

Abstract
Background: Geranylgeranylacetone (GGA) has been recently reported to be centrally active after oral administration and protect against ischemic brain injury. This study was aimed to investigate the underlying mechanism of the protective effect of GGA. Methods: In this study, transient middle cerebral artery occlusion (tMCAO) was established. Neurological score and brain water content were adopted to investigate the role of GGA in vivo. Evans-blue (EB), western blot and immunofluorescence staining of tight junction proteins were performed to evaluate blood brain barrier (BBB) permeability. Inflammation response was assessed by immunofluorescence staining of MPO and Iba-1 and quantitative real-time polymerase chain reaction (qRT-PCR) of proinflammatory cytokines. In in vitro experiment, after oxygen-glucose deprivation (OGD), transepithelial electrical resistance (TEER) and endothelial cell monolayer permeability assay were conducted to examine the effects of GGA on barrier integrity. Furthermore, heat shock protein (HSP) 70 expression was knockdown by RNA interference in bEnd.3 cells to verify the involvement of HSP70 in the action of GGA. TEER, endothelial cell monolayer permeability, CCK8 and flow cytometry were performed. Expression of caspase-3 was detected by western blot and immunofluorescence staining. Results: Our results indicated that pretreatment with a single oral GGA dose (800 mg/kg) reduced the infarct volume and prevented the neurological impairments after tMCAO. Importantly, GGA ameliorated cerebral ischemia/reperfusion (I/R) induced BBB breakdown and rescued tight junction proteins (TJPs). GGA also profoundly decreased neutrophil infiltration, inhibited glial activation and reduced the expression of proinflammatory cytokines. Consistently, GGA significantly decreased OGD-induced BBB hyper-permeability in vitro. Consistent with the previous studies, GGA promoted HSP70 induction after I/R insult. Mechanistic study showed that GGA inhibited OGD-induced apoptosis of bEnd.3 cells. Genetic inhibition of HSP70 attenuated GGA’s anti-apoptotic effect and reversed the protective effects of GGA.
Keywords: GGA, cerebral ischemia/ischemia, BBB, HSP70, anti-apoptosis
Introduction
Ischemic stroke is a prevalent destructive cerebrovascular disorder with immense socioeconomic impact [1]. Reperfusion has been proven to be the most effective therapeutic intervention against ischemic damage. However, restoration of blood supply frequently causes deterioration of brain injury, which is called reperfusion injury. BBB breakdown has been considered as the basic pathological change that occurs during cerebral I/R injury. BBB is a highly dynamic and specialized structure between the blood and brain, which regulates the entry of molecules into the brain parenchyma and maintains optimal neuronal microenvironment [2]. Disruptions of the BBB and edema formation contribute to stroke progression and neurodegeneration, leading to the severity of patient outcome [3,4]. Therefore, finding an effective drug to prevent BBB breakdown could be an efficient approach to halt ischemic stroke progression.
Cellular up-regulation of HSPs, especially HSP70, has been studied for its protective potential against cerebral I/R injury. Mounting evidence supports that HSP70 could confer neuroprotection through multiple mechanisms including anti-apoptotic effect [5]. Therefore, HSP70-overexpression strategies may hold great promise in attenuating cerebral I/R injury. Geranylgeranylacetone (GGA), is an acyclic isoprenoid compound with low toxicity [6]. Via inducing HSP70, GGA mediates cytoprotection against numerous stressors in different cells and tissues [7-9]. Moreover, the ability of GGA to penetrate the BBB makes it centrally active after oral administration [6,10]. Preponderance of evidence suggests that oral GGA administration induces HSP70 expression in the central nervous system (CNS) and mitigates acute brain ischemic damage [9,10]. However, how much of the beneficial effect of GGA may be due to HSP70 induction remains elusive. In the work presented here, we aimed to test the hypothesis that GGA may protect against cerebral I/R-induced BBB disruption through HSP70 anti-apoptosis mechanism.
Materials and methods
Experimental design
In experiment 1, 144 rats were randomly allocated to the following groups: (1) sham group, (2) vehicle + tMCAO group (vehicleischemia group), and (3) GGA + tMCAO group (GGA-ischemia group). GGA (G5048, Sigma-Aldrich) (800 mg/kg in 5% gum arabic) was given orally 48 hours before surgery. The time and dose of GGA administration were designed based on previous study [11]. Vehicle group was given an equal volume of gum arabic.
In experiment 2, bEnd.3 cells were randomly assigned to the five groups: (1) control group; (2) vehicle-OGD group; (3) GGA-OGD group; (4) scrambled siRNA-GGA-OGD group and (5) HSP70 siRNA-GGA-OGD group. Cells in scrambled siRNA-GGA-OGD group and HSP70 siRNA-GGA-OGD group were transfected with scrambled siRNA or HSP70 siRNA, respectively. 48 hours after transfection, cells were subjected to GGA administration (10 μmol/l GGA 2 h before OGD exposure) and subsequent OGD treatment.
Transient middle cerebral artery occlusion (tMCAO)
Adult Sprague-Dawley (SD) rats (260-280 g) (Cat# 10395233, Shanghai Laboratory Animal Center) were used in our study. All the procedures were approved by the Animal Research Committee of the 2nd Xiangya Hospital, Central South University, Hunan, China. Reporting results conform to the ARRIVE guidelines. As described previously, we induced tMCAO in rat models [12]. After anesthetized, a silicone-coated 4-0 suture was gently inserted into the internal cerebral artery (ICA) and advanced until the rounded tip occluded the beginning of the middle cerebral artery (MCA). After occlusion for 120 minutes, the suture was withdrawal to allow reperfusion. Sham-operated rats received the same surgery without suture insertion. The animals were sacrificed 24 h after reperfusion.
Infarct volume measurement
After being sacrificed by decapitation under intraperitoneal injection of 10% chloral hydrate anesthesia (4 ml/kg), the brain samples were sliced into coronal sections (approximately 2-mm-thick). The brain sections were then stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC) (T8877, Sigma-Aldrich) at 37°C in the dark for 15 min. Infarct size was measured using the image analysis software (Image J, NIH, MD, USA) as previously reported [13]. Infarction volume was expressed as (infarct volume/volume of contralateral hemisphere) × 100%. Laser Doppler flowmetry was used for real-time monitoring of the regional cerebral blood flow (rCBF) in the MCA territory. The success of occlusion was characterized as CBF reduction ≥80% during the ischemic period. The mortality in our study was less than 5%.
Neurobehavioral assessments
Neurobehavioral assessments were assessed at 24 h after MACO by two individuals blind to the assignment of rats and determined using the modified neurological severity score (mNSS) [14]. The NSS was graded on a scale from 1 to 18.
Brain water content assessment
After deep anesthesia, rats were sacrificed by cervical dislocation. Brain specimens were quickly removed and divided into the left (non-ischemic) and right (ischemic) hemispheres. Dissected hemispheres were weighed wet immediately and reweighed after an oven dry at 80°C overnight. The brain water content was calculated as ([wet weight - dry weight]/wet weight) × 100%.
Evans blue dye extravasation
Sample collection and measure protocol were carried out following the previous study [15]. Briefly, 2 hours before euthanasia, rats were anesthetized and received jugular vein injection of evans blue (EB) (2% in saline, 4 ml/kg). Rats were then transcardially perfused with ice-cold PBS to extract the EB. After decapitation, brains were surgically removed and dissected into right and left hemispheres. The brain tissues were then homogenized in trichloroacetic acid (1 ml). After centrifugation (21 000 g for 20 minutes), the homogenates were diluted with ethanol (1:3) and were added to a 96-well plate for fluorescence spectroscopy. Measure fluorescence intensity at 620/680 nm.
Cell cultures and OGD treatment
The mouse brain endothelial cell line (bEnd.3) cells were purchased from the American Type Culture Collection (Manassas, VA) and cultured in DMSO (Gibco Laboratories) supplemented with 10% fetal bovine serum, 1% streptomycin, and 1% penicillin [16].
For OGD treatment, cell cultures were incubated in a glucose-free medium in an anaerobic chamber (5% CO2, 95% N2) at 37°C for 6 hours.
Knockdown of HSP70 with siRNA
bEnd.3 cells were grown to 60-70% confluence on 24-well plates and then transfected with 50 nM HSP70 siRNA or scrambled control siRNA using siRNA transfection reagent (sc-29528). After 48 h of transfection, the bEnd.3 cells were harvested for the following experiments.
Transendothelial electrical resistance measurement (TEER)
A Millicell ERS-Volt-Ohm Meter was employed each time to measure TEER according to manufacturer’s protocol [17]. TEER values of the cell layers was obtained through sample TEER subtracting the background TEER values (the resistance of bank filters). TEER value of untreated monolayers was served as a blank control.
Endothelial cell monolayer permeability assay
Monolayer inserts were transferred to upper chamber Transwell inserts containing 0.4 μm pores. The upper chamber was filled with 200 μL of X-DMEM media, and the lower chamber was filled with 850 μL of media. After OGD, 1 mg/ml FITC-dextran was added to the upper culture medium at 37°C for 2 hours. Using a Chameleon microplate spectrophotoflurometer, relative fluorescence passed through the chamber (in the lower chambers) was determined (492/518 nm absorption/emission wavelengths).
Cell viability assay
The viability of bEnd.3 cells was assessed by Cell Counting Kit-8 (CCK8) (Solarbio, Beijing, China) according to the manufacturer’s protocol. bEnd.3 cells were cultured in 96-well plates and exposed to experimental conditions. After OGD insult, 10 μl CCK-8 solution was added to each culture well for 2 h at 37°C. The optical density (OD) value at the wavelength of 450 nm was recorded using a microplate reader.
Immunostaining
Anaesthetized rats were perfused with 100 mL of 4% paraformaldehyde. Brains were removed rapidly and frozen over dry ice in isopentane in the embedding media OCT. The brain cryosections (20 μm in thickness) were processed using conventional immunostaining as described in previous publications. Brain sections were incubated overnight at 4°C with primary antibodies including: occludin (1:100, ab216327, Abcam), ZO-1 (1:100, ab190085, Abcam), CD31 (1:200, ab28364, Abcam), Iba-1 (1:40, ab107159, Abcam), MPO (1:100, ab9535, Abcam). Sections were then incubated in appropriate fluorescently conjugated secondary antibodies. In in vitro experiments, after various treatments, bEnd.3 cells were fixed in 4% paraformaldehyde (PFA) for 15 minutes. To conduct immunofluorescence staining, cells were incubated anti-caspase-3 (1:1,000, Abcam) overnight at 4°C. After being washed in PBST, cells were incubated with secondary antibody: Alexa 488-conjugated (1:200, Life Technologies) for 1 hour. Cell nuclei were stained with 3,3’-diaminobenzidine for 3 min at room temperature.
Western blot analysis
In in vivo experiment, the peri-infarct brain tissues from ipsilateral cortex were used for western blotting. Tissues were homogenized in cold radioimmunoprecipitation assay buffer. In in vitro experiment, cells were lysed in cell lysis buffer within 20 min. Protein concentrations were determined with BCA assay kit (QPBCA, Thermo Scientific). Protein extracts were electrophoresed by 10% SDS-PAGE. The separated protein was then electrotransferred onto PVDF membranes and blocked with 10% skimmed milk at 37°C for 2 hours. The membranes were incubated with primary antibodies with gentle agitation at 4°C overnight. The primary antibodies were occludin (1:500, 710192, Invitrogen), ZO-1 (1:500, ab190085, Abcam), HSP70 (1:1000, 4872, Cell Signaling Technology). β-actin (1:5000, MABT825, Millipore Sigma) was used as a loading control. Afterwards, the membranes were washed and transferred into a buffer with appropriate secondary antibodies for 1 h at 37°C. An ECL detection kit was used to visualize the protein bands.
Real-time PCR (RT-PCR)
Total RNA was isolated from bEnd.3 cultures or tissue samples from the cortex ischemic penumbra. Total RNA extraction was performed using Trizol Reagent (15596018, Thermo Fisher). Reverse transcription was performed using a reverse transcription kit (4366597, Thermo Fisher). Primers are listed in Table 1.
Table 1.
Primer sequences for target genes
| Gene | Primer sequence | |
|---|---|---|
|
| ||
| Forward | Reverse | |
| IL-6 | 5’-TAGTCCTTCTACCCCAATTTCC-3’ | 5’-TTGGTCCTTAGCCACTCCTTC-3’ |
| IL-1β | 5’-GCAACTGTTCCTGAACTCAACT-3’ | 5’-ATCTTTTGGGGCGTCAACT-3’ |
| TNF-α | 5’-CCCTCACACTCAGATCATCTTCT-3’ | 5’-GCTACGACGTGGGCTACAG-3’ |
Flow cytometry
Apoptotic ratio of bEnd.3 cells was analyzed by flow cytometry using an assay kit (BD Biosciences Pharmingen) as previously described (23). Briefly, bEnd.3 cells were lysed with 0.25% trypsin without EDTA. After centrifugation (2000 rpm × 5 min), cells were resuspended in 200 ml binding buffer. Cells were then labeled with 5 ml FITC-annexin V and 5 ml propidium iodide (PI). After incubated for 10 minutes at room temperature, cells were collected for flow cytometry. Stained cells were detected on the flow cytometer. FlowJo software was used for flow cytometry analysis.
Statistical analysis
All quantitative values were presented as the mean ± standard error (SEM). Differences were evaluated using one-way ANOVA (Tukey’s multiple comparison test) or Student’s t-test (two groups). All statistical analyses were made using GraphPad Prism 7 (GraphPad Software). Values of P<0.05 was considered significant.
Results
GGA decreased infarct size and improved neurological outcome after tMCAO
Figure 1A presents representative brain slices stained with TTC 24 h after reperfusion in separated group. A characteristically large TTC-defined lesion was observed in the ipsilateral hemisphere of vehicle-treated rats, while the infarct volume was noticeably decreased by GGA prtreatment (Figure 1A, 1B). To further explore the function of GGA, neurological deficits were assessed at 24 h perfusion after MCAO. Vehicle-treated animals exhibited severe neurological deficits after tMCAO throughout the survival period compared with sham-operated rats. The neurobehavior score of rats in GGA-ischemia group declined significantly compared with vehicle-ischemia group (Figure 1C). Meanwhile, no significant region of cerebral blood flow (rCBF) changes were observed between vehicle-ischemia group and GGA-ischemia group (Figure 1D), indicating little relation between improvement of CBF and the neuroprotective effect of GGA.
Figure 1.
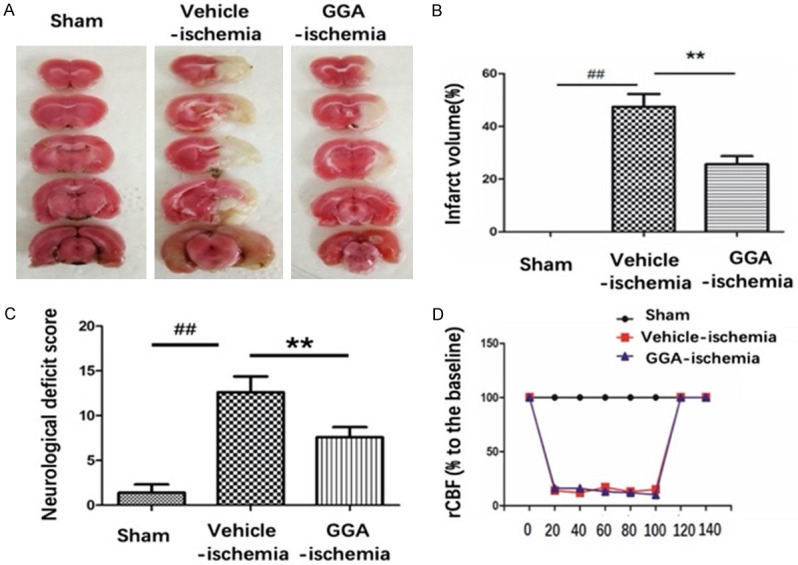
Pretreatment with single dose (800 mg/kg) oral administration of GGA reduced stroke severity at 24 h after perfusion following 2 h MCAO. A. Representative images of TTC staining. B. Brain infarct volumes were quantified (mean ± SEM, n=6). C. Neurological scores of rats in sham, GGA-ischemia and vehicle-ischemia group at 24 h perfusion after MCAO, respectively (mean ± SEM, n=6). D. Laser Doppler measurements revealed regional cerebral blood flow of different groups upon middle cerebral artery occlusion and reperfusion. #P<0.05 versus sham group; *P<0.05 versus vehicle-ischemia group; ##P<0.01 versus sham group; **P<0.01 versus vehicle-ischemia group.
GGA attenuated brain edema and alleviated BBB disruption after tMCAO
We measured brain water content of both hemispheres among groups. The water content was not significantly different between ipsilateral and contralateral hemispheres of sham group nor was it significantly different between contralateral hemisphere of rats treated with tMCAO and vehicle only. In vehicle-ischemia group, brain water content of ipsilateral hemisphere was significantly increased compared with sham group, which was markedly reduced by GGA pretreatment (Figure 2A). In line with these findings, the EB concentration of the ipsilateral hemisphere of sham group was almost close to 0. Greater EB leakage was seen in the ipsilateral hemisphere in the vehicle-ischemia group than in the sham group, indicating BBB leakage in tMCAO-treated rats. The volume of EB extravasation within ischemic brain parenchyma was dramatically lower in GGA-ischemia group compared with vehicle-ischemia group, which suggested that GGA treatment reduced cerebral I/R-induced BBB hyper-permeability (Figure 2B, 2C).
Figure 2.

GGA attenuated tMCAO-induced disruption of BBB and TJPs degradation and disorganization. A. Brain water content in left side and right side of brains in sham-operated rats, vehicle-ischemia rats and GGA-pretreated ischemic rats, respectively (mean ± SEM, n=5). B. Quantitative analysis of EB extravasation (mean ± SEM, n=5). C. Representative immunoblots depicting protein expression levels of occludin and ZO-1 in the cerebral cortex from rats of the sham, vehicle-ischemia and GGA-ischemia groups, respectively. D, E. Bar graphs represent the densitometry analysis of immunoblots (mean ± SEM, n=5). F. Representative fluorescence photographs of CD31 (green)/occludin (red) at 24 h after reperfusion following MCAO. G. Representative fluorescence photographs of CD31 (green)/ZO-1 (red) at 24 h after reperfusion following MCAO. Scale bars =20 μm. ##P<0.05 compared with sham group; *P<0.05 compared with vehicle-ischemia group; **P<0.01 versus vehicle-ischemia group
Western blot was adopted to assess the protein expression of tight junction proteins (TJPs), occludin and ZO-1. Our results revealed that occludin and ZO-1 protein expressions were markedly decreased in vehicle-ischemia group compared with sham group, while GGA-pretreatment significantly rescued the TJPs loss (Figure 2D, 2E). To further evaluate the effect of GGA on the TJPs rearrangement, occludin/CD31 double staining was conducted at 24 h reperfusion after tMCAO. Occludin positive staining was observed continuously located on the margin of endothelial cells (ECs) in sham group, while in vehicle-ischemia group, occludin signal was reduced with the continuity being disrupted. GGA pretreatment greatly reversed the changes (Figure 2F). Similarly, GGA pretreatment attenuated ZO-1 discontinuity on ECs (Figure 2G).
GGA inhibited inflammatory response after cerebral I/R
BBB breakdown and the subsequent neuroinflammatory response after BBB breakdown are critical determinants of stroke progression. Therefore, we examined infiltrating neutrophils and activated microglia through anti-MPO and anti-Iba-1 immunofluorescence staining, respectively. Results revealed that few MPO+ cells were detectable in sham group. Rats in vehicle-ischemia group showed a higher number of MPO+ cell infiltration compared with that in sham group. GGA pretreatment notably inhibited I/R-induced increase of MPO+ cell number (Figure 3A, 3B). Similarly, there were more Iba-1 positive cells in penumbra at 24 hours after tMCAO compared with sham group, which was markedly abolished by GGA pretreatment (Figure 3C, 3D). Activated neutrophils and phagocytes in turn result in the production of proinflammatory cytokines. Therefore, we further evaluated pro-inflammatory cytokines expressions (TNFα, IL-1β and IL-6) using RT-PCR. We observed that notable increase of these cytokines in vehicle-ischemia group compared with sham group, which was inhibited by GGA (Figure 3E-G). Collectively, these results illustrated that GGA significantly attenuated neuroinflammation following cerebral I/R insult.
Figure 3.
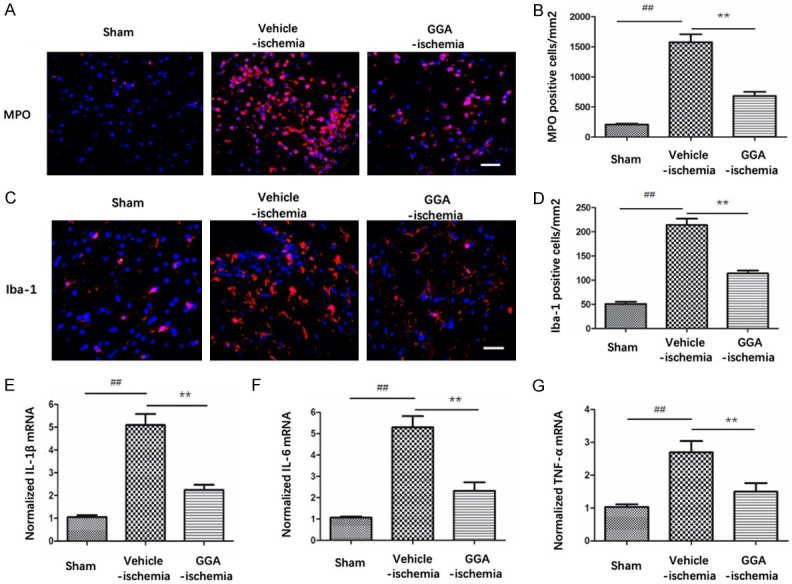
GGA reduced cerebral I/R-induced neuroinflammation. A. Representative immunofluorescence images of infiltrated neutrophils (MPO+ cells), counterstained with DAPI in ipsilateral cortex peri-ischemic area 24 h after perfusion following MCAO. B. Quantification of MPO-positive cells in the ischemic border (mean ± SEM, n=5). C. Representative photographs of Iba-1 immunofluorescence staining in ipsilateral cortex peri-ischemic area from sham, vehicle-ischemia and GGA-ischemia groups 24 h after reperfusion. D. Quantification of Iba-1-positive cells in the ischemic border of ipsilateral cortex (mean ± SEM, n=5). Scale bar 20 μm. E-G. Bar graph shows the mRNA levels of TNF-α, IL-1β and IL-6 in ipsilateral cortex 24 h after reperfusion following MCAO. Data were expressed as average copies per copy of GAPDH, and normalized to sham group (mean ± SEM, n=5). ##P<0.01 versus sham group; *P<0.05 versus vehicle-ischemia group; **P<0.01 versus vehicle-ischemia group.
GGA reverted OGD-induced barrier leakage in bEnd.3 cells
Then, we investigated the involvement of GGA in BBB permeability in an in vitro BBB model. The TEER was used to evaluate the barrier function of bEnd.3 monolayer. After 7 days of culture, bEnd.3 monolayer showed high TEER, which indicated that the tight junctions (TJ) between endothelial cells were formed, thus creating an intact barrier (Figure 4A). We subsequently investigated the effect of GGA on paracellular permeability of bEnd.3 monolayer under OGD condition. Compared with control group, 6 h OGD caused a drastic reduction of TEER value compared with control group, indicating OGD condition increased the permeability of endothelial monolayers. Pretreatment with GGA, but not vehicle, significantly increased TEER value compared with vehicle-OGD group (Figure 4B). Correspondingly, 6 h OGD noticeably increased the permeability of FITC dextran across bEND3 monolayers, while GGA pretreatment significantly abrogated OGD-induced barrier hyper-permeability (Figure 4C). Collectively, these results indicated GGA’s capacity to preserve BBB function under ischemic conditions in vitro.
Figure 4.
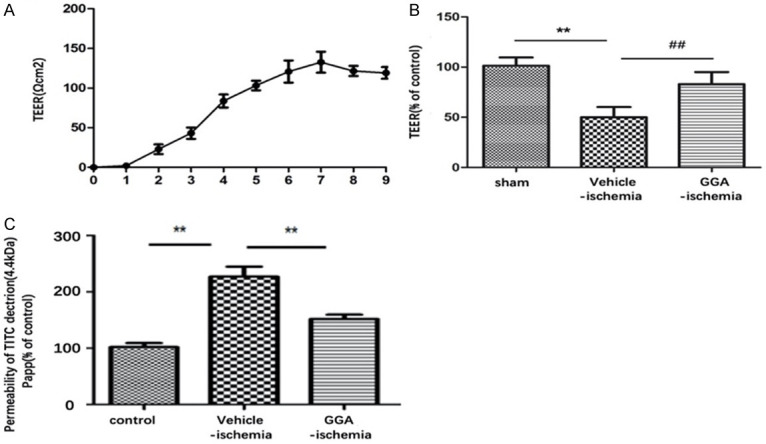
GGA protected OGD-induced disruption of in vitro BBB integrity. A. TEER value of bEnd.3 was measured at indicated days. B. TEER assay of mono-culture in different groups (mean ± SEM, n=5). C. The permeability of fluorescein isothiocyanate (FITC)-dextran across bEnd.3 (mean ± SEM, n=5). ##P<0.05 compared with sham group; *P<0.05 compared with indicated group; **P<0.01 versus indicated group group.
GGA induced HSP70 expression after ischemic injury in vivo and in vitro
To evaluate the effect of GGA on HSP70 expression during tMCAO, western blot was performed. HSPB70 protein expressions by western blot are shown in Figure 5A, 5B. The striking increase of HSP70 expression in ischemic penumbra was observed in vehicle-ischemia group compared with sham group, which was even higher in GGA-ischemia group. However, almost equal expressions of HSP70 were found in the three groups. In vitro, pretreatment with GGA also prominently enhanced HSP70 induction in cultured bEnd.3 caused by OGD (Figure 5C, 5D). The results strongly suggested that GGA inhanced HSP70 induction after tMCAO both in vivo and in vitro.
Figure 5.
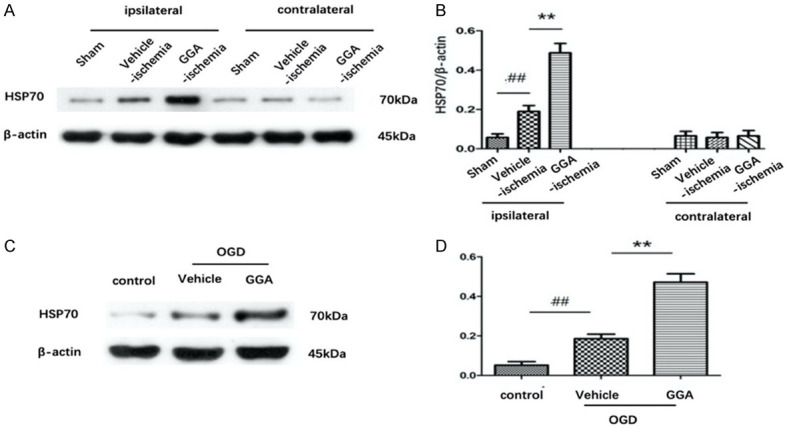
GGA upregulated heat-shock protein 70 (HSP70) expression in vivo and in vitro. A. Representative immunoblots depicting HSP70 protein levels. B. The band intensity of HSP70 was determined, corrected to that of actin (mean ± SEM, n=5). C, D. Representative western blots showing the protein level of HSP70 in bEnd.3 cells of control, OGD+ vehicle and OGD+GGA groups. The band intensities were assessed through scanning densitometry (mean ± SEM, n=3). #P<0.05 versus sham group; *P<0.05 versus vehicle-ischemia group.
HSPB8 restored BBB integrity partially via a HSP70-mediated antiapoptotic mechanism
To clarify the role of HSP70 in GGA-mediated protection against ischemic BBB injury, we adopted in vitro experimental paradigms. bEnd.3 cells were first transfected with HSP70 siRNA or scramble siRNA for 48 h and then treated with GGA before OGD insult. As shown in Figure 6A, 6B, protein expression of HSP70 was clearly suppressed by HSP70 siRNA-transfection. Silencing HSP70 significantly reversed the effect of GGA on barrier function (Figure 6C, 6D). Cell Counting Kit-8 (CCK-8) assay was performed to measure cell viability. Compared with control group, OGD treatment reduced cell viability in the medium significantly, while GGA pretreatment obviously restored the cell viability. However, HSP70 siRNA, but not the scramble-siRNA, abolished the GGA’s protective effect against OGR injury significantly (Figure 7A).
Figure 6.
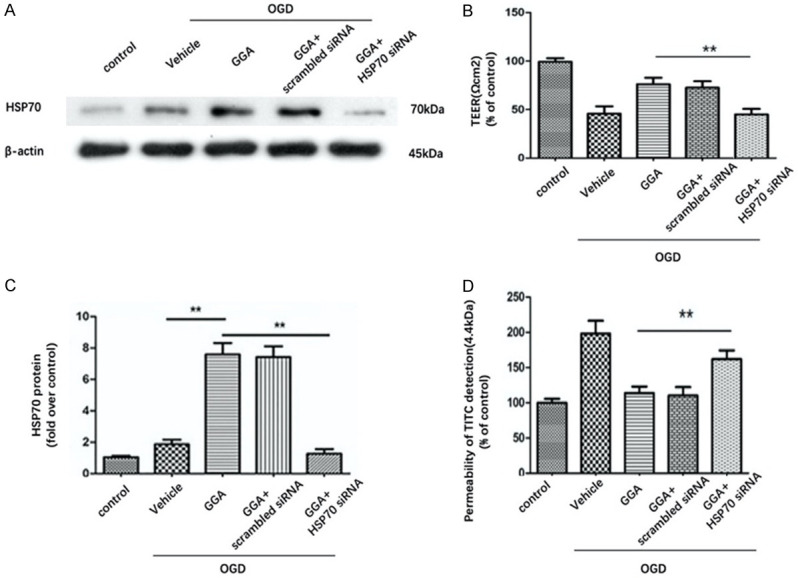
HSP70 suppression abolished the effect of GGA on BBB integrity. A. Representative western blot showing the levels of HSP70 in control, OGD+ vehicle, OGD+GGA, OGD+GGA+ scramble siRNA and OGD+GGA+HSP70 siRNA groups. B. Densitometric analyses of HSP70 expression (mean ± SEM, n=3). C. TEER assay to evaluate the protective effect of GGA against OGD-induced disruption of barrier function in different groups (mean ± SEM, n=3). D. The paracellular diffusion assay was performed 6 h after OGD treatment (mean ± SEM, n=3).
Figure 7.

HSP70 suppression abolished the effect of GGA on endothelial apoptosis. A. Cell viability was detected with CCK-8 (mean ± SEM, n=3). B. The apoptotic cell death was detected by flow cytometry. The cell distributions in flow cytometric histograms are as follow: Lower left quadrants: viable cells. Upper left quadrants: necrotic cells. Upper right quadrants: late apoptotic cells. Lower right quadrants: early apoptotic cells. C. Quantitative analysis of apoptotic rate (mean ± SEM, n=3). *P<0.05 compared with indicated group; **P<0.01 versus indicated group.
The apoptosis of bEnd.3 was examined via flow cytometry in different groups. The results showed that OGD significantly increased both early and late apoptotic cell death, which were attenuated by GGA. Consistently, co-treatment of HSP70 siRNA, but not scramble siRNA, impaired the anti-apoptotic effect of GGA on bEnd.3 apoptosis after OGD (Figure 7B, 7C). We also used cleaved caspase-3 to determine apoptosis. As shown in Figure 8A, 8B, protein expression of cleaved caspase-3 was drastically increased in bEnd.3 exposed to OGD. GGA pretreatment significantly reduced the level of cleaved caspase-3. We also observed significant increase in caspase-3-positive cells in OGD-vehicle group compared with control group. During OGD, co-culture with GGA reduced the number of caspase-3+ cell, whereas HSP70 depletion abrogated this effect (Figure 8C). Collectively, these data suggested that GGA reversed the BBB compromise after I/R injury through HSP70’s anti-apoptosis action.
Figure 8.
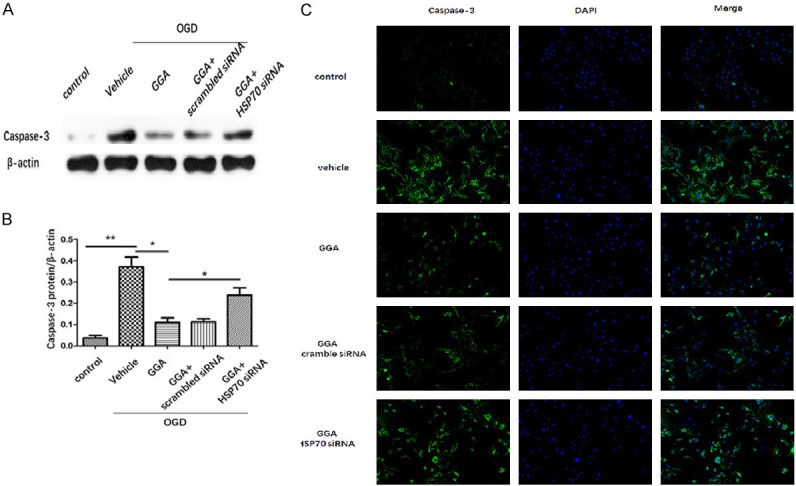
HSP70 suppression abrogated GGA’s effect on caspase-3. A, B. Representative protein bands and quantified evaluation of cleavedcaspase-3 (mean ± SEM, n=3). C. Representative microphotographs of immunofluorescence staining of caspase-3. Caspase-3 (green) and the nuclei (DAPI, blue). *P<0.05 compared with indicated group; **P<0.01 versus indicated group.
Discussion
In the present study, we used the well-established MCAO/R model to investigate the neuroprotective role of GGA. We demonstrated that GGA afforded robust protective effects against cerebral I/R injury, consistent with the previous reports. Specifically, pretreatment with 800 mg/kg of GGA orally significantly reduced infarct volume, improved behavioral dysfunction and attenuated cerebral edema in a tMCAO-induced brain injury rat model. Mounting evidence has shown that post-ischemic cerebral edema is related to the increased BBB permeability [18]. Consistently, our results showed that the extravasation of EB in the vehicle-ischemia group was markedly higher compared with sham group, while GGA significantly reduced tMCAO-induced EB leakage, indicating that GGA prevented I/R induced BBB integrity loss. In an in vitro BBB model, we also demonstrated that GGA significantly adverted OGD-induced barrier leakage in bEnd.3 cell monolayer.
BBB is the pivotal barrier for central nervous system. When BBB is dirupted, blood cells leak into brain, which leads to the recruitment of inflammatory cells in the CNS, and consequently results in neuronal injury and neuron death [19,20]. The barrier is primarily formed by specialized brain ECs. TJs between adjacent ECs generate a rate-limiting restrictive barrier that prevents bloodborne substances from entering the brain [21,22]. Occludin is one of the predominant TJ proteins, which interacts with cytoskeletal scaffolding proteins such as ZO-1, contributing to paracellular “tightness” between adjacent BBB endothelial cells [23,24]. Therefore, in the study described herein, we focused on these two proteins. Our study prevailed that tMCAO resulted in severe loss of occludin and ZO-1 and disrupted their continuous, linear distribution in the vessels. GGA pretreatment obviously protected against these proteins reduction and disorganization. This suggests that GGA protected against I/R induced BBB disruption is associated with an effective improvement in TJ associated proteins.
Following I/R injury, activated microglia and infiltrating leukocytes release proinflammatory mediators including TNF-α, IL-1β and IL-6. These cytokines generally hold responsibility for the destructive opening of BBB following stroke. As a result, various types of inflammatory cells are allowed to migrate across BBB to the ischemic region, which further secrete cytokines and activate glial cells [25]. Amplification of the inflammatory response finally leads to the disruption of BBB [26]. In our present study, we found that GGA markedly reduced neutrophil infiltration and microglia activation, as well as decreased pro-inflammatory cytokine production. Altogether GGA treatment attenuated neuroinflammation.
Previous studies have revealed that GGA exerted neuroprotection against focal ischemia and the neuroprotective effect of GGA may due to its amplification of HSP70 expression [27,28]. In good agreement with the previous studies, the present study showed that pretreatment with a single oral GGA dose (800 mg/kg) induced HSP70 expression in ipsilateral hemisphere after 24 h reperfusion post-MCAO without inducement of HSP70 in contralateral hemisphere. In all published reports, HSP70 induction by GGA was observed in the disease models but not in control rats. It could thus be interpreted that GGA may enhance the stressor-induced HSP expression in the brain. Recently, HSP70 has been demonstrated to exert anti-apoptotic activity [29,30]. A previous study has revealed that HSP70 silencing promoted apoptosis in hypoxia-reoxygenation cell model [31]. Therefore, we presumed that GGA may provide protection against cerebral I/R-induced BBB breakdown through the anti-apoptotic action of HSP70. Using an in vitro BBB model, we indicated that genetic silence of HSP70 significantly abrogated the effect of GGA on the endothelial permeability. Moreover, GGA suppressed the endothelial apoptosis induced by OGD, which reversed by HSP70 siRNA. These results suggested that the anti-apoptosis action in bEnd.3 of GGA was HSP70 dependent.
Conclusion
The results from our study supported the idea that GGA efficiently exerted a protective effect on BBB integrity during cerebral I/R. GGA protected against BBB destruction via HSP70 anti-apoptosis effect. In conclusion, GGA, as an apoptosis suppressor, may be a promising pharmacotherapy for cerebral I/R injury.
Acknowledgements
This work was supported by Natural Science Foundation of Hunan Province (2018JJ3749) and the National Natural Science Foundation of China (No. 81801205).
Disclosure of conflict of interest
None.
References
- 1.Feigin VL, Norrving B, George MG, Foltz JL, Roth GA, Mensah GA. Prevention of stroke: a strategic global imperative. Nat Rev Neurol. 2016;12:501–512. doi: 10.1038/nrneurol.2016.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 3.Guell K, Bix GJ. Brain endothelial cell specific integrins and ischemic stroke. Expert Rev Neurother. 2014;14:1287–1292. doi: 10.1586/14737175.2014.964210. [DOI] [PubMed] [Google Scholar]
- 4.Brouns R, Wauters A, De Surgeloose D, Mariën P, De Deyn PP. Biochemical markers for blood-brain barrier dysfunction in acute ischemic stroke correlate with evolution and outcome. Eur Neurol. 2011;65:23–31. doi: 10.1159/000321965. [DOI] [PubMed] [Google Scholar]
- 5.del Zoppo GJ, Sharp FR, Heiss WD, Albers GW. Heterogeneity in the penumbra. J Cereb Blood Flow Metab. 2011;31:1836–1851. doi: 10.1038/jcbfm.2011.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sinn DI, Chu K, Lee ST, Song EC, Jung KH, Kim EH, Park DK, Kang KM, Kim M, Roh JK. Pharmacological induction of heat shock protein exerts neuroprotective effects in experimental intracerebral hemorrhage. Brain Res. 2007;1135:167–76. doi: 10.1016/j.brainres.2006.11.098. [DOI] [PubMed] [Google Scholar]
- 7.Wang X, Yuan B, Dong W, Yang B, Yang Y, Lin X, Gong G. Induction of heat-shock protein 70 expression by geranylgeranylacetone shows cytoprotective effects in cardiomyocytes of mice under humid heat stress. PLoS One. 2014;9:e93536. doi: 10.1371/journal.pone.0093536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ikeyama S, Kusumoto K, Miyake H, Rokutan K, Tashiro S. A non-toxic heat shock protein 70 inducer, geranylgeranylacetone, suppresses apoptosis of cultured rat hepatocytes caused by hydrogen peroxide and ethanol. J Hepatol. 2001;35:53–61. doi: 10.1016/s0168-8278(01)00053-8. [DOI] [PubMed] [Google Scholar]
- 9.Zhao Z, Faden AI, Loane DJ, Lipinski MM, Sabirzhanov B, Stoica BA. Neuroprotective effects of geranylgeranylacetone in experimental traumatic brain injury. J Cereb Blood Flow Metab. 2013;33:1897–908. doi: 10.1038/jcbfm.2013.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fujiki M, Kobayashi H, Abe T, Ishii K. Astroglial activation accompanies heat shock protein upregulation in rat brain following single oral dose of geranylgeranylacetone. Brain Res. 2003;991:254–257. doi: 10.1016/s0006-8993(03)03540-6. [DOI] [PubMed] [Google Scholar]
- 11.Fujiki M, Hikawa T, Abe T, Uchida S, Morishige M, Sugita K, Kobayashi H. Role of protein kinase C in neuroprotective effect of geranylgeranylacetone, a noninvasive inducing agent of heat shock protein, on delayed neuronal death caused by transient ischemia in rats. J Neurotrauma. 2006;23:1164–1178. doi: 10.1089/neu.2006.23.1164. [DOI] [PubMed] [Google Scholar]
- 12.Yan BC, Park JH, Shin BN, Ahn JH, Kim IH, Lee JC, Yoo KY, Hwang IK, Choi JH, Park JH, Lee YL, Suh HW, Jun JG, Kwon YG, Kim YM, Kwon SH, Her S, Kim JS, Hyun BH, Kim CK, Cho JH, Lee CH, Won MH. Neuroprotective effect of a new synthetic aspirin-decursinol adduct in experimental animal models of ischemic stroke. PLoS One. 2013;8:e74886. doi: 10.1371/journal.pone.0074886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang J, Li Y, Tang Y, Tang G, Yang GY, Wang Y. CXCR4 antagonist AMD3100 protects blood-brain barrier integrity and reduces inflammatory response after focal ischemia in mice. Stroke. 2013;44:190–197. doi: 10.1161/STROKEAHA.112.670299. [DOI] [PubMed] [Google Scholar]
- 14.Zheng J, Shi L, Liang F, Xu W, Li T, Gao L, Sun Z, Yu J, Zhang J. Sirt3 ameliorates oxidative stress and mitochondrial dysfunction after intracerebral hemorrhage in diabetic rats. Front Neurosci. 2018;12:414. doi: 10.3389/fnins.2018.00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujimoto M, Takagi Y, Aoki T, Hayase M, Marumo T, Gomi M, Nishimura M, Kataoka H, Hashimoto N, Nozaki K. Tissue inhibitor of metalloproteinases protect blood-brain barrier disruption in focal cerebral ischemia. J Cereb Blood Flow Metab. 2008;2859:1674–1685. doi: 10.1038/jcbfm.2008.59. [DOI] [PubMed] [Google Scholar]
- 16.Omidi Y, Campbell L, Barar J, Connell D, Akhtar S, Gumbleton M. Evaluation of the immortalised mouse brain capillary endothelial cell line, b. End3, as an in vitro blood-brain barrier model for drug uptake and transport studies. Brain Res. 2003;990:95–112. doi: 10.1016/s0006-8993(03)03443-7. [DOI] [PubMed] [Google Scholar]
- 17.Cao GS, Chen HL, Zhang YY, Li F, Liu CH, Xiang X, Qi J, Chai CZ, Kou JP, Yu BY. YiQiFuMai powder injection ameliorates the oxygen-glucose deprivation-induced brain microvascular endothelial barrier dysfunction associated with the NF-κB and ROCK1/MLC signaling pathways. J Ethnopharmacol. 2016;183:18–28. doi: 10.1016/j.jep.2016.02.028. [DOI] [PubMed] [Google Scholar]
- 18.Haley MJ, Lawrence CB. The blood-brain barrier after stroke: structural studies and the role of transcytotic vesicles. J Cereb Blood Flow Metab. 2017;37:456–470. doi: 10.1177/0271678X16629976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Borlongan CV, Glover LE, Sanberg PR, Hess DC. Permeating the blood brain barrier and abrogating the inflammation in stroke: implications for stroke therapy. Curr Pharm Des. 2012;18:3670–3676. doi: 10.2174/138161212802002841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mills JH, Alabanza L, Weksler BB, Couraud PO, Romero IA, Bynoe MS. Human brain endothelial cells are responsive to adenosine receptor activation. Purinergic Signal. 2011;7:265–73. doi: 10.1007/s11302-011-9222-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bazzoni G. Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev. 2004;84:869–901. doi: 10.1152/physrev.00035.2003. [DOI] [PubMed] [Google Scholar]
- 22.Sandoval KE, Witt KA. Blood-brain barrier tight junction permeability and ischemicstroke. Neurobiol Dis. 2008;32:200–219. doi: 10.1016/j.nbd.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 23.Morita K, Sasaki H, Furuse M, Tsukita S. Endothelial claudin: claudin-5/TMVCF constitutes tight junction strands in endothelial cells. J Cell Biol. 1999;147:185–194. doi: 10.1083/jcb.147.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luissint AC, Federici C, Guillonneau F, Chrétien F, Camoin L, Glacial F, Ganeshamoorthy K, Couraud PO. Guanine nucleotide-binding protein Gαi2: a new partner of claudin-5 that regulates tight junction integrity in human brain endothelial cells. J Cereb Blood Flow Metab. 2012;32:860–873. doi: 10.1038/jcbfm.2011.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stanimirovic D, Satoh K. Inflammatory mediators of cerebral endothelium: a role in ischemic brain inflammation. Brain Pathol. 2006;10:113–126. doi: 10.1111/j.1750-3639.2000.tb00248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu T, Zhang T, Yu H, Shen H, Xia W. Adjudin protects against cerebral ischemia reperfusion injury by inhibition of neuroinflammation and blood-brain barrier disruption. J Neuroinflammation. 2014;11:107. doi: 10.1186/1742-2094-11-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yasuda H, Shichinohe H, Kuroda S, Ishikawa T, Iwasaki Y. Neuroprotective effect of a heat shock protein inducer, geranylgeranylacetone in permanent focal cerebral ischemia. Brain Res. 2005;1032:176–182. doi: 10.1016/j.brainres.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 28.Fujiki M, Kobayashi H, Inoue R, Tatsuya A, Ishii K. Single oral dose of geranylgeranylacetone for protection against delayed neuronal death induced by transient ischemia. Brain Res. 2004;1020:210–213. doi: 10.1016/j.brainres.2004.06.031. [DOI] [PubMed] [Google Scholar]
- 29.Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62:670–84. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ravagnan L, Gurbuxani S, Susin SA, Maisse C, Daugas E, Zamzami N, Mak T, Jäättelä M, Penninger JM, Garrido C, Kroemer G. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat Cell Biol. 2001;3:839–43. doi: 10.1038/ncb0901-839. [DOI] [PubMed] [Google Scholar]
- 31.Zhai C, Lv J, Wang K, Li Q, Qu Y. HSP70 silencing aggravates apoptosis induced by hypoxia/reoxygenation in vitro. Exp Ther Med. 2019:1013–1020. doi: 10.3892/etm.2019.7697. [DOI] [PMC free article] [PubMed] [Google Scholar]