

This cohort study describes the association between an autosomal recessive macular dystrophy with a recurrent variant in CLN5.
Key Points
Question
Are variants in the neuronal ceroid lipofuscinosis type 5 (CLN5) gene, which is known to cause a variant of autosomal recessive neuronal ceroid lipofuscinosis, associated with a retinal dystrophy with relatively mild neurologic manifestations?
Findings
In this cohort study of 7 patients affected with a retinal dystrophy with a prominent macular involvement, all patients carried the homozygous missense variant c.415T>C (p.Phe139Leu) in CLN5.
Meaning
These results suggest that CLN5, similar to CLN7 and CLN3, may be associated with a retinal dystrophy as well as neuronal ceroid lipofuscinosis.
Abstract
Importance
Homozygous variants in the neuronal ceroid lipofuscinosis type 5 (CLN5) gene are associated with neuronal ceroid lipofuscinosis, a progressive neurologic disorder that leads to ataxia, seizures, and early death. The association between a homozygous variant in this gene and a macular dystrophy is described here.
Objective
To describe an autosomal recessive macular dystrophy associated with a recurrent variant in CLN5.
Design, Setting, and Participants
This cohort study took place at a national referral center and had a follow-up duration ranging between 1 and 5 years. All patients who were identified to carry a specific homozygous missense variant in CLN5, among more than 2000 patients who were diagnosed with or suspected to have retinal dystrophies, who did not carry this variant, were included. Data were collected between June 2014 and September 2020.
Exposures
All patients who were sampled for DNA analysis due to molecularly unconfirmed retinal dystrophy and who were subsequently identified to carry the homozygous missense variant c.415T>C (p.Phe139Leu) in CLN5 were included, while patients who did not carry the variant were excluded.
Main Outcomes and Measures
Retinal phenotype associated with this specific homozygous missense variant in CLN5.
Results
Seven affected patients (mean [SD] age, 43 [18] years; age range, 33-52 years; 5 male) carried the homozygous missense in CLN5. All patients were diagnosed as having a macular dystrophy. Four patients had mild electroretinographic alterations. All patients had hypoautofluorescent maculas with retinal thinning (central subfield thickness, 80 µm). Visual acuity ranged between 2/200 and 20/100. Neurologic symptoms were mild (dizziness) in 5 patients and absent in 2 patients. Neuroimaging demonstrated cerebellar atrophy and white matter lesions, respectively, in 2 patients.
Conclusions and Relevance
These results suggest that CLN5, similar to CLN7, may be associated with isolated macular dystrophy as well as neuronal ceroid lipofuscinosis. The variant c.415T>C p.Phe139Leu does not seem to be associated with any prominent neurologic disease at least until the fourth to sixth decades of life. These findings may imply a specific role of CLN5 in macular neurons. Additional study is suggested, such as molecular screening for this variant in cohorts of patients with undiagnosed macular dystrophies and biological studies of its molecular effects.
Introduction
Neuronal ceroid lipofuscinoses (NCLs) are a group of lysosomal storage disorders that are typically recessive and frequently manifest in childhood, early adulthood, or rarely during late adulthood. Neuronal ceroid lipofuscinosis is a progressive neurodegenerative disease causing ataxia, seizures, mental deterioration, and often early death.1,2 Some forms of NCLs encompass additional features including rapidly progressive generalized retinal degeneration (eg, NCLs associated with CLN3 [Batten disease, CLN3 gene] or CLN7 [MFSD8 gene], often leading to early-onset childhood blindness).3,4 The disease mechanism responsible for the neurodegenerative disease is thought to be lysosomal dysfunction, which manifests as ultrastructural intracytoplasmic lysosomal accumulations.1,2
Recently, specific recessive variants in the MFSD8 gene were shown to be associated with a spectrum of nonsyndromic retinal degeneration, including isolated macular dystrophy without any neurologic findings.5,6,7 Because of its location in the outer plexiform layer of the retina, the protein product of MFSD8 was suggested to form parts of synaptic vesicles in the photoreceptor terminals, leading to photoreceptor synaptic dysfunction when altered.7 The higher content of synaptic ribbons for vesicle docking in peripheral cones compared with central cones could explain a selective macular impairment caused by certain variants.7 Similarly, isolated nonsyndromic retinal dystrophy has been associated with certain variants in CLN3.8
Recently, we discovered a specific recessive missense variant (c.415T>C [p.Phe139Leu]) in CLN5 in patients with nonsyndromic retinal dystrophies. However, variants in this gene, similar to CLN3 and MSFD8, are known to cause NCL.2 We speculate that this CLN5 variant, in analogy with certain variants in CLN3 and MFSD8, is associated with an isolated retinopathy with macular involvement, which we set out to describe.
Methods
This study was approved by the institutional research ethics committee at King Khaled Eye Specialist Hospital in Riyadh, Saudi Arabia, and adhered to the tenets of the Declaration of Helsinki.9 Written informed consent was obtained from the study participants and none of the participants received any stipend.
Investigations included next-generation sequencing, multimodal retinal imaging, electrophysiology, and neurologic assessment. Genetic testing and clinical investigations are described in the eMethods in the Supplement. Data were collected from June 2014 to September 2020.
Results
Within a cohort of 2086 patients with retinal dystrophy analyzed by a customized retinal dystrophy panel, 7 affected patients from Saudi Arabia (mean [SD] age, 43 [18] years; age range, 33-52 years; 5 men and 2 women) carried the homozygous missense variant c.415T>C (p.Phe139Leu) (NM_006493.4, chr.13: 77570112 in hg19) in CLN5 (Figure 1, Figure 2, and Figure 3; eTables 1 and 2 and eFigures 1-12 in the Supplement). To our knowledge, the CLN5 variant c.415T>C has not been described previously in association with any other health condition and has no frequency in public databases (gnomAD). Of 21 bioinformatic in silico programs, 17 were associated with having a pathogenic effect for this variant. Furthermore, high conservation was shown by 7 of 8 bioinformatic conservation tools (eMethods in the Supplement). In addition, 1 patient had 2 missense variants in USH2A of unclear significance (eTable 1 in the Supplement).
Figure 1. Clinical Findings in Patient 4 With a Characteristic Macular Degeneration and a Homozygous c.415T>C (p.Phe139Leu) Missense Variant in CLN5.

Patient 4 is in her 30s. A and B, Unremarkable peripheral retina with central macular degeneration and temporally pale and excavated discs. Note tiny yellowish crystals in the macula of the left eye. C and D, Fundus autofluorescence shows a characteristic central macular hypoautofluorescence.
Figure 2. Transfoveal Horizontal Spectral-Domain Optical Coherence Tomography Scans in Patient 4 With a Homozygous c.415T>C (p.Phe139Leu) Missense Variant in CLN5.

Patient 4 is in her 30s. There is a central loss of photoreceptors and outer retinal hyperreflective deposits that are more prominent in the left eye, compared with the right eye. A and C, The fundus image shows the location of the optical coherence tomography scan.
Figure 3. Segregation Analysis in the Family of Patient 4 With a Characteristic Nonsyndromic Macular Degeneration Associated With the Homozygous c.415T>C (p.Phe139Leu) Missense Variant in CLN5.
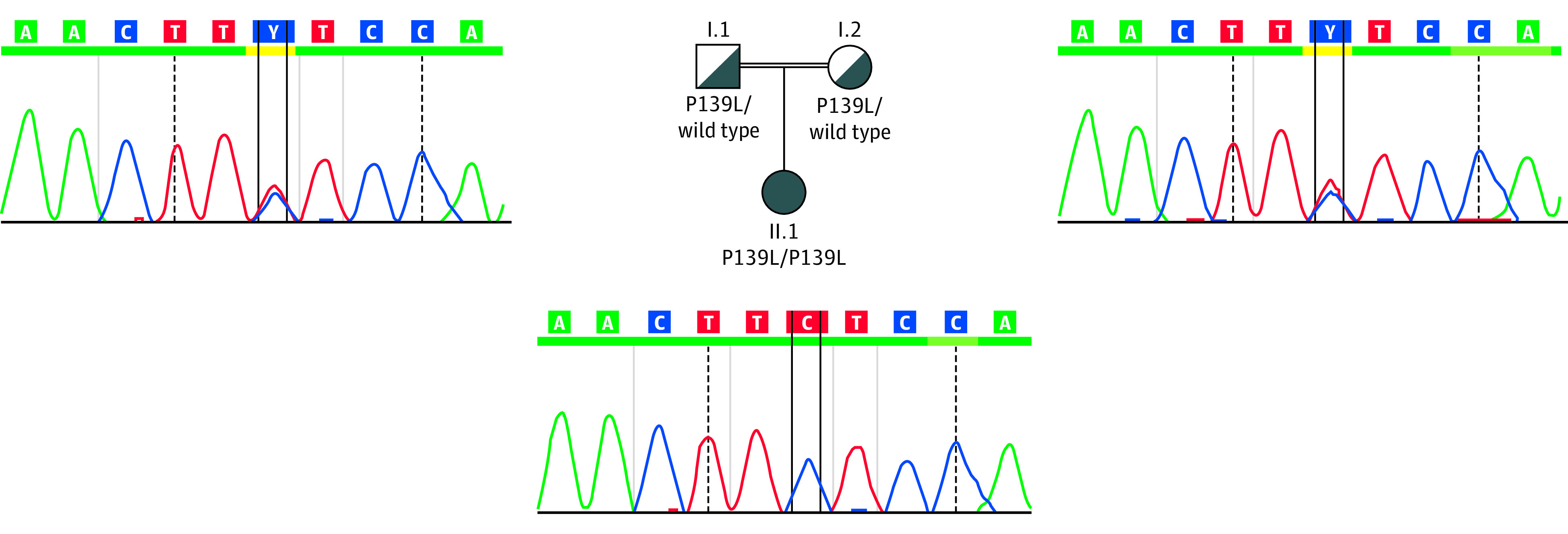
The segregation analysis confirms the recessive inheritance of the variant, which segregates with disease.
No other causative variant could be found in the 7 patients carrying the homozygous missense variant c.415T>C. For 2 patients (patients 4 and 5), whole-exome sequencing was performed without revealing any further potentially causative variants. Segregation analysis for patient 4 confirmed homozygosity of the variant (Figure 3) and, because of the heterozygous carriership in the parents, excluded a deletion in trans to this CLN5 variant. Two additional patients, among the 2086 patients screened with the next-generation sequencing, carried the CLN5 variant c.415T>C (p.Phe139Leu) in a heterozygous state, but other clearly causative variants were found (data not shown). Therefore, the heterozygous CLN5 variant was not considered to be relevant in these 2 cases.
All 7 affected patients were unrelated and were diagnosed as having an isolated macular dystrophy with normal (n = 3) or mildly reduced (n = 4) electroretinography (eTable 2 and eFigures 2, 4, 6, 8, 10, and 12 in the Supplement). The reduction in the electroretinography was noted in the light-adapted single-flash cone and 30 Hz flicker responses. The latter was in addition delayed in 3 of 7 patients (eTable 2 in the Supplement). All had symptoms of gradually progressive loss of central vision, with an onset ranging between age 16 and 42 years (median age, 34 years). All patients had specific clinical features including hypoautofluorescent maculas with retinal thinning. Mean central subfield thickness was 80 µm (eTable 1 in the Supplement; Figure 2). Mean change of the optical coherence tomography central subfield thickness over time was 10 μm (eTable 1 in the Supplement). Visual acuity ranged between 2/200 and 20/100, and 2 of the patients had a progression equal to or more than 3 lines during follow-up in 1 eye each. The other patients and eyes remained largely stable over time.
All patients had an area of macular hypoautofluorescence ranging between half of a disc diameter to 1 disc diameter, corresponding to an area of loss of the outer segment ellipsoid zone. In addition, all patients had temporally pale and excavated discs; however, intraocular pressure was within the normal limits in all patients (mean [SD] intraocular pressure, 16 [4] mm Hg) without any treatment, except in patient 3 who was diagnosed previously as having primary open-angle glaucoma. Patient 2 had keratoconus and had received intrastromal corneal rings in both eyes. Additional results including neurologic examination are given in the eResults in the Supplement.
Discussion
We describe 7 patients with a macular dystrophy associated with the homozygous c.415T>C (p.Phe139Leu) missense variant in CLN5. Interestingly, one of the patients (patient 7; eFigure 11 in the Supplement) was initially diagnosed as having type 2 macular telangiectasia, which also may show macular hypoautofluorescence, outer retinal cysts, and outer retinal hyperreflective deposits such as those seen in some of the patients in this study.
At least 2 patients were investigated further with magnetic resonance imaging, which showed mild cerebellar atrophy in 1 patient with a history of possible hypertension-related stroke and possible microvascular white-matter disease in the other patient (eResults in the Supplement). This is in contrast to a previous description of marked cerebellar and cortical atrophy in CLN5 disease.2 These results suggest that CLN5, similar to CLN7, may cause a retinal dystrophy with prominent macular involvement, as well as NCLs. Additional study is suggested, such as molecular screening for this variant in cohorts of patients with undiagnosed macular dystrophies, type 2 macular telangiectasia, and possibly also in patients with normal tension glaucoma. Furthermore, functional biological studies such as animal studies or in vitro studies, of the molecular effects of this variant, are warranted.
Limitations and Strengths
Albeit the study has several limitations such as retrospective design, limited cohort size, and lack of segregation analysis in all but one of the families, several other pieces of information strongly support that this variant is indeed disease causing. These include the absolutely symmetrical, characteristic, and homogenous clinical appearance of the condition among all patients, the lack of identification of the homozygous variant in any other affected or unaffected patients and the lack of identification of any other disease-causing genes/variants in these patients, including 2 patients with negative whole-exome sequencing. Furthermore, all patients had normal gait and none of the patients had any tremor or ataxia.
Conclusions
We conclude that this specific variant in CLN5 was associated with relatively mild neurologic findings in the context of CLN5 disease, at least until the fourth to sixth decades of life. These findings suggest a specific role of CLN5 in macular neurons, perhaps similar to MFSD8, whose protein product is thought to form part of synaptic vesicles in the photoreceptor terminals, leading to photoreceptor synaptic dysfunction when altered.7 Identification of the potential role of CLN5 in macular neurons might lead to the development of novel therapeutic strategies for macular disorders.
eMethods
eTable 1. Clinical findings in a cohort of patients with a nonsyndromic macular dystrophy
eTable 2. Full-field electroretinogram (ERG) and optical coherence tomography (OCT) findings in 7 patients with a nonsyndromic macular dystrophy and the homozygous c.415T>C (p.Phe139Leu) missense variant in CLN5
eFigure 1. Multimodal retinal imaging in Patient 1 with a nonsyndromic macular dystrophy and the homozygous c.415T>C (p.Phe139Leu) missense variant in CLN5
eFigure 2. Electroretinogram of Patient 1
eFigure 3. Multimodal retinal imaging in Patient 2 with a nonsyndromic macular dystrophy and the homozygous c.415T>C (p.Phe139Leu) missense variant in CLN5
eFigure 4. Electroretinogram of Patient 2 with the homozygous c.415T>C (p.Phe139Leu) missense variant in CLN5
eFigure 5. Color fundus image in the right eye of Patient 3 with a nonsyndromic macular dystrophy and the homozygous c.415T>C (p.Phe139Leu) missense variant in CLN5
eFigure 6. Electroretinogram of Patient 4 with the homozygous c.415T>C (p.Phe139Leu) missense variant in CLN5
eFigure 7. Multimodal retinal imaging in Patient 5 with a nonsyndromic macular dystrophy and the homozygous c.415T>C (p.Phe139Leu) missense variant in CLN5
eFigure 8. Electroretinogram of Patient 5 with the homozygous c.415T>C (p.Phe139Leu) missense variant in CLN5
eFigure 9. Multimodal retinal imaging in Patient 6 with a nonsyndromic macular dystrophy and the homozygous c.415T>C (p.Phe139Leu) missense variant in CLN5
eFigure 10. Electroretinogram of Patient 6 with the homozygous c.415T>C (p.Phe139Leu) missense variant in CLN5
eFigure 11. Multimodal retinal imaging in Patient 7 with a nonsyndromic macular dystrophy and the homozygous c.415T>C (p.Phe139Leu) missense variant in CLN5
eFigure 12. Electroretinogram of Patient 7 with the homozygous c.415T>C (p.Phe139Leu) missense variant in CLN5
eResults
eReferences
References
- 1.Mole SE, Anderson G, Band HA, et al. Clinical challenges and future therapeutic approaches for neuronal ceroid lipofuscinosis. Lancet Neurol. 2019;18(1):107-116. doi: 10.1016/S1474-4422(18)30368-5 [DOI] [PubMed] [Google Scholar]
- 2.Mancini C, Nassani S, Guo Y, et al. Adult-onset autosomal recessive ataxia associated with neuronal ceroid lipofuscinosis type 5 gene (CLN5) mutations. J Neurol. 2015;262(1):173-178. doi: 10.1007/s00415-014-7553-y [DOI] [PubMed] [Google Scholar]
- 3.Masten MC, Williams JD, Vermilion J, et al. The CLN3 Disease Staging System: a new tool for clinical research in Batten disease. Neurology. 2020;94(23):e2436-e2440. doi: 10.1212/WNL.0000000000009454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Magliyah M, AlRaddadi O, Balbaid A, Schatz P. Multimodal retinal imaging in MFSD8-neuronal ceroid lipofuscinosis. Ophthalmic Genet. 2019;40(6):588-590. doi: 10.1080/13816810.2019.1709125 [DOI] [PubMed] [Google Scholar]
- 5.Roosing S, van den Born LI, Sangermano R, et al. Mutations in MFSD8, encoding a lysosomal membrane protein, are associated with nonsyndromic autosomal recessive macular dystrophy. Ophthalmology. 2015;122(1):170-179. doi: 10.1016/j.ophtha.2014.07.040 [DOI] [PubMed] [Google Scholar]
- 6.Zare-Abdollahi D, Bushehri A, Alavi A, et al. MFSD8 gene mutations; evidence for phenotypic heterogeneity. Ophthalmic Genet. 2019;40(2):141-145. doi: 10.1080/13816810.2019.1592200 [DOI] [PubMed] [Google Scholar]
- 7.Khan KN, El-Asrag ME, Ku CA, et al. ; for NIHR BioResource-Rare Diseases and UK Inherited Retinal Disease Consortium . Specific alleles of CLN7/MFSD8, a protein that localizes to photoreceptor synaptic terminals, cause a spectrum of nonsyndromic retinal dystrophy. Invest Ophthalmol Vis Sci. 2017;58(7):2906-2914. doi: 10.1167/iovs.16-20608 [DOI] [PubMed] [Google Scholar]
- 8.Ku CA, Hull S, Arno G, et al. Detailed clinical phenotype and molecular genetic findings in CLN3-associated isolated retinal degeneration. JAMA Ophthalmol. 2017;135(7):749-760. doi: 10.1001/jamaophthalmol.2017.1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.World Medical Association . World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310(20):2191-2194. doi: 10.1001/jama.2013.281053 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eMethods
eTable 1. Clinical findings in a cohort of patients with a nonsyndromic macular dystrophy
eTable 2. Full-field electroretinogram (ERG) and optical coherence tomography (OCT) findings in 7 patients with a nonsyndromic macular dystrophy and the homozygous c.415T>C (p.Phe139Leu) missense variant in CLN5
eFigure 1. Multimodal retinal imaging in Patient 1 with a nonsyndromic macular dystrophy and the homozygous c.415T>C (p.Phe139Leu) missense variant in CLN5
eFigure 2. Electroretinogram of Patient 1
eFigure 3. Multimodal retinal imaging in Patient 2 with a nonsyndromic macular dystrophy and the homozygous c.415T>C (p.Phe139Leu) missense variant in CLN5
eFigure 4. Electroretinogram of Patient 2 with the homozygous c.415T>C (p.Phe139Leu) missense variant in CLN5
eFigure 5. Color fundus image in the right eye of Patient 3 with a nonsyndromic macular dystrophy and the homozygous c.415T>C (p.Phe139Leu) missense variant in CLN5
eFigure 6. Electroretinogram of Patient 4 with the homozygous c.415T>C (p.Phe139Leu) missense variant in CLN5
eFigure 7. Multimodal retinal imaging in Patient 5 with a nonsyndromic macular dystrophy and the homozygous c.415T>C (p.Phe139Leu) missense variant in CLN5
eFigure 8. Electroretinogram of Patient 5 with the homozygous c.415T>C (p.Phe139Leu) missense variant in CLN5
eFigure 9. Multimodal retinal imaging in Patient 6 with a nonsyndromic macular dystrophy and the homozygous c.415T>C (p.Phe139Leu) missense variant in CLN5
eFigure 10. Electroretinogram of Patient 6 with the homozygous c.415T>C (p.Phe139Leu) missense variant in CLN5
eFigure 11. Multimodal retinal imaging in Patient 7 with a nonsyndromic macular dystrophy and the homozygous c.415T>C (p.Phe139Leu) missense variant in CLN5
eFigure 12. Electroretinogram of Patient 7 with the homozygous c.415T>C (p.Phe139Leu) missense variant in CLN5
eResults
eReferences