
Figure 4.
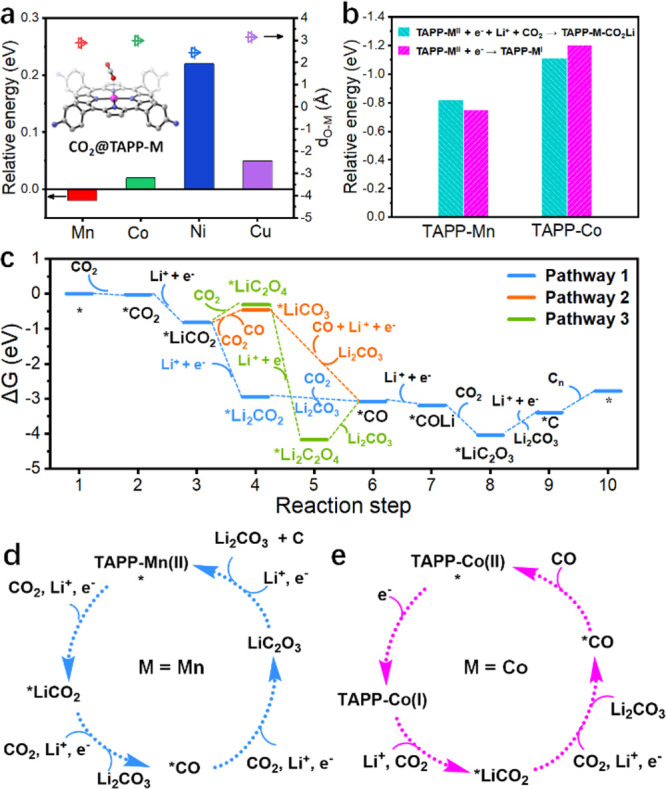
DFT calculations. (a) Energy profiles of CO2 adsorption on TAPP-M (M = Mn, Co, Ni, Cu) molecules. dO–Mn is the distance between the O atom in CO2 and the metal atom in TAPP-M. The inset is the corresponding molecular schematic illustration. (b) Energy profiles of the first electron accepted by TAPP-M (M = Mn, Co) molecules in two different pathways. (c) Energetic profiles (ΔG) for Li2CO3 nucleation and Cn formation with the three reaction pathways on the TAPP-Mn molecule at U = 0 V. (d) Four-electron pathway occurs on the TAPP-Mn site. (e) Two-electron pathway occurs on the TAPP-Co site.