed] [Google Scholar]
- Poulin P, Theil FP. (2002) Prediction of pharmacokinetics prior to in vivo studies. 1. Mechanism-based prediction of volume of distribution. J Pharm Sci 91:129–156. [DOI] [PubMed] [Google Scholar]
- Qaseem A, Yost J, Etxeandia-Ikobaltzeta I, Miller MC, Abraham GM, Obley AJ, Forciea MA, Jokela JA, Humphrey LL. (2020) Should clinicians use chloroquine or hydroxychloroquine alone or in combination with azithromycin for the prophylaxis or treatment of COVID-19? Living practice points from the American College of Physicians (Version 1) [published correction appears in Ann Intern Med (2020) 173:166]. Ann Intern Med 173:137–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers T, Leahy D, Rowland M. (2005) Physiologically based pharmacokinetic modeling 1: predicting the tissue distribution of moderate-to-strong bases. J Pharm Sci 94:1259–1276. [DOI] [PubMed] [Google Scholar]
- Rodgers T, Rowland M. (2006) Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci 95:1238–1257. [DOI] [PubMed] [Google Scholar]
- Savarino A, Boelaert JR, Cassone A, Majori G, Cauda R. (2003) Effects of chloroquine on viral infections: an old drug against today’s diseases? Lancet Infect Dis 3:722–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada Y, Hanano M, Sugiyama Y, Harashima H, Iga T. (1984) Prediction of the volumes of distribution of basic drugs in humans based on data from animals. J Pharmacokinet Biopharm 12:587–596. [DOI] [PubMed] [Google Scholar]
- Schrezenmeier E, Dörner T. (2020) Mechanisms of action of hydroxychloroquine and chloroquine: implications for rheumatology. Nat Rev Rheumatol 16:155–166. [DOI] [PubMed] [Google Scholar]
- Schmitt MV, Lienau P, Fricker G, Reichel A. (2019) Quantitation of lysosomal trapping of basic lipophilic compounds using in vitro assays and in silico predictions based on the determination of the full pH profile of the endo-/lysosomal system in rat hepatocytes. Drug Metab Dispos 47:49–57. [DOI] [PubMed] [Google Scholar]
- Schroeder RL, Gerber JP. (2014) Chloroquine and hydroxychloroquine binding to melanin: some possible consequences for pathologies. Toxicol Rep 1:963–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi TT, Yu XX, Yan LJ, Xiao HT. (2017) Research progress of hydroxychloroquine and autophagy inhibitors on cancer. Cancer Chemother Pharmacol 79:287–294. [DOI] [PubMed] [Google Scholar]
- Tett SE, Cutler DJ, Day RO, Brown KF. (1988) A dose-ranging study of the pharmacokinetics of hydroxy-chloroquine following intravenous administration to healthy volunteers. Br J Clin Pharmacol 26:303–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tietz PS, Yamazaki K, LaRusso NF. (1990) Time-dependent effects of chloroquine on pH of hepatocyte lysosomes. Biochem Pharmacol 40:1419–1421. [DOI] [PubMed] [Google Scholar]
- Trapp S, Rosania GR, Horobin RW, Kornhuber J. (2008) Quantitative modeling of selective lysosomal targeting for drug design. Eur Biophys J 37:1317–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker O, Birkett DJ, Alván G, Gustafsson LL, Sjöqvist F. (1983) Characterization of chloroquine plasma protein binding in man. Br J Clin Pharmacol 15:375–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe J, Kozaki A. (1978) Relationship between partition coefficients and apparent volumes of distribution for basic drugs. I. Chem Pharm Bull (Tokyo) 26:665–667. [DOI] [PubMed] [Google Scholar]
- White NJ. (1985) Clinical pharmacokinetics of antimalarial drugs. Clin Pharmacokinet 10:187–215. [DOI] [PubMed] [Google Scholar]
- Yokogawa K, Ishizaki J, Ohkuma S, Miyamoto K. (2002) Influence of lipophilicity and lysosomal accumulation on tissue distribution kinetics of basic drugs: a physiologically based pharmacokinetic model. Methods Find Exp Clin Pharmacol 24:81–93. [DOI] [PubMed] [Google Scholar]
- Yu DY, Alder VA, Cringle SJ. (1991) Measurement of blood flow in rat eyes by hydrogen clearance. Am J Physiol 261:H960–H968. [DOI] [PubMed] [Google Scholar]
- Zheng N, Zhang X, Rosania GR. (2011) Effect of phospholipidosis on the cellular pharmacokinetics of chloroquine. J Pharmacol Exp Ther 336:661–671. [DOI] [PMC free article] [PubMed] [Google Scholar]